
Abstract
A dinickel complex-enabled, enantioselective reductive alkenylation reaction of N-Ts imines with vinyl chlorides is reported. This method is effective with a wide array of (hetero)aryl and aliphatic imines bearing diverse functional groups. Under mildly reductive conditions, synthetically valuable allylic amines are obtained in good yields with excellent enantioselectivities. Furthermore, this catalytic system can be extended to aldehydes, offering an efficient route to access chiral allylic alcohols from readily available starting materials. The potential applications of this method are demonstrated by its use in the late-stage functionalization of biologically active molecular derivatives and the manipulation of reaction products. Initial mechanistic investigations and DFT calculation lend support to a catalytic addition pathway.
Similar content being viewed by others
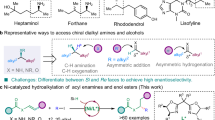
Introduction
α-Chiral amines are ubiquitous in natural products, pharmaceuticals, and other biologically active molecules, making their enantioselective synthesis a critical and enduring pursuit in synthetic chemistry (Fig. 1a)1,2,3,4,5,6. In fact, nearly 40% of the top 200 best-selling small molecule drugs in 2023 contain α-stereogenic amines7. Among the diverse methodologies developed for α-chiral amine synthesis, the direct enantioselective addition of imines with carbon nucleophiles stands out as a particularly direct and efficient approach (Fig. 1b). A broad spectrum of carbon nucleophiles, such as organo-magnesium8,9,10, -zinc11,12,13,-aluminum14 and -boron15,16,17,18,19, has been successfully employed in these addition reactions, facilitated by various chiral catalysts. However, the inherent challenges associated with the preparation and handling of these air- and moisture-sensitive organometallic reagents underscore the need for alternative strategies. One such strategy involves the use of readily available, bench-stable carbon electrophiles in direct addition reactions with imines under reductive conditions20,21. This approach not only mitigates the practical limitations of organometallic reagents but also offers a more step-economic pathway, given that many organometallic reagents are synthesized from carbon electrophiles.

a Biologically active α-chiral amines. b Comparison between classic organometallic addition and reductive addition to imines. c Specific imines have been successfully applied in reductive addition reactions and the challenges of simple N-protected imines. d Dinickel-catalyzed reductive addition of N-Ts imines with vinyl chlorides (this work).
Asymmetric reductive carbonyl addition reactions with various carbon electrophiles have been extensively studied and notable advancements have been achieved through various transition-metal catalysis, such as chromium (known as the Nozaki–Hiyama–Kishi (NHK) reaction)22,23,24,25,26, nickel27,28,29,30,31,32, and cobalt33,34,35,36,37,38. In contrast, the exploration of analogous reductive addition reactions of imines remains limited (Fig. 1c). For instance, in 2023, the Zhou group innovatively reported a nickel-catalyzed enantioselective reductive addition of aldimines with a wide range of aryl halides and sulfonates, utilizing the chelating properties of an azaaryl protecting group, which is not readily deprotected39. Concurrently, the Shi group disclosed a chiral cobalt-bisphosphine catalyst enabled the asymmetric reductive (hetero)arylation of cyclic N-sulfonyl imines with aryl halides, though this method was restricted to cyclic imine substrates40. The Chen group further contributed by reporting a cobalt-catalyzed enantioselective reductive vinyl addition of α-imino esters with alkenyl halides, leading to the formation of enantioenriched α-vinylic amino acids featuring quaternary carbon centers41,42,43,44. Despite these efforts, transition-metal-catalyzed aza-NHK-type reactions, particularly those involving simple acyclic N-sulfonyl imines, remain underexplored due to several challenges: 1) Limited bonding-site of imine which can’t form chelation interaction with catalyst causes difficulty for stereoinduction; 2) Simple imines are unable to form favorable 6-membered cyclic transition states; 3) Aliphatic imines easily undergo isomerization to unreactive enamines.
Our previous work demonstrated that a dinickel complex45,46,47 could catalyze the enantioselective α-alkylation of acyclic ketones using unactivated alkyl iodides48. Mechanistic investigations revealed that the low-valent organonickel species generated in this system exhibited significant nucleophilicity, consistent with other findings27,28,29,30,31,32,49,50,51. Motivated by these observations, we hypothesized that this dinickel complex could also facilitate the reductive addition of imines with vinyl halides (Fig. 1d). Success in this approach would enable the efficient synthesis of enantiomerically enriched allylic amines52,53,54,55,56, valuable intermediates in organic synthesis.
Results
Optimization reaction conditions
We initiated our investigation by examining the reductive alkenyl addition of N-tosyl imine 1a with styrenyl chloride 2a. The reaction was conducted in the presence of NiBr2·glyme as the catalyst, zinc dust as the reductant, and sodium iodide (NaI) as an additive, using a selection of bimetallic naphthyridine-bis(oxazoline) (napbox) ligands46,57 in tetrahydrofuran (THF) (Table 1, entries 1−3). Initial screening identified several promising ligands (L1−L3) that facilitated the reaction with moderate yields and high enantioselectivities. Among these, napbox L1 provided the highest enantioselectivity (entry 1). Extending the reaction time to 48 h increased the yield to 79% (entry 4). However, employing Mn instead of Zn as the reductant resulted in a slight reduction in both yield and enantioselectivity (entry 5). Notably, increasing the reaction concentration further improved the yield to 91% without affecting enantioselectivity (entry 6). Control experiments showed the significant role of NaI in the reaction to receive a high yield (entry 7). Despite establishing optimal conditions, we also evaluated several monomeric ligands commonly used in combination with Ni catalysts (entries 8−14). The Box ligand L4 and BiOx ligand L5 were ineffective in this transformation (entries 8−9). Additionally, Pyox L7, Quinox L8, Napox L9, and Pybox L10, which possessed the same oxazoline ring substitutions as L1, produced the addition products with markedly lower efficiencies (entries 11−14). These results underscored the superior performance of bimetallic ligands in this transformation.
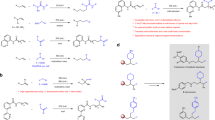
Substrate scope
Under the optimized reaction conditions, we explored the substrate scope of this transformation, as summarized in Fig. 2. Initially, imines bearing various sulfonyl protecting groups underwent reductive vinylic addition with high yields, though the choice of protecting group notably influenced enantioselectivity (4−8). Subsequently, a range of imines, including both aromatic and aliphatic variants, were subjected to the reaction (9−33). Aromatic imines with substituted phenyl rings generally provided the corresponding allylic amines in good yields (9−19), though ortho-substitution led to reduced yield and enantioselectivity (13). Imine bearing a strongly electron-deficient substituent showed lower conversion, albeit with preserved enantioselectivity (19). Additionally, naphthyl and heteroaryl imines proved to be compatible with the reaction conditions, affording chiral amines in moderate to high yields and excellent enantioselectivities (20−25). The reaction also demonstrated broad tolerance toward aliphatic imines, which were typically challenging due to undesired deprotonation when using organometallic reagents, yielding allylic amines in high yields with remarkable enantioselectivities (26−33). Finally, the scope of vinyl chlorides was examined (34−43). Various trans-styrenyl chlorides with different substitutions on the phenyl ring reacted efficiently, regardless of the substitution patterns (ortho, meta, or para) and electronic properties (electron-withdrawing or -donating). Notably, aryl bromide, which offered further functionalization opportunities, remained intact (38). However, cis-styrenyl chloride and alkyl-substituted vinyl chloride were unsuitable substrates for this transformation (see Supplementary Information (SI) for details).

a Scope of different sulfonyl protecting groups. b Scope of aryl imines. c Scope of aliphatic imines. d Scope of vinyl chlorides. aReaction condition as shown in Table 1. bZnBr2 (20 mol%) was added. cImine 1 (1.5 equiv), vinyl chloride 2 (0.2 mmol) was reacted in THF (0.2 M) at 40 °C for 48 h. d40 °C, 48 h. eZnBr2 (1.0 equiv) was added, L3 as ligand, in THF (0.1 M) at 25 °C for 24 h. f40 °C for 24 h. TBDPS t-butyl-diphenylsilyl.
Furthermore, the dinickel catalyst also proved effective in the reductive vinylic addition of aldehydes, which were less electrophilic compared to N-Ts imines. Recently, both the Meng36 and Shi29 groups independently reported enantioselective reductive alkenylation of aldehydes using chiral Co and Ni catalysts, respectively. However, in the Ni-catalyzed system, only moderate enantioselectivity (71:29 e.r.) was achieved with trans-styrenyl halides when using a monomeric ligand. In contrast, employing the bimetallic ligand (see SI for the optimization) resulted in significantly enhanced enantioselectivity with this type of substrates (Fig. 3). A diverse range of substituted benzaldehydes (44−58), irrespective of the electronic nature of the substitution (electron-donating or withdrawing) and substitution patterns (ortho, meta, para), naphthyl (59) and heteroaryl aldehydes (60−63) underwent reductive transformation efficiently, yielding allylic alcohols in moderate to high yields with excellent enantioselectivities. Furthermore, the reaction exhibited broad functional group tolerance, including aryl bromide (48), aryl iodide (49), ether (50−52), thioether (54), boronic ester (55), ester (57), cyano (58), and carbamate (62) functionalities. However, when aliphatic aldehydes were used as substrates, the reaction afforded low yields, though enantioselectivity was retained (see SI for details). This observation aligned with previous reports on Ni-catalyzed reductive addition reactions of aldehydes27,28,29.

a Scope of aldehydes. b Scope of vinyl chlorides. aUnder N2, aldehydes (0.2 mmol, 1.0 equiv), vinyl chloride 2 (1.5 equiv), NiBr2•glyme (5 mol%), L11 (2.5 mol%), NaI (2.0 equiv), ZnBr2 (20 mol%), NEt3 (2.0 equiv) and Zn (3.0 equiv) were stirred in 2-Me-THF (0.2 M) at 25 °C for 16 h. b24 h. cNiBr2∙glyme (10 mol%), L11 (5 mol%), 16 h. dNiBr2∙glyme (10 mol%), L11 (5 mol%), 24 h. eNiBr2∙glyme (10 mol%), L11 (5 mol%), 48 h. fNiBr2∙glyme (10 mol%), L11 (5 mol%), 40 °C, 24 h. gNiBr2∙glyme (10 mol%), L11 (5 mol%), 0 °C, 48 h. Bpin pinacolato-boron. Boc t-butyloxy carbonyl.
Synthetic applications
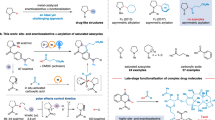
To demonstrate the practical utility of this methodology, several key transformations were carried out (Fig. 4). First, enantioselective reductive vinylic addition reactions were performed on aldehydes derived from various biologically active compounds, enabling the late-stage installation of allylic alcohols with excellent diastereoselectivities under mild conditions (Fig. 4a). Additionally, the chiral allylic amines generated from the reductive vinylic addition of imines proved versatile for further functionalizations (Fig. 4b). The tosyl protecting group was readily removed, affording primary amine 78 with high enantiospecificity. Moreover, N-tosyl amine 3 was efficiently converted to N-Boc amine 79, highlighting the ease of nitrogen elaboration in allylic amines. The olefin functionality also demonstrated significant reactivity, undergoing hydrogenation to yield 80 and epoxidation to produce 81, both with retention of enantioselectivities. Notably, reaction of product 9 with bis(collidine)bromonium(I) hexafluorophosphate (BBH) furnished azetidine 82, containing three contiguous stereocenters. These examples underscored the synthetic versatility and broad applicability of the products obtained via this methodology.

a Late-stage functionalization of complex aldehydes. b Product manipulation on allylic amines.
Mechanistic study
To gain insights into the reaction mechanism, several experiments were conducted (Fig. 5). First, to determine whether the active catalytic species was bimetallic or monomeric, we synthesized a dinickel complex stabilized by an acetate ion following the method reported by the Uyeda group46. This dinickel complex efficiently catalyzed the reductive alkenylation of imines, producing the desired product with enantioselectivity comparable to that observed in the original reaction (Fig. 5a). This result strongly suggested that the dinickel species functioned as the active catalyst. Regarding the C–C bond formation, two potential mechanisms were considered: (a) a Ni-catalyzed cross-coupling pathway, where the imine underwent single-electron reduction to generate an α-amino radical58,59,60 that subsequently recombined with Ni, followed by reductive elimination, and (b) a 1,2-addition process. To rule out the cross-coupling mechanism, we introduced a radical probe as an additive, but no radical adduct was detected (Fig. 5b). Instead, the imine alkenylation product was formed with high efficiency. Additionally, when an olefin-tethered imine 2b was subjected to the reaction conditions, no cyclization product was observed, and the direct addition product 83 was isolated in moderate yield with high enantioselectivity (Fig. 5c). Moreover, when cyclopropyl imine 1c was employed, the desired addition product 84 was isolated, without observation of ring-opening cross-coupling product. These findings were inconsistent with a radical cross-coupling mechanism. In order to support a 1,2-addition pathway, a competition reaction between imine and aldehyde was conducted under the reductive alkenylation condition (Fig. 5d). The addition to more electrophilic imine predominated the reaction, consistent with 1,2-addition mechanism.

a Reaction catalyzed by performed dinickel complex. b Radical probe experiment. c Ring-closing and opening experiments. d Competing experiment between aldehyde and imine.
DFT calculation
To further unveil the enantioselective mechanism of the dinickel-catalyzed reductive addition of imines with vinyl halides, unrestricted density functional theory calculations were performed at SMD-M06L-D3/def2-TZVP//M06L/def2-TZVP/def2-SVP level of theory (see computational details and benchmark test in SI and Supplementary Data 1). The different spin states of calculated models had been evaluated for the energy profile, indicating the reaction mechanism should majorly occur in the favorable triplet states, although selected intermediates might transfer to quintet state as shown in Supplementary Fig. S8. The triplet state of dinuclear (napbox)Ni2 (3[Ni]2) as its ground state can facilitate the activation of styrenyl chloride 2a to form a stable π-complex with C1 coordinated to both Ni1 and Ni2, supporting by an exothermic energy of −43.7 kcal/mol (Supplementary Fig. S7). Thus, this predominant dinickel π-complex 3int1 achieves the favorable energy profile as shown the key pathway in Fig. 6a. Furthermore, the following oxidative addition of the C–Cl bond give a barrierless step via 3ts1, indicating the high reactivity of the triplet species. Both Ni co-participate in the oxidative addition generated the bridging species 3int2 via the vinyl and chloride group. Under the excess NaI, the halide anion exchange occurs from Cl- to I- driven by favorable thermodynamical energy (−5.0 kcal/mol in triplet states). The formed iodide intermediate 3int3 undergoes the direct imine migration insertion of N-Ms imine by a majorly reactive Ni and a synergistic Ni. This enantioselective step is determined by the addition from the Re or Si face of the imine. Calculated results indicate that the Si face attacking majorly on the Ni1 reaction site achieve the most stable transition state 3ts2-S (ΔG‡ = 23.7 kcal/mol, 2.2 kcal/mol lower than that of 5ts2-S), leading to S-configured intermediate 3int4 in agreement with the experimental observations S-allylic amines. The other Re face attacking and the alternative Ni2 reaction site have also been evaluated as shown in 3ts2-R, 3ts2’-R and 3ts2’-S, respectively (the energy profile dominated by the Ni2 reaction site as shown in Supplementary Fig. S9). Considering the Boltzmann distribution, the predicted ee value is 87% (exp. 76%). A closer inspection of the most corresponding 3ts2-S and 3ts2’-R indicates that Ni1 as the reactive site can strongly bind with the imine substrate and facilitate the addition comparing with Ni2 site (2.11 Å in 3ts2-S vs. 2.15 Å in 3ts2’-R). The relatively early transition state in 3ts2-S further support the high reactivity (C2-C3: 2.17 Å). For the chiral control, there are apparently CH-π, polar O-π and non-classical O-H weak interactions between the imine and the chiral ligand in 3ts2-S, supporting by the spatial distances and IGMH analysis. The illustrated 3D space-filling mode also shows the matched chiral cavity of catalyst with the reactive substrates in 3ts2-S. Benefited from the binuclear Ni complex, their synergistic interaction enhances the reactivity and enantio-selectivity. The R, R-napbox ligand achieved the pronounced weak interaction with imine substrate to regulate the S-enantioselectivity.

a Computed key reaction profile. b Stereoselectivity analysis. Bond length (Å) in black. IGMH analysis of noncovalent interaction with δginter = 0.004. The values in parentheses represent the electron energy.
Our plausible mechanism was depicted in Fig. 7. Active π-complex int1, formed via the coordination of styrenyl chloride 2a with dinuclear Ni, undergoes C–Cl bond oxidative addition to produce a vinyl-bridged intermediate int2. After the ligand exchange by NaI to the iodide intermediate int3, the nucleophilic addition of the Si-face imine substrate controlled by R, R-napbox ligand occurs to generate the S-intermediate int4. Finally, releasing addition product via ligand exchange by ZnI₂ can form int5, which undergo the reduction by Zn and the coordination of another 2a to regenerate int1 for the next catalytic cycle.

Proposed catalytic cycle for the dinickel-catalyzed reductive alkenylation of imines with vinyl halides.
Discussion
In summary, we have developed a dinickel-catalyzed reductive alkenylation reaction of imines and aldehydes using vinyl chloride as the alkenylation agent. This approach enables the efficient synthesis of allylic amines and allylic alcohols in good yields with excellent enantioselectivities. The mild reaction conditions exhibit broad functional group tolerance, accommodating substrates that are often incompatible with traditional organometallic reagents. The unique efficacy of the dinickel catalyst, particularly in suppressing side reactions and enhancing chiral induction, underscores the utility of bimetallic systems in asymmetric catalysis.
Methods
Representative procedure for the synthesis of compound 3
The NiBr2•glyme (6.1 mg, 0.02 mmol, 10 mol%), L1 (6.6 mg, 0.01 mmol, 5 mol%), NaI (60.0 mg, 0.4 mmol, 2.0 equiv) and Zn powder (26.0 mg, 0.4 mmol, 2.0 equiv) were introduced into a flame-dried Schlenk tube in a N2-filled glove box. After removal from the glove box, Schlenk tube was connected to Schlenk line under N2. Dry THF (1 mL) was injected into the Schlenk tube, the mixture was stirred at r.t. for 30 min. The vinyl chloride (0.3 mmol, 1.5 equiv) and imine (0.2 mmol, 1.0 equiv) were sequentially added under nitrogen. The resulting mixture was stirred under particular temperature for 48 h. The reaction mixture was cooled to room temperature, quenched with saturated NH4Cl (aq.) solution and extracted with EtOAc (3x). The organic phase was washed with brine, dried over anhydrous Na2SO4, filtered, and concentrated under reduced pressure. The crude material was purified using column chromatography to give the pure product.
Data availability
The data reported in this paper are available within the article and its Supplementary Information files (including experimental details, NMR data, computational details, NMR and HPLC spectrums). All data are also available from the corresponding author upon request.
References
Shibasaki, M. & Kanai, M. Asymmetric synthesis of tertiary alcohols and α-tertiary amines via Cu-catalyzed C−C bond formation to ketones and ketimines. Chem. Rev. 108, 2853–2873 (2008).
Nugent, T. C. Chiral amine synthesis (Wiley-VCH, 2010).
Kobayashi, S., Mori, Y., Fossey, J. S. & Salter, M. M. Catalytic enantioselective formation of C−C bonds by addition to imines and hydrazones: a ten-year update. Chem. Rev. 111, 2626–2704 (2011).
Mailyan, A. K. et al. Cutting-edge and time-honored strategies for stereoselective construction of C–N bonds in total synthesis. Chem. Rev. 116, 4441–4557 (2016).
Trowbridge, A., Walton, S. M. & Gaunt, M. J. New strategies for the transition-metal catalyzed synthesis of aliphatic amines. Chem. Rev. 120, 2613–2692 (2020).
Lupidi, G., Palmieri, A. & Petrini, M. Enantioselective catalyzed synthesis of amino derivatives using electrophilic open-chain N-activated ketimines. Adv. Synth. Catal. 363, 3655–3692 (2021).
Vitaku, E., Smith, D. T. & Njardarson, J. T. Analysis of the structural diversity, substitution patterns, and frequency of nitrogen heterocycles among U.S. FDA approved pharmaceuticals. J. Med. Chem. 57, 10257–10274 (2014).
Rong, J. et al. Catalytic enantioselective addition of Grignard reagents to aromatic silyl ketimines. Nat. Commun. 7, 13780 (2016).
Ortiz, P., Collados, J. F., Jumde, R. P., Otten, E. & Harutyunyan, S. R. Copper-catalyzed enantioselective alkylation of enolizable ketimines with organomagnesium reagents. Angew. Chem. Int. Ed. 56, 3041–3044 (2017).
Zhang, L.-M., Luo, W., Fu, J., Liu, Y. & Zhang, J. W-Phos ligand enables copper-catalyzed enantioselective alkylation of N-sulfonyl ketimines with Grignard reagents. ACS Catal. 13, 8830–8837 (2023).
Fujihara, H., Nagai, K. & Tomioka, K. Copper-amidophosphine catalyst in asymmetric addition of organozinc to imines. J. Am. Chem. Soc. 122, 12055–12056 (2000).
Fu, P., Snapper, M. L. & Hoveyda, A. H. Catalytic asymmetric alkylations of ketoimines. Enantioselective synthesis of N-substituted quaternary carbon stereogenic centers by Zr-catalyzed additions of dialkylzinc reagents to aryl-, alkyl-, and trifluoroalkyl-substituted ketoimines. J. Am. Chem. Soc. 130, 5530–5541 (2008).
Yamada, K. -i. & Tomioka, K. Copper-catalyzed asymmetric alkylation of imines with dialkylzinc and related reactions. Chem. Rev. 108, 2874–2886 (2008).
Hirner, S. et al. Rhodium-catalyzed enantioselective addition of organoaluminum reagents to N-tosyl ketimines. Org. Lett. 16, 3162–3165 (2014).
Tokunaga, N. et al. C2-symmetric bicyclo[2.2.2]octadienes as chiral ligands: their high performance in rhodium-catalyzed asymmetric arylation of N-tosylarylimines. J. Am. Chem. Soc. 126, 13584–13585 (2004).
Trincado, M. & Ellman, J. A. Enantioselective synthesis of α-aryl alkylamines by Rh-catalyzed addition reactions of arylboronic acids to aliphatic imines. Angew. Chem. Int. Ed. 47, 5623–5626 (2008).
Marques, C. S. & Burke, A. J. Advances in the catalytic asymmetric arylation of imines using organoboron reagents: an approach to chiral arylamines. ChemCatChem 3, 635–645 (2011).
Yang, G. & Zhang, W. A palladium-catalyzed enantioselective addition of arylboronic acids to cyclic ketimines. Angew. Chem. Int. Ed. 52, 7540–7544 (2013).
Wu, C., Qin, X., Moeljadi, A. M. P., Hirao, H. & Zhou, J. S. Copper-catalyzed asymmetric arylation of N-heteroaryl aldimines: Elementary step of a 1,4-insertion. Angew. Chem. Int. Ed. 58, 2705–2709 (2019).
Knappke, C. E. I. et al. Reductive cross-coupling reactions between two electrophiles. Chem. Eur. J. 20, 6828–6842 (2014).
Poremba, K. E., Dibrell, S. E. & Reisman, S. E. Nickel-catalyzed enantioselective reductive cross-coupling reactions. ACS Catal. 10, 8237–8246 (2020).
Hargaden, G. C. & Guiry, P. J. The development of the asymmetric Nozaki–Hiyama–Kishi reaction. Adv. Synth. Catal. 349, 2407–2424 (2007).
Tian, Q. & Zhang, G. Recent advances in the asymmetric Nozaki–Hiyama–Kishi reaction. Synthesis 48, 4038–4049 (2016).
Gil, A., Albericio, F. & Álvarez, M. Role of the Nozaki–Hiyama–Takai–Kishi reaction in the synthesis of natural products. Chem. Rev. 117, 8420–8446 (2017).
Takai, K. et al. Reactions of alkenylchromium reagents prepared from alkenyl trifluoromethanesulfonates (triflates) with chromium(II) chloride under nickel catalysis. J. Am. Chem. Soc. 108, 6048–6050 (1986).
Guo, H. et al. Toolbox approach to the search for effective ligands for catalytic asymmetric Cr-mediated coupling reactions. J. Am. Chem. Soc. 131, 15387–15393 (2009).
Zhang, S. et al. Design and synthesis of tunable chiral 2,2’-bipyridine ligands: application to the enantioselective nickel-catalyzed reductive arylation of aldehydes. Angew. Chem. Int. Ed. 61, e202117843 (2022).
Zhu, Z., Xiao, J., Li, M. & Shi, Z. Nickel-catalyzed intermolecular asymmetric addition of aryl iodides across aldehydes. Angew. Chem. Int. Ed. 61, e202201370 (2022).
Gu, P. et al. Chromium- and metal-reductant-free asymmetric Nozaki–Hiyama–Kishi (NHK) reaction enabled by metallaphotoredox catalysis. Angew. Chem. Int. Ed. 63, e202408195 (2024).
Huang, S. & Zhou, J. S. Nickel-catalyzed enantioselective reductive arylation of common ketones. J. Am. Chem. Soc. 146, 12895–12900 (2024).
Hu, L., Le Blanc Lele Fosso, J., Guillot, R., Mellah, M. & Schulz, E. Electrochemical enantioselective nickel-catalyzed cross-coupling of aldehydes with aryl iodides. Chem. Eur. J. 30, e202403432 (2024).
Ding, L. et al. Stereoselective vinylic C−H addition via metallaphotoredox migration. Angew. Chem. Int. Ed. 64, e202413557 (2025).
Jiang, X. et al. Photoassisted cobalt-catalyzed asymmetric reductive Grignard-type addition of aryl iodides. J. Am. Chem. Soc. 144, 8347–8354 (2022).
Jiang, H. et al. Photoinduced cobalt-catalyzed desymmetrization of dialdehydes to access axial chirality. J. Am. Chem. Soc. 145, 6944–6952 (2023).
Kokić, B. et al. Strategies for carbon electrophile addition to carbonyls and imines by cobalt catalysis. Eur. J. Org. Chem. 26, e202300997 (2023).
Lin, C. et al. Cobalt-catalyzed enantioselective alkenylation of aldehydes. Angew. Chem. Int. Ed. 63, e202405290 (2024).
Chen, J. et al. Cobalt-catalyzed asymmetric migratory Nozaki–Hiyama–Kishi coupling. J. Am. Chem. Soc. 146, 26223–26232 (2024).
Shao, Y.-P. & Liang, Y.-M. Dynamic kinetic reductive Grignard-type addition for the construction of axial and central chirality. ACS Catal. 15, 1147–1157 (2025).
Zhang, L. et al. Nickel-catalyzed enantioselective reductive arylation and heteroarylation of aldimines via an elementary 1,4-addition. J. Am. Chem. Soc. 145, 8498–8509 (2023).
Xiao, J. et al. Enantioselective reductive (hetero)arylation of cyclic N-sulfonyl imines by cobalt catalysis. Angew. Chem. Int. Ed. 62, e202300743 (2023).
Xia, T. et al. Cobalt-catalyzed asymmetric aza-Nozaki–Hiyama–Kishi (NHK) reaction of α-imino esters with alkenyl halides. Angew. Chem. Int. Ed. 63, e202316012 (2024).
Wu, X. et al. Modular α-tertiary amino ester synthesis through cobalt-catalysed asymmetric aza-Barbier reaction. Nat. Chem. 16, 398–407 (2024).
Zhang, C., Wu, X., Qu, J. & Chen, Y. A general enantioselective α-alkyl amino acid derivatives synthesis enabled by cobalt-catalyzed reductive Addition. J. Am. Chem. Soc. 146, 25918–25926 (2024).
Xia, T., Wu, W., Wu, X., Qu, J. & Chen, Y. Cobalt-catalyzed enantioselective reductive α-chloro-carbonyl addition of ketimine to construct the β-tertiary amino acid analogues. Angew. Chem. Int. Ed. 63, e202318991 (2024).
Zhou, Y.-Y. & Uyeda, C. Catalytic reductive [4 + 1]-cycloadditions of vinylidenes and dienes. Science 363, 857–862 (2019).
Behlen, M. J. & Uyeda, C. C2-symmetric dinickel catalysts for enantioselective [4 + 1]-cycloadditions. J. Am. Chem. Soc. 142, 17294–17300 (2020).
Uyeda, C. & Farley, C. M. Dinickel active sites supported by redox-active ligands. Acc. Chem. Res. 54, 3710–3719 (2021).
Wang, P., Zhu, L., Wang, J. & Tao, Z. Catalytic asymmetric α-alkylation of ketones with unactivated alkyl halides. J. Am. Chem. Soc. 145, 27211–27217 (2023).
Li, Y., Li, W., Tian, J., Huang, G. & Lv, H. Nickel-catalyzed asymmetric addition of aromatic halides to ketones: highly enantioselective synthesis of chiral 2,3-dihydrobenzofurans containing a tertiary alcohol. Org. Lett. 22, 5353–5357 (2020).
Zhou, P. & Xu, T. Nickel-catalyzed intramolecular desymmetrization addition of aryl halides to 1, 3-diketones. Chem. Commun. 56, 8194–8197 (2020).
Sun, B. et al. Parallel paired electrolysis-enabled asymmetric catalysis: Simultaneous synthesis of aldehydes/aryl bromides and chiral alcohols. Sci. Bull. 68, 2033–2041 (2023).
Johannsen, M. & Jørgensen, K. A. Allylic amination. Chem. Rev. 98, 1689–1708 (1998).
Patel, S. J. & Jamison, T. F. Asymmetric catalytic coupling of organoboranes, alkynes, and imines with a removable (trialkylsilyloxy)ethyl group–direct access to enantiomerically pure primary allylic amines. Angew. Chem. Int. Ed. 43, 3941–3944 (2004).
Zhou, C.-Y., Zhu, S.-F., Wang, L.-X. & Zhou, Q.-L. Enantioselective nickel-catalyzed reductive coupling of alkynes and imines. J. Am. Chem. Soc. 132, 10955–10957 (2010).
Li, L., Liu, Y.-C. & Shi, H. Nickel-catalyzed enantioselective α-alkenylation of N-sulfonyl amines: modular access to chiral α-branched amines. J. Am. Chem. Soc. 143, 4154–4161 (2021).
Bishop, H. D., Zhao, Q. & Uyeda, C. Catalytic asymmetric synthesis of zinc metallacycles. J. Am. Chem. Soc. 145, 20152–20157 (2023).
Fahrni, C. J. & Pfaltz, A. Synthesis of chiral C2-symmetric binucleating ligands. Helv. Chim. Acta 81, 491–506 (1998).
Turro, R. F., Brandstätter, M. & Reisman, S. E. Nickel-catalyzed reductive alkylation of heteroaryl imines. Angew. Chem. Int. Ed. 61, e202207597 (2022).
Hu, H. & Wang, Z. Cr-catalyzed asymmetric cross aza-pinacol couplings for β-amino alcohol synthesis. J. Am. Chem. Soc. 145, 20775–20781 (2023).
Hu, H., Shi, Z., Guo, X., Zhang, F.-H. & Wang, Z. A radical approach for asymmetric α-C–H addition of N-sulfonyl benzylamines to aldehydes. J. Am. Chem. Soc. 146, 5316–5323 (2024).
Acknowledgements
The authors are grateful for the financial support from the National Key R&D Program of China (2021YFA1500100, Q.P.), the Natural Science Foundation of China (22471067, Z.T., 92156017, Q.P. and 22403053, H.Z.), the Natural Science Foundation of Hunan Province (2022JJ20006, Z.T.), Natural Science Foundation of Tianjin (24JCZDJC00750, Q.P.), “Frontiers Science Center for New Organic Matter”, Nankai University (63181206, Q.P.), Haihe Laboratory of Sustainable Chemical Transformation of Tianjin (24HHWCSS00019, Q.P.), the Postdoctoral Fellowship Program of China Postdoctoral Science Foundation (GZC20240750, H.Z.) and the Fundamental Research Funds for the Central Universities. We also thank the Analytical Instrumentation Center of Hunan University for mass spectrometry analysis.
Author information
Authors and Affiliations
Contributions
Z.T. conceived and designed the project. P.Z., P.W. and J.Z. conducted the experiments. H.Z. conducted the DFT calculations. Q.P. and Z.T. wrote the manuscript. All authors contributed to analyse the data and edit the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewers for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Zhou, P., Wang, P., Zhu, H. et al. Dinickel-catalyzed enantioselective reductive addition of imines with vinyl halides. Nat Commun 16, 8871 (2025). https://doi.org/10.1038/s41467-025-63940-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-025-63940-y
