
Abstract
Astrocytes are critical contributors to brain disorders, yet the mechanisms underlying their selective vulnerability to specific diseases remain poorly understood. Here, we demonstrate that NR3C1 acts as a key regulator of early postnatal astrocyte development, shaping long-term immune responses in mice. Through integrative analyses of gene expression, chromatin accessibility, and long-range chromatin interactions, we identify 55 stage-specific TFs, with NR3C1 uniquely associated with early postnatal maturation. Although mice lacking astrocytic NR3C1 exhibit no detectable developmental abnormalities, these mice display heightened susceptibility to exacerbated immune responses following adult-onset experimental autoimmune encephalomyelitis (EAE). Many of the dysregulated EAE response genes are linked to candidate cis-regulatory elements altered by early NR3C1 loss, driving exacerbated inflammatory responses. Notably, only NR3C1 depletion during early, but not late, astrocyte development induces long-lasting epigenetic reprogramming that primes astrocytic immune responses.
Similar content being viewed by others


Introduction
Astrocytes are prominent glial cells within the mammalian brain that perform critical functions, including metabolic support of neurons, synapse remodeling and plasticity, maintaining blood–brain barrier, and immune responses1. Therefore, astrocytic dysfunction contributes significantly to the progression of both neurodevelopmental and neurodegenerative diseases2,3,4,5,6. However, the mechanism by which astrocytic dysfunction initially arises and the role of developmental gene regulatory networks in this process remain largely unexplored.
Astrocyte differentiation begins in the late stage of embryogenesis from radial glial cells, triggered by a gliogenic switch that redirects their fate from neurogenesis to gliogenesis7. Several transcription factors (TFs) have been identified to induce astrocyte differentiation. NFIA and SOX9 play key roles by regulating chromatin structure to induce gene expression related to glial specification8. STAT3 forms a complex with the co-activator p300 to facilitate astrocyte differentiation9,10. Recent genome-wide transcriptome and epigenome characterization during in vitro differentiation or in vivo maturation (postnatal day 4 and 2-month-old mice) has revealed key TFs involved in astrocyte development, such as ATF3, RUNX2, RORB, DBX2, and LHX211,12. Despite these advances, our understanding of the gene regulatory programs during astrocyte development remains limited due to several reasons.
First, there is a lack of characterization with precise temporal resolution of each developmental stage. Particularly, the dynamic regulatory programs during the first postnatal month—a crucial period of developmental progression13—are largely unknown. Second, stage-specific gene expression in brain development often involves dynamic reorganization of chromatin architecture14,15. However, the long-range chromatin interactions between enhancers and their putative target genes during astrocyte development have been largely unexplored, creating a significant gap in our understanding of epigenetic regulation. Lastly, due to the aforementioned limitations, the function of TFs specific to each astrocytic perinatal developmental stage remains elusive.
To address these challenges, we conducted a comprehensive investigation of dynamic gene regulatory programs from embryogenesis to postnatal mature stages of astrocytes at precise temporal resolution. Using Aldh1l1-EGFP transgenic mouse, we collected astrocytes and their progenitors from embryonic day 16 (E16), marking the initiation stage of gliogenesis, through to postnatal day 30 (P30), representing the mature stage of astrocyte development. We employed Hi-C on accessible regulatory DNA (HiCAR)16 along with ATAC-seq and RNA-seq, and identified a total of 55 TFs involved in various astrocyte developmental stages. Among these, NR3C1 emerged as a key regulator during the early postnatal stage. Interestingly, depletion of NR3C1 in astrocytes did not lead to any observable phenotype. However, upon the induction of experimental autoimmune encephalomyelitis (EAE) in adult stages, NR3C1-deficient mice exhibited a significantly exacerbated immune response, which turned out to be largely driven by the epigenetic priming mechanism of NR3C1.
Results
Precise temporal transcriptome profiling during perinatal astrocytic development
To investigate the dynamic gene regulation during perinatal astrocyte development, we utilized Aldh1l1-EGFP transgenic mice, since ALDH1L1 is a well-established pan-astrocytic marker known for its stable expression across astrocyte populations and developmental stages17. EGFP-positive astrocytes were isolated from mouse cortical tissue using fluorescence-activated cell sorting (FACS). The perinatal period is critical for astrocyte development, so we sampled across a broad temporal window, from embryonic day 16 (E16), when radial glial cells initiate differentiation into glial cells, to postnatal day 30 (P30) when astrocytes reach a mature morphology (Fig. 1a). We employed RNA-seq, ATAC-seq, and HiCAR to capture transcriptome, epigenome, and long-range chromatin contact dynamics occurring throughout the astrocyte development (Supplementary Data 1).

a Schematic of the multi-omics analyses of astrocyte development. Astrocytes were isolated from E16, E18, P1, P3, P7, P17, and P30 mouse cortices with two biological replicates. b Principal component analyses based on RNA-seq (left), ATAC-seq (middle), and HiCAR (right) representing astrocyte developmental trajectory throughout each dataset. The color indicates the time point of each developmental stage. c Heatmap showing the expression patterns of developmental DEGs (left) and accessibility patterns of their linked cCREs (right). Representative genes and ontologies are displayed to the right of the heatmap. d Boxplots representing significant long-range chromatin interaction scores between DEGs and Differentially Accessible Peaks (DAPs). The box indicates the interquartile range (IQR), and the whiskers represent the highest and lowest points within 1.5 × IQR. P values of differences of interaction between early DEG and DAP (n = 438) were P = 0.029 for Early vs Mid, P = 0.016 for Early vs Late, P = 0.332 for Mid vs Late; between mid DEG and DAP (n = 106) were P = 0.014 for Early vs Mid, P = 0.239 for Early vs Late, P = 0.505 for Mid vs Late; between late DEG and DAP (n = 572) were P = 1.24 × 10−3 for Early vs Mid, P < 2.20 × 10−16 for Early vs Late, P < 2.20 × 10−16 for Mid vs Late (two-sided Kolmogorov–Smirnov test). Early, E16 and E18; Mid, P1, P3, and P7; Late, P17 and P30. e–g Genome browser snapshots of representative DEGs, including Neurod1 (e), Gfap (f), and Slc1a2 (g) with gene expression, chromatin accessibility, and significant long-range chromatin interactions.
We first confirmed the developmental specificity of the collected astrocytes by examining the expression of known marker genes. As expected, the expression of Aldh1l1, the marker used for astrocyte sorting, remained stable from late embryogenesis through the first postnatal month (Supplementary Fig. 1a). Further, the expression levels of immature astrocyte markers, including Nes and Vim, were decreased from P7 and P17 (Supplementary Fig. 1a)18. Conversely, the mature astrocyte markers, including Gfap, Aqp4, Aldoc, and Slc1a2, were gradually upregulated starting at P1 (Supplementary Fig. 1a)19. Lastly, while expression of other cell type markers was rarely observed, astrocyte-specific genes were consistently upregulated across all developmental time points (Supplementary Fig. 1b).
Next, we performed principal component analysis (PCA) to map the developmental trajectory of transcriptome profiles. The trajectory analysis well aligned the samples along the developmental stages, with replicates grouping together, validating that our dynamic transcriptome landscapes accurately reflect developmental progression (Fig. 1b). Our analysis further grouped the time points into three major stages: early (E16 to E18), mid (P1 to P7), and late (P17 to P30) (Fig. 1b and Supplementary Fig. 1c). We then identified 4198 stage-specific differentially expressed genes (DEGs) (Fig. 1c, Supplementary Data 2). The early-stage genes such as Nes, Meis2, Neurog2, and Mki67 suggest that characteristics of proliferating radial glial cells persist throughout embryogenesis18. In contrast, late-stage genes, including S100b, Rora, and Il33, were enriched for gene ontology terms related to immune response pathways, consistent with previously reported transcriptome profiles of mature astrocytes12. Importantly, key regulators of the gliogenic switch, such as Nfia, Jak2, and Stat3, were highly expressed during the mid-stage, highlighting this period as a critical window for astrocyte specification.
A landscape of long-range cCRE-promoter contacts during astrocyte development
To dissect stage-specific gene regulatory programs, we mapped the candidate cis-regulatory elements (cCREs) and identified their putative target genes based on long-range chromatin interactions (see Methods). Using ATAC-seq results across all developmental time points, a total of 171,571 cCREs were defined. Additionally, HiCAR analysis captured 167,559 significant long-range interactions based on covNorm20 (Supplementary Data 3). Consistent with the observed transcriptome dynamics, the trajectories of both cCRE accessibility and long-range chromatin interactions were well aligned along the developmental time points with high reproducibility (Fig. 1b, Supplementary Fig. 1c).
To characterize stage-specific gene regulatory programs, we applied the ‘activity-by-contact’ (ABC) model21, which quantified the contribution of each cCRE to its putative target gene based on the cCRE accessibility weighted by the contact frequencies with the promoter. As a result, a total of 268,446 cCRE-promoter pairs were defined throughout astrocyte development. Multiple lines of evidence support the regulatory potential of the identified cCRE-gene pairs. First, the DEG-associated cCREs exhibited chromatin accessibility, positively correlating with the gene expression patterns (Fig. 1c). Second, the promoter regions of stage-specific DEGs demonstrated a high level of chromatin contacts with cCREs that were specifically active during the corresponding developmental stages (Fig. 1d), as exemplified by the loci of Neurod1, Gfap, and Slc1a2 (Fig. 1e–g). Collectively, our multi-omics analyses provide a comprehensive view of the transient and dynamic epigenetic regulatory landscapes during perinatal astrocyte development, offering precise temporal resolution of gene regulatory networks.
NR3C1 is a key regulator of early postnatal astrocyte development
The high-resolution gene regulatory networks led to the identification of key TFs involved in astrocyte development. Using integrative analyses, we predicted TF activities at each developmental stage based on the PageRank algorithm22. As expected, known developmental TFs such as NFIA8 and STAT39 exhibited high activities during mid-stage (Supplementary Fig. 2a). To focus on stage-specific TFs, we selected 55 TFs showing high specificity in terms of both TF activities and expression levels across developmental stages (Early, n = 26; Mid, n = 16; Late, n = 13) (Fig. 2a). During the early stage, TFs involved in chromatin structure and neurogenesis, such as CTCF, YY1, and NEUROD1, highlighted the maintenance of neuronal pluripotency during late embryogenesis. On the other hand, TFs associated with immune responses, including ETS1, FOXO1, and RORA, showed strong activities during the late stage. Notably, given precise temporal analyses, we were also able to identify developmental TFs with strong activities during the mid-stage, including intermediate-early genes (IEGs) such as EGR1, FOS, and JUN, as well as nuclear receptors like NR4A3, NR4A1, and NR3C1.

a Integrated analysis for the identification of developmental TFs. The color represents the scaled gene expression of each TF. The size of the square indicates the normalized PageRank score of each TF estimated by Taiji. The maximum activity time point for each TF is highlighted by a black box. b, c UMAP projection of 72,666 cells of neurogenic and astrocytic lineages from mouse developing cortices. The plots were colored by annotated cell types (b) or pseudotime (c). d Relative expression patterns of developmental TFs identified in Fig. 2a. The TFs expressed with more than a twofold change in astrocytic lineages are colored blue (n = 6), while in neurogenic lineages, colored green (n = 7). e, f UMAP plots colored by the normalized expression of Nr3c1 (e) and Neurod6 (f). g Scatter plot representing the effect of each TF in silico perturbation within both neurogenic and astrocytic lineages. The color indicates the developmental stage at which each TF shows the highest PageRank scores. The x and y axes indicate the effects of TF perturbation at astrocyte and neurogenic lineages, respectively. TFs that are highly expressed in astrocytic lineages are marked with black circles.
To verify the specificity of TF activities in each developmental stage, we analyzed a single-cell RNA-seq (scRNA-seq) dataset of the mouse developing cortex23. We focused on cells belonging to the neurogenic and astrocytic lineages, defined by their marker gene expression. Analyses of 50,722 neurogenic and 21,944 astrocytic lineage cells revealed shared progenitors in the early developmental stage, which then diverged into distinct trajectories, consistent with known stages of mouse brain development, as shown by Uniform Manifold Approximation and Projection (UMAP) (Fig. 2b, c, Supplementary Fig. 2b).
We then examined whether the 55 TFs were differentially expressed between the two lineages along their developmental trajectories. Comparison of expression patterns between the neurogenic and astrocytic lineages revealed that 13 of the 55 lineage-specific TFs exhibited a greater than two-fold change during development (Fig. 2d). Notably, IEGs such as Jun, Fos, Egr1, and Junb were specifically expressed in the astrocytic lineage. In addition, the expression of TFs, including Stat3 and Nr3c1, had increased during the later developmental stages (Fig. 2d, e). On the other hand, Sox4, Sox11, Bcl11a, and Neurod6 showed higher expression levels in the neurogenic lineage compared to the astrocytic lineage (Fig. 2d, f). Further, we performed in silico perturbation analysis using CellOracle24 to estimate the perturbation impact of each TF in both lineages (Fig. 2g). The in silico perturbation analyses revealed distinct lineage-specific patterns. Early-stage TFs such as NEUROD6 and SOX11 predominantly affected the neurogenic lineage, as expected. By contrast, late-stage TFs, including FOXO1 and RORA, had a greater impact on the astrocytic lineage. Although several TFs that are highly expressed in astrocytes (Jun, Fos, Egr1, Junb, and Stat3) were predicted to influence both lineages, NR3C1 uniquely demonstrated both high expression and a lineage-specific perturbation effect in astrocytes, suggesting its critical role in early postnatal astrocyte development.
NR3C1 target genes are associated with immune-related brain dysfunctions
Brain developmental gene regulatory networks are frequently implicated in various neurological disorders25. To explore the pathological associations of NR3C1 along with five astrocyte mid-stage specific TFs (FOS, EGR1, JUN, JUNB, and STAT3), we performed gene set enrichment analyses using DisGeNET26 on the predicted target genes of each TF. To identify TF target genes, we integrated chromatin accessibility data from cCREs harboring the corresponding TF motif with HiCAR-derived interaction frequencies, following an approach similar to the ABC model (see Methods). Using the resulting cCRE-promoter pairs (Fig. 1c), we assigned gene-level scores by summing ABC scores of linked cCREs harboring each TF motif to consolidate the sparse TF bound enhancer signal, defining candidate genes predominantly regulated by each TF. Our analyses revealed distinct associations between target genes of each TF and various neurological disorders (Fig. 3a). For instance, we observed an enrichment of genes associated with bipolar disorder, where the rapamycin (mTOR) pathway is known to be involved through target genes of STAT3 (Fig. 3b, P = 0.010)27. Notably, the target genes of NR3C1 exhibited a significant association with multiple sclerosis (MS), autoimmune diseases, and Alzheimer’s disease, suggesting potential roles of astrocytic NR3C1 in these immune-related neurological disorders (Fig. 3c, P = 0.012).

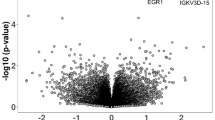
a A heatmap displaying the enrichment scores of the target genes regulated by each TF for brain disorders. The NES score was obtained from GSEA analysis, indicating the degree of disease association for the target genes of each TF. Bidirectional hierarchical clustering was performed based on Euclidean distances of NES scores. *FDR < 0.05, **FDR < 0.01, ***FDR < 0.001. b, c GSEA for STAT3 and NR3C1 to disease-associated genes for ‘Bipolar disorder’ (b P = 0.010) and ‘Multiple sclerosis’ (c P = 0.012), respectively.
NR3C1 depletion exacerbates the immune response upon EAE induction
A recent CRISPRi screening has also reported that NR3C1 plays a role in regulating immune responses in astrocytes28. However, its regulatory function and physiological role in early postnatal astrocytic development are unknown. To investigate the potential role of NR3C1 in this context, we generated astrocyte-specific inducible NR3C1 conditional knockout (NR3C1 cKO) mice using the Cre-lox system, crossing Aldh1l1-creERT2 mice with loxP-floxed Nr3c1 (Nr3c1f/f) mice (Fig. 4a, see Methods). On the day of delivery, 4-hydroxy tamoxifen (4-OHT) was injected subcutaneously to induce NR3C1 depletion prior to its estimated active stage around P7-P17. NR3C1 cKO mice exhibited a significantly reduced number of NR3C1-positive astrocytes at P17 compared to Cre−-control mice (Fig. 4b, two-sided t test, P < 1 × 10−4). Furthermore, we observed a significant decrease in NR3C1-positive astrocytes starting from P5, and this reduction remained consistent until P53 (Supplementary Fig. 3a, b).

a Schematics of experiments for the generation of NR3C1 cKO mice and autoimmune challenges. b The number of NR3C1+ astrocytes in Cre− control and NR3C1 cKO mice at P17. Mice were used with n = 3 for Cre− control, n = 5 for NR3C1 cKO. Data are shown as mean ± standard error of the mean (SEM) and are analyzed by one-sided unpaired t test (P < 0.0001). c Heatmap representing DEGs of NR3C1 cKO astrocytes at P17 under naive conditions. d Bar graphs showing the overlapping ratio between DEGs of NR3C1 cKO astrocytes and astrocyte developmental stage-specific DEGs. Hypergeometric p value are shown together (Up-P7, P = 1.64 × 10−5; Up-P17, P = 0.010; Down-P17, P = 2.72 × 10−13; Down-P30, P = 1.31 × 10−7). e Top 5 enriched MSigDB Hallmark pathways of DEGs in NR3C1 cKO astrocytes. f GFAP expression in the corpus callosum. Bar graphs showing the area of GFAP expression compared between Cre− control and NR3C1 cKO mice under naive conditions (P = 0.749) and at the peak of EAE (P = 4.65 × 10−2). Mice were used with n = 3 or n = 7 for Cre− control and n = 4 or n = 7 for NR3C1 cKO at naive or EAE peak stage, respectively. Data are shown as mean ± SEM and were analyzed by a two-sided unpaired t test. g EAE clinical score in Cre− control mice (n = 12) and NR3C1 cKO mice (n = 10). Two-way ANOVA, P < 0.0001. Data are shown as mean ± SEM. Scale bars, 20 μm. h IBA1 expression in corpus callosum at the EAE peak (P = 0.056). Mice were used with n = 7 for Cre− control and 7 for NR3C1 cKO. Data are shown as mean ± SEM and were analyzed by two-sided unpaired t test. i The number of CD4+ T cells in corpus callosum at the EAE peak (P = 1.6 × 10−3). Mice were used with n = 7 for Cre− control and 7 for NR3C1 cKO. Data are shown as mean ± SEM and were analyzed by two-sided unpaired t test. Scale bars, 40 μm. j, k GFAP expression (j P = 2.09 × 10−2) and the number of CD45+ cells (k P = 6.50 × 10−3) in the spinal cord at the peak of EAE. Mice were used with n = 3 for Cre− control and n = 4 for NR3C1 cKO. Data are shown as mean ± SEM and were analyzed by two-sided unpaired t test. Scale bars, 100 μm (j) and 50 μm (k). Source data are provided as a Source Data file.
After confirming the KO efficiency, we performed RNA-seq using astrocytes isolated from both Cre− control and NR3C1 cKO mice at P17. The transcriptome profiles of Cre− control and NR3C1 cKO astrocytes exhibited high reproducibility between replicates (Supplementary Fig. 3c). DEG analysis identified 2256 downregulated and 1895 upregulated genes in NR3C1 cKO astrocytes (Fig. 4c, Supplementary Data 4). The DEGs showed significant overlap with genes expressed from P7 to P30 during astrocyte development (Fig. 4d). Specifically, the upregulated genes in NR3C1 cKO astrocytes largely overlapped with developmental genes expressed at P7 and P17, whereas downregulated genes were enriched among genes expressed at P17 and P30, supporting the functional implication of NR3C1 in the early postnatal stage. Despite these significant changes in gene expression, surprisingly, NR3C1 cKO mice exhibited no observable phenotypic changes under the normal developmental process. Astrocyte-specific NR3C1 depletion did not affect astrocytic population, morphology, or marker gene expression (Supplementary Fig. 3d, e). Given the association between NR3C1 and immune-related neurological disorders, we also examined the expression of immune-responsive genes in NR3C1 cKO astrocytes. Unexpectedly, the DEGs in NR3C1 cKO astrocytes did not exhibit significant enrichment for immune response pathways among either upregulated or downregulated genes (Fig. 4e). Moreover, NR3C1 cKO astrocytes showed no changes in the expression of reactive astrocyte markers (top in Fig. 4f, Supplementary Fig. 3f).
Given that NR3C1 target genes appear to be associated with MS and autoimmune diseases, as shown in Fig. 3a, we hypothesized that NR3C1 depletion might have physiological consequences when additional immune stimulus is introduced. To test our hypothesis, we again induced astrocyte-specific NR3C1 depletion by injecting 4-OHT into Aldh1l1-creERT2; Nr3c1f/f mice at P0, and then applied an immune challenge at 9 weeks of age by inducing EAE using MOG35–55 peptide, a well-established mouse model for MS as demonstrated in Fig. 4a. We found that NR3C1 expression in astrocytes remained unchanged following EAE induction compared to non-EAE (naive) controls (Supplementary Fig. 4a). Nonetheless, we observed that astrocytic NR3C1 cKO significantly worsened the progression of EAE, with significantly exacerbated clinical scores (Fig. 4g, two-way ANOVA, P < 1 × 10−4). Approximately half of the NR3C1 cKO mice reached a clinical score of 5, characterized by sudden death or severe paralysis. Notably, while the expression level of reactive astrocyte marker, GFAP, was comparable between Cre− control and NR3C1-depleted astrocytes under naive conditions, NR3C1 cKO mice showed a significant increase in GFAP expression during EAE progression (bottom in Fig. 4f). NR3C1 cKO mice with EAE also showed increased IBA1 expression compared to Cre−-control mice (Fig. 4h), although the increase was less pronounced than that of GFAP. Interestingly, this was accompanied by a higher number of brain-infiltrating CD4+ T cells (Fig. 4i, Supplementary Fig. 4b, c), which are the main effector cells during EAE progression. We observed a similar increase in GFAP and IBA1 expression in the spinal cord and cerebellum of NR3C1 cKO mice following EAE induction (Fig. 4j, Supplementary Fig. 4d, e). Furthermore, NR3C1 cKO mice exhibited increased numbers of CD45+ immune cells and CD4+ T cells in the spinal cord (Fig. 4k, Supplementary Fig. 4b, c, f), indicating that NR3C1 regulates astrocyte-mediated immune responses across multiple CNS regions beyond the cortex. Taken together, these data suggest that astrocytic NR3C1 deficiency contributes to a more inflammatory immune environment.
snRNA-seq analysis reveals NR3C1-mediated immune responses at the peak of EAE
To elucidate how NR3C1 cKO leads to an exacerbated EAE phenotype, we conducted single-nucleus RNA-seq (snRNA-seq) using the 10X Genomics v3.1 platform on cortices collected at the peak of EAE with two biological replicates for each of Cre− control and NR3C1 cKO astrocytes (see Methods). Using the canonical Scanpy framework29, a total of 44,548 nuclei were retained and clustered into eight distinct cell types without a sample bias (Supplementary Fig. 5a–d, Supplementary Data 5). Based on known marker genes, eight distinct major brain cell types were annotated, including astrocytes (Aldh1l1, Gfap), microglia (C1qa, Tmem119), neurons (Slc17a7, Rbfox3), oligodendrocyte precursor cells (Pdgfra), ependymal cells (Tmem212), meningeal cells (Slc47a1), and two endothelial cell types (Cldn5 and Rgs5) (Supplementary Fig. 5e).
To uncover molecular mechanisms underlying exacerbated EAE phenotype in NR3C1 cKO mice, we focused on the astrocyte population. In NR3C1 cKO astrocytes, 1271 and 756 downregulated and upregulated EAE-responsive genes were identified compared to Cre− control at the peak of EAE (Fig. 5a, Supplementary Data. 6). To determine whether these genes were altered specifically within a distinct astrocyte subpopulation, we conducted subclustering analysis, which revealed five distinct astrocyte subpopulations (Fig. 5b, Supplementary Fig. 5f, g). Although subpopulation C2, marked by expression of Gfap and Tnc, showed a slight expansion in NR3C1 cKO mice at the peak of EAE, the EAE-responsive genes were globally dysregulated across all astrocyte subpopulations (Fig. 5c, Supplementary Fig. 5h). For example, astrocytic immune response genes, such as Igfbp5, Notch1, Stat2 and Gfap, were consistently upregulated across all subpopulations (Fig. 5d). To further assess the pathological relevance of the EAE-responsive genes, we compared our data with transcriptome datasets obtained from MS patients30,31. Genes upregulated or downregulated at NR3C1 cKO astrocytes at the peak of EAE showed concordant higher or lower expression, respectively, in active MS lesions compared to inactive lesions (Fig. 5e). Stratified analysis of MS lesions, including normal-appearing white matter (NAWM), active lesions (AL), chronic active lesions (CAL), chronic inactive lesions (CIL), and remyelinated lesions (RL), further confirmed that genes upregulated in NR3C1 cKO astrocytes were more highly expressed in inflammatory lesion types (AL and CAL), while downregulated genes showed reduced expression in these same regions compared to less inflammatory lesions (CIL and RL) (Supplementary Fig. 5i). Collectively, these findings suggest that NR3C1 in astrocytes modulates the expression of genes relevant to immune-related neurological disorders, such as MS.

a Heatmap representing DEGs between astrocytes from Cre− control and NR3C1 cKO mice at the EAE peak. b t-SNE projections illustrating subpopulation information of astrocytes (C0-C4). c Dot plots showing the expression patterns of DEGs in astrocyte subpopulations. Dot size indicates the average ratio of expressed gene numbers per cell. Color represents the average expression level of genes in subpopulations. d Scatter plots of representative DEG expressions in astrocytes from Cre− control and NR3C1 cKO mice. e Violin plots showing expression levels of DEGs at the EAE peak in active or inactive brain lesions of multiple sclerosis patients (two-sided Kolmogorov–Smirnov test, Up, P < 2.20 × 10−16; Down, P < 2.20 × 10−16). f Top 5 enriched MSigDB Hallmark pathways of upregulated or downregulated DEGs in astrocytes from NR3C1 cKO mice at the EAE peak. P values were calculated by one-sided hypergeometric test. g Violin plots showing gene expression scores of TNF-α signaling (P = 2.89 × 10−15), IFN-γ signaling (P = 0.015), and IL-2/STAT5 signaling (P = 8.77 × 10−5). The box indicates the IQR, and the whiskers represent the highest and lowest points within 1.5 × IQR. h Distribution of fold change values between Cre− control and NR3C1 cKO for GR-suppressing LPS responsive genes (P = 9.66 × 10−15). P values were calculated by two-sided Kolmogorov–Smirnov test.
To further characterize the immune dysregulation in NR3C1 cKO mice with EAE, we analyzed the enrichment of Hallmark pathways in EAE-responsive DEGs (see Methods). We found that TNF-α signaling genes were downregulated, whereas type II interferon, IL-2, and IL-6-responsive genes were upregulated in NR3C1 cKO astrocytes (Fig. 5f). Consistently, expression patterns of TNF-α signaling, type II interferon, and IL-2/STAT5-responsive genes in active MS lesions mirrored those observed in NR3C1 cKO astrocytes (Fig. 5g), indicating a global immune dysregulation upon NR3C1 depletion. Lastly, NR3C1 is known to bind to the glucocorticoid response element (GRE) to activate target genes, while also tethering to the chromatin through the interaction with AP-1 or NF-κB proteins to mediate negative regulation32. Given the dual functional roles of NR3C1, we explored genes repressed by NR3C1 in mouse embryonic fibroblasts (MEFs) following lipopolysaccharide (LPS) and dexamethasone treatment33. Interestingly, genes that were repressed by NR3C1 upon immune challenge were upregulated in NR3C1 cKO mice at the peak of EAE (Fig. 5h). These findings suggest that the exacerbated immune responses observed in NR3C1 cKO astrocytes during EAE reflect the molecular signatures of MS, likely resulting from the loss of the anti-inflammatory function of NR3C1-dependent gene regulation.
NR3C1 depletion epigenetically predisposes inflammatory response genes
Despite the lack of observable phenotypes in NR3C1 cKO astrocytes in non-EAE conditions, NR3C1 cKO astrocytes exhibited an exacerbated EAE phenotype characterized by upregulation of inflammatory genes, such as IFN-γ and IL-2. To explain this phenomenon, we hypothesized that NR3C1 depletion may epigenetically prime these inflammatory genes during the early postnatal stage, thereby creating selective vulnerabilities to EAE induction. To test our hypothesis, we examined gene activity scores and cCRE accessibility by performing ATAC-seq with non-EAE astrocytes purified from P17 Cre− control and NR3C1 cKO mice (see Methods, Supplementary Fig. 6a).
As expected, commonly up- or downregulated genes in NR3C1 cKO mice with or without EAE induction showed concordant up or down gene expression and gene accessibility before EAE induction (Supplementary Fig. 6b, c). However, surprisingly, a substantial proportion of EAE-specific upregulated genes exhibited higher gene accessibility in NR3C1 cKO astrocytes even before EAE induction, although their expression levels were not distinguishable at this stage (top in Fig. 6a, b). In contrast, both gene expression and accessibility of EAE-specific downregulated genes were already altered by NR3C1 depletion before EAE induction (bottom in Fig. 6a, b). Next, we examined the cCRE accessibility linked to the up- or downregulated EAE-specific responsive genes. Using long-range cCRE-promoter interactions defined by ABC score in P17 astrocytes, we identified 936 and 1580 cCREs linked to up- and downregulated EAE-responsive genes, respectively (see Methods). Sorting these cCREs based on the change of accessibility with or without NR3C1 cKO, we observed that many of these cCREs were either opened or closed, concordantly, prior to EAE induction (Fig. 6c), suggesting epigenetic predisposition of EAE-responsive genes. As exemplified by Pxdn, an upregulated gene following EAE induction, the accessibility of the gene body and the linked cCREs were increased even before EAE induction, while its expression was comparable in NR3C1 cKO astrocytes at the naive condition (Fig. 6d).

a Heatmap showing gene expression and gene body accessibility of EAE-specific DEGs (n = 526 for upregulated and n = 1019 for downregulated). Expression fold changes at EAE peak, expression levels at naive, and accessibility of gene body in naive P17 astrocytes were shown. b Boxplots showing the gene expression and chromatin accessibility of EAE-specific DEGs shown in Fig. 6a. Two-sided Kolmogorov–Smirnov test, Up-accessibility P < 2.20 × 10−16, Down-expression P < 2.20 × 10−16, Down-accessibility P < 2.20 × 10−16. The box indicates the IQR, and the whiskers represent the highest and lowest points within 1.5 × IQR. c Line plots represent the ratio of open-to-closed cCREs linked to EAE-specific up- and downregulated genes. A value of 1 (or 0) indicates that all linked cCREs are upregulated (or downregulated) in NR3C1 cKO mice. d Genome browser snapshots of representative EAE-responsive upregulated genes (Pxdn). e Heatmap showing gene expression and gene body accessibility of EAE-specific upregulated genes (n = 526). Expression levels and accessibility of the gene body in astrocytes from 12 W naive mice following P0-induced NR3C1 cKO were shown (left). Boxplots showing the gene expression and chromatin accessibility of EAE-specific upregulated genes (right). Two-sided Kolmogorov-Smirnov test, accessibility P = 9.59 × 10−8. The box indicates the IQR, and the whiskers represent the highest and lowest points within 1.5 × IQR. f Venn diagram showing the overlap between differentially accessible peaks in three groups: astrocytes from P17 naive NR3C1 cKO mice (P0-induced NR3C1 cKO), astrocytes from 12 W naive NR3C1 cKO mice (P0-induced or P30-induced NR3C1 cKO) (left). Bar graph showing the overlap of DAPs linked to EAE-specific upregulated genes with DAPs in astrocytes from 12 W naive NR3C1 cKO mice (P0-induced or P30-induced NR3C1 cKO) (right).
Next, to determine whether NR3C1-dependent epigenetic predisposition persists into later stages, we performed RNA-seq and ATAC-seq using astrocytes from 12-week-old (W) mice following NR3C1 cKO at P0 (P0-induced) (Supplementary Fig. 6d). Interestingly, we found that gene expression patterns and altered gene accessibility persisted even at 12 W (Fig. 6e). Further, of the 581 differentially accessible peaks linked to EAE-specific upregulated genes at P17, 33.6% retained their altered chromatin accessibility at 12 W (Fig. 6f), supporting the long-lasting effect of early NR3C1 depletion on the epigenetic priming of immune response genes in EAE.
We further assessed the developmental stage-specificity of this effect by comparing NR3C1 cKO astrocytes induced at P0 (early postnatal) versus P30 (mature stage). RNA-seq and ATAC-seq analyses at 12 W revealed that P30-induced NR3C1 cKO failed to recapitulate the epigenetic alterations observed in P0-induced NR3C1 cKO (Supplementary Fig. 6e, f). Only 5.5% of the differentially accessible peaks linked to EAE-specific upregulated genes were retained in P30-induced NR3C1 cKO (Fig. 6f). Furthermore, the differentially expressed genes at 12 W in P30-induced cKO mice exhibited distinct functional enrichment patterns compared to those observed in P0-induced cKO mice at the same time point (Supplementary Fig. 6g). These findings underscore a critical developmental window in which NR3C1 regulates long-lasting epigenetic programming of astrocytic immune-responsive genes. Collectively, our findings suggest that NR3C1 depletion in early postnatal stages alters the epigenetic state of inflammatory genes in astrocytes, thereby predisposing them for future activation.
Context-dependent regulatory role of NR3C1 in epigenetic predisposition
Previous studies have suggested that NR3C1 dimerization may activate gene expression, while its interaction with other TFs, such as AP-1 and NF-κB, may repress gene expression, although both functions require NR3C1 binding to the genome33,34. To test this possibility and confirm that the observed epigenetic predispositions were directly driven by NR3C1 depletion, we performed NR3C1 and c-JUN ChIP-seq on astrocytes (see Methods, Fig. 7a, Supplementary Fig. 7a, b). NR3C1 ChIP-seq peaks were significantly enriched in cCREs linked to both up- and downregulated genes, indicating that the abnormal EAE-responsive gene expression is directly associated with NR3C1 binding (Fig. 7b).

a Genome browser snapshot of representative EAE-responsive upregulated genes (Mxi1) with NR3C1 and c-JUN binding in cultured primary astrocytes. b Pie charts illustrating the proportion of NR3C1-bound cCREs associated with upregulated genes (left), downregulated genes (middle), and cCREs not linked to dysregulated genes (right) at the peak of EAE. c Boxplot showing the distance between dysregulated genes (n = 704 for upregulated, n = 1160 for downregulated) and the nearest NR3C1-only binding sites or NR3C1/c-JUN co-bound regions. Two-sided Mann–Whitney test, NR3C1-only, upregulated P = 9.04 × 10−2, downregulated P = 2.82 × 10−23; NR3C1/c-JUN co-bound, upregulated P = 5.36 × 10−3, downregulated P = 1.23 × 10−25. The box indicates the IQR, and the whiskers represent the highest and lowest points within 1.5 × IQR. d Top 5 enriched TF motif sequences in cCREs for EAE-responsive genes. P values were calculated by a one-sided hypergeometric test. e A schematic overview illustrating the impact of NR3C1 depletion-induced epigenetic predisposition on the selective vulnerability of immune responses in astrocytes.
To investigate the context-dependent regulatory role of NR3C1, we categorized genomic regions into NR3C1-only and NR3C1/c-JUN co-bound sites, where c-JUN is a core component of the AP-1 complex. We found that downregulated genes were located near both NR3C1-only and co-bound regions, whereas upregulated genes were predominantly associated with NR3C1/c-JUN co-bound regions, compared to control regions (Fig. 7c, see Methods). These results suggest that NR3C1 may exert distinct regulatory effects in astrocytes depending on its binding partners35. We also performed motif analysis of cCREs linked to EAE-upregulated and -downregulated genes. Notably, cCREs associated with upregulated genes were enriched for STAT5, BCL6, and STAT6 motifs, implying the involvement of additional immune-regulatory factors cooperating with NR3C1 in astrocytes (Fig. 7d; Supplementary Data 7 and 8).
Collectively, our data suggest that NR3C1 functions as a key TF for modulating astrocytic immune response from the early postnatal developmental stages. Although NR3C1 depletion alone does not result in observable phenotypes, it epigenetically predisposes astrocytic immune-responsive genes, which persist in later stages (Fig. 7e, left). Upon immune stimulation, these primed cCREs rapidly induce the expression of inflammatory genes, triggering heightened immune responses in the context of EAE (Fig. 7e, right). Furthermore, the subtle reduction of NR3C1 in the corpus callosum of MS patients suggests that the role of NR3C1 in brain immune responses extends beyond the mouse EAE model (Supplementary Fig. 7c, P = 7.93 × 10−2)36.
Discussion
We explored how astrocytes acquire their functional identities through stage-specific gene regulatory networks during development. Leveraging a multi-omics approach, including transcriptome, cCRE activity, and long-range cCRE-promoter interactions, we delineated the dynamic regulatory landscape across multiple developmental stages (E16 to P30), uncovering critical temporal windows of astrocyte maturation. Our integrative profiling revealed key transcriptional and epigenetic transitions, including the identification of NR3C1 and other TFs such as IEGs and nuclear receptors that are transiently active during the mid-developmental window (P1-P7). These TFs likely contribute to establishing diverse astrocytic functions. Among these, NR3C1 (encoding the glucocorticoid receptor, GR) emerged as a central regulator. Its depletion during early postnatal development led to persistent epigenetic dysregulation and heightened immune responses upon EAE induction, despite the absence of observable phenotypes at baseline. These findings highlight a broader principle that TF activity during discrete developmental windows can establish long-term epigenetic predispositions that increase disease susceptibility later in life. Our finding is consistent with recent studies implicating astrocyte epigenetic memory in CNS pathology37.
Our study highlights the pathological relevance and therapeutic implication of NR3C1 encoding the GR, a TF activated by GC binding that regulates anti-inflammatory transcriptional programs38. Given the widespread clinical use of GCs to manage acute MS relapses, understanding the cellular and molecular mechanisms of NR3C1 in the CNS is of critical therapeutic importance. In humans, methylation of NR3C1 has been associated with perinatal stress, such as maternal psychological problems, parental divorce, or hospitalization39. The heightened DNA methylation reduces NR3C1 expression in both peripheral blood and brain tissues40. Consistently, NR3C1 reduced expression has also been observed in MS patients compared to healthy controls, suggesting a potential link between NR3C1 dysregulation and MS pathology41.
Beyond its diagnostic relevance, NR3C1 may be subject to long-lasting modulation following GC therapy, requiring careful consideration in treatment contexts. GC treatment is known to induce lasting epigenetic changes in immune cells. For instance, dexamethasone exposure causes transient demethylation at FKBP5 enhancer regions, upregulating its expression42. Elevated FKBP5, in turn, impairs GR nuclear translocation and reduces its transcriptional activity, thereby contributing to GC resistance43. In MS patients, the high-dose methylprednisolone treatment triggers the persistent hypomethylation of ZBTB16 in the peripheral blood, further supporting the idea that GC-induced epigenetic imprints can be long-lasting in patients44. These findings collectively suggest that GC therapy can reprogram immune cells through durable epigenetic mechanisms. Given that NR3C1 similarly governs immune regulation in astrocytes, it is plausible that repeated GC exposure may induce comparable epigenetic modifications in astrocytes. However, further research is needed to demonstrate whether GC treatment induces epigenetic changes in astrocytes.
Several challenges still remain in fully elucidating the molecular mechanisms of astrocyte development. Firstly, our findings were based on mouse models, which may not fully reflect the complexity of human astrocyte development and disease etiology. Future investigations should focus on validating our results in in vitro differentiated human astrocytes or human brain organoids. Second, the precise epigenetic mechanisms underlying NR3C1-dependent immune suppression remain to be clarified. While we show that the combinatorial activities between NR3C1 and c-JUN have distinct regulatory effects on EAE-responsive genes (Fig. 7c), a more comprehensive analysis is required to identify additional cofactors that interact with NR3C1. These further analyses would help explain how early postnatal NR3C1 depletion leads to long-lasting epigenetic priming of immune-responsive genes. Additionally, while our study emphasizes the role of early postnatal NR3C1 on susceptibility to exacerbated immune responses, further investigation is needed to determine the exact duration and impact of the epigenetic priming in astrocytes across various immune-related diseases beyond MS. Lastly, although we focused on the NR3C1 depletion, our study also revealed the involvement of 16 other TFs during the mid-stage development. Also, NR3C1 belongs to the nuclear receptor subfamily 3, group C, which also includes mineralocorticoid receptor (MR, NR3C2), androgen receptor (AR), and progesterone receptor (PGR). These receptors share structural similarities and chromatin-binding sequences, often interacting in a context-dependent manner. Recent studies have highlighted that NR3C2 functions as a TF during astrocyte development45 and an immune modulator46. Given these findings, further studies are needed to determine the potential functional interplay between these nuclear receptors and other mid-stage-specific TFs in shaping astrocyte identity and immune responses.
Methods
Aldh1l1-EGFP and NR3C1 cKO mice generation
All experimental procedures performed in the study were approved by the Institutional Animal Care and Use Committee (IACUC, KA2024-074) of the Korea Advanced Institute of Science and Technology (KAIST). Aldh1l1-EGFP mice generated by the GENSAT project were obtained from the Barres laboratory17. FVB-Tg(Aldh1l1-cre/ERT2)1Khakh/J mice and B6.Cg-Nr3c1tm1.1Jda/J mice were obtained from the Jackson Laboratory. Tg(Aldh1l1-cre/ERT2)1Khakh/J mice were crossed with B6.Cg-Nr3c1tm1.1Jda/J mice to generate astrocyte-specific NR3C1 conditional knockout (NR3C1 cKO) mice. Then, to induce NR3C1 deletion during the early developmental stage, 65 μg of 4-Hydroxytamoxifen (Sigma-Aldrich) per 1 g of body weight was administered to all littermates by subcutaneous (s.c.) injection for two consecutive days from postnatal day 0 (P0) to P1. For 4-Hydroxytamoxifen treatment, 50 mg of 4- Hydroxytamoxifen (Sigma-Aldrich) was dissolved in 1.25 ml of absolute ethanol (Merck) and 1.25 ml of Koliphor EL (Sigma) to make a stock of 20 mg/ml and stored at −20 °C. Before use, the 100 μl of the stock solution was diluted in 200 μl of 1× DPBS (Welgene). Both sexes of Aldh1l1-EGFP and NR3C1 cKO mice were used for multi-omics analyses of developmental astrocytes. FVB/N female mice were purchased from Daehan BioLink (DBL) and KOATECH. FVB/N female mice were used for in vitro fertilization with Aldh1l1-EGFP mice. Aldh1l1-creERT2 mice were backcrossed with BL6/J mice more than 10 generations to minimize genetic variability and reduce the potential effects of mixed background. To minimize variability in disease onset and severity, only female mice were used for EAE induction. All mice were housed in specific pathogen-free (SPF) conditions, in a temperature-controlled (20–24 °C) and humidity-controlled (40–60%) environment, under a 12 h light-dark cycle with free access to chow and water.
Astrocyte collection during developmental stages
Purified astrocytes from Aldh1l1-gfp;FVB/N mice (total of four mice, comprising both males and females) were used for multi-omics analyses and WT;FVB/N littermates were used for FACS sorting as a negative control. Astrocytes were isolated from E16.5, E18.5, P1, P3, P7, P17, and P30 mice by modified immunopanning47 and FACS sorting. Briefly, for embryonic mice, pregnant female mice were anesthetized with 2% isoflurane, and the abdomen was surgically opened to collect embryos. The cortex of each embryonic pup was prepared by decapitation. After surgery, the pregnant females were euthanized by carbon dioxide (CO2) inhalation. P0-P3 pups were placed on ice for ~10 minutes for anesthesia. P17 and adult mice were euthanized by CO2 inhalation before cortical tissue collection. Then, the cerebral cortices were dissected, and the meninges were removed. To dissociate cortices, they were chopped and enzymatically digested with papain (Worthingtons). Then, the cortices were dissociated into single cells through serial trituration. To remove macrophages and microglia, the single cell suspension was incubated on Donkey anti-goat IgG (H + L) (Jackson Laboratories, 705-005-003)-coated dish. Then, the supernatant (single-cell suspension) was collected for FACS sorting. To avoid tube clogging during FACS sorting, myelin debris was removed from samples over P7 using myelin debris removal beads (Miltenyi Biotec), following the manufacturer’s instructions.
Astrocyte collection from NR3C1 cKO mice
Magnetic separation was used to obtain astrocytes from Cre− control and NR3C1 cKO mice. Briefly, mice were euthanized by CO2 inhalation before cortical tissue collection. Then, cerebral cortices from P17 mice were dissected out and enzymatically digested in papain solution (Worthingtons). Then, the cortices were mechanically dissociated through serial trituration. After dissociation, myelin was removed using the myelin removal beads (Miltenyi Biotec). The single-cell suspension was incubated with anti-ACSA-2 microbeads (Miltenyi Biotec), loaded on columns, and collected to obtain astrocytes. The collected astrocytes were subjected to small-scale RNA-seq and ATAC-seq.
Small-scale RNA sequencing and data processing
Total RNA was extracted from 100,000 purified cells using the NucleoSpin RNA XS kit (Macherey-Nagel, 740902). The isolated RNA was then converted into cDNA utilizing the Ovation RNA-Seq System (TECAN, M01206), following the manufacturer’s instructions. To generate RNA-seq libraries, the cDNA underwent fragmentation, targeting lengths of 200 to 600 base pairs by sonication (Covaris, S220). RNA-seq libraries were manually generated through a series of steps, including DNA end repair (Lucigen, ER0720), adenosine addition at the 3’ end (NEB, M0212), and ligation of indexed adapters (NEB, M2200). The libraries were subjected to PCR amplification (ThermoFisher Scientific, F549), with the number of cycles optimized based on DNA quantification via qPCR (KAPA, KK4854). Finally, we sequenced the RNA-seq libraries with paired-end mode using the Illumina NextSeq 550 platform (San Diego).
The sequenced reads were aligned to the mouse reference genome (GRCm38.p6, GENCODE) using TopHat (v2.1.1)48 incorporating gene annotation information. We then quantified gene expression with StringTie (v1.3.5)49, generating a gene count matrix. Differential expression analysis during astrocyte development was conducted using EdgeR (v3.32.1)50. Briefly, genes with low expression levels were filtered, and scaling factors were calculated to normalize library sizes. We then modeled gene expression patterns across seven time points using a spline curve with 3 degrees of freedom. The genes were defined as DEGs when the FDR was less than 0.05.
Bulk ATAC sequencing and data processing
The 50,000 purified cells were lysed with a Resuspension buffer composed of 10 mM Tris-HCl (pH 7.5), 10 mM NaCl, 3 mM MgCl2, 0.1% Tween-20 (Sigma-Aldrich, P7949), and 1% BSA, supplemented with 0.1% IGEPAL CA-630 (Sigma-Aldrich, 18896), 0.01% digitonin (Invitrogen, BN2006). The detergents were diluted within the Resuspension buffer immediately after incubation on ice. Subsequently, the buffer was removed from the nuclei through a washing step, and a transposition mix (Illumina, 15027865) was applied for digestion and ligation of adapters at 37 °C for 30 minutes. The fragmented DNA was purified (QIAGEN, 28004) and amplified using indexed adapter primers (NEB, M0541). The number of PCR cycles was optimized based on qPCR DNA quantification (KAPA, KK4854). Finally, we sequenced the ATAC-seq libraries with paired-end mode using the Illumina NextSeq 550 platform.
For the data processing, we initially trimmed the Nextera adapter sequences from the raw sequencing reads utilizing Cutadapt (v1.16). Then, the reads were aligned to the mouse reference genome (GRCm38.p6) using Bowtie2 (v2.3.4)51. The low-quality alignments (MAPQ < 30) and mitochondrial reads were removed, and PCR duplicates were eliminated using Picard (v2.6.0). To correct the position of Tn5 transposase insertion sites, reads were shifted to +4 or −5 base pairs for the positive or negative strand, respectively. We defined peaks using MACS2 (v2.2.7.1)52 in each sample with default parameters, except for q value = 0.01, nomodel, and shift = −100. After combining the peaks, we quantified the accessibility using featureCounts (v2.0.0)53 function. Differential accessibility analysis followed the same procedures as RNA-seq analysis, utilizing EdgeR.
HiCAR experiment and data processing
The experimental procedures of HiCAR were adapted from a previous study with slight modifications16. Initially, 100,000 cells were crosslinked with 1% formaldehyde at room temperature for 10 mins, followed by quenching of the crosslinking activity with 0.2 M glycine for 10 mins. After centrifugation, nuclei were extracted by treatment with NPB (PBS containing 5% BSA, 1 mM DTT, 0.2% IGEPAL, Roche Complete Protease Inhibitor, 12.5 μl RNaseOUT) and incubated at 4 °C for 15 mins. The extracted nuclei were then treated with 50 μl 2× TB buffer (66 mM Tris-AC pH 7.8, 132mM K-AC, 20 mM Mg-AC, 32% DMF), 15 μl water, and 5 μl Tn5 for HiCAR. Tagmentation reaction was activated at 37 °C for 1 h with rotation, followed by termination with 350 μl EDTA (40 mM) treatment. After washing the nuclei with PBS containing 0.075% BSA, the nuclei were resuspended in 36.5 μl water, 1 μl 5 M NaCl, and 12.5 μl 2% SDS, and incubated at 62 °C for 10 mins.
To deactivate the SDS activity, a mixture of 112 μl water, 2.8 μl 5 M NaCl, and 25 μl 10% Triton X-100 was added and incubated at 37 °C for 15 mins with rotation. The nuclei were then washed with 300 μl 1.1× NEBuffer 3.1, followed by treatment with 90 μl 1.1× NEBuffer 3.1, 100U CviQI, and 2 μl splint oligo (20 μM, ACACGACGCTCTTCCGATCT) at room temperature for 2 h with rotation. For in situ ligation, a mixture comprising 48 μl of 10× T4 ligation buffer, 6 μl of T4 DNA ligase, 2.4 μl 20 mg/ml BSA, 40 μl 10% Triton X-100, and 283.6 μl H2O was added, and the nuclei were rotated at room temperature for 4 hours. To reverse crosslink, 2× RCB buffer (100 mM Tris-HCl pH 8.0, 100 mM NaCl, 0.4% SDS) was mixed and incubated at 68 °C for at least 1.5 h.
Subsequently, DNA was precipitated by treating with 2 μl glycogen, 20 μl NaAC, and 500 μl ethanol and incubating at −80 °C for at least 30 mins. The DNA was centrifuged at 14,000 × g for 7 min at 4 °C. After washing the DNA pellet twice with 75% ethanol, it was dissolved in 21 μl 10 mM Tris-HCl (pH 8.0). DNA concentration was measured and adjusted to 1 ng/μl for the following steps. 10× T4 ligation buffer and T4 DNA ligase were added in amounts equivalent to 0.05× and 0.012× of the volume of DNA, respectively, and circularization was activated at room temperature for 2 h. The circularized DNA was purified using a DNA clean & concentrator kit, eluting in 16 μl 10 mM Tris-HCl (pH 7.5). Then, HiCAR libraries were constructed using the purified DNA and sequenced, skipping three cycles for read 1, using the MGI DNBSEQ-T7 platform.
The datasets were processed according to a previously reported pipeline. In summary, the sequencing outputs were aligned to the reference genome (mm10) using BWA-mem54 with “-SP” option. Subsequently, the aligned reads with low mapping quality or those with the same genomic coordinate were filtered out. PCR duplicates were removed, and the uniquely aligned pairs were rearranged based on genomic coordinates. The normalized Hi-C contact maps were generated using covNorm20 (ver. 1.0.0) to find the significant chromatin contacts at 5 kb resolution.
Prediction of cCREs and their target genes using ABC model
We employed the ABC model to predict the putative cCREs for each gene and their relative activity21. The ABC model evaluates the regulatory effect on a gene by considering both the strength of cCRE activity and the contact frequency with a target gene. Briefly, we defined cCRE activity as the normalized chromatin accessibility and chromatin contact frequency as KR-normalized HiCAR contact frequency between the cCRE and the promoter at 5 kb resolution. ABC scores were derived by multiplying the cCRE activity and chromatin contacts. Subsequently, the ABC scores were normalized by the sum of all ABC scores within 1 Mb from the target gene. The putative cCREs were defined if the ABC scores were greater than 0.02.
Prediction of TF activity scores
We employed the PageRank algorithm via Taiji22 (v1.3.0) to predict TF activity. In brief, we defined promoters and cCREs using ATAC-seq peaks called by MACS with a P value cutoff of 5 × 10−3. To predict TF binding sites, we searched for 1,346 mouse motifs from chromVARmotifs (https://github.com/GreenleafLab/chromVARmotifs) within each peak using the FIMO algorithm55. Subsequently, the cCREs harboring each TF motif sequence were assigned to the nearest genes to create a regulatory network. Finally, TF activity scores were calculated within the network using a personalized PageRank algorithm implemented in Taiji’s analysis. For further analysis, we focused on the TFs exhibiting (1) significant differential expression throughout astrocyte development, (2) significant TF activity, (3) high maximum activity score (1.5 × 10−3), and (4) high variability of TF activities (higher than twofold changes between the maximum and minimum activities across different time points).
Analyses of scRNA-seq dataset of mouse developing cortices
We collected publicly available scRNA-seq datasets of mouse developing cortices23. To align with our datasets, we isolated cortical cells from mice within the age range of E13 to P7. We further normalized the gene expression using total read counts and then log-transformed. The highly variable genes were selected using ‘highly_variable_genes’ function implemented in scanpy with default parameters. Using the variable genes, the normalized data were scaled, and PCA was conducted. The bbknn algorithm56 was applied to align the data from each sample based on the top 50 principal components (PCs). Finally, the cells were clustered by unsupervised clustering and visualized by the UMAP with default parameters. For the comparative analysis between neurogenic and astrocytic lineages, we focused on cell types, including radial glia, glioblasts, and neuroblasts in the glutamatergic lineage, as well as glutamatergic neurons, as annotated in previous studies23.
Further analysis was conducted separately for the neurogenic lineage and astrocytic lineage. Gene count matrices were normalized and scaled, followed by performing PCA. To minimize experimental bias in clustering analysis within a cell type, cells from different samples were aligned using harmony based on the top 10 PCs. Then, trajectory analysis was performed using paga algorithm in scanpy. A cell from the radial glial cell cluster was designated as the root cell, and diffusion maps and pseudotime of each lineage were calculated. To compare the expression between the neurogenic and astrocytic lineages, the expression pattern of each TF along the pseudotime was fitted to a spline function.
in silico TF perturbation analysis
We performed in silico TF perturbation analysis using CellOracle24. The processed scRNA-seq data was integrated with a gene regulatory network (GRN) derived from the mouse sci-ATAC-seq atlas57. We then conducted PCA and KNN imputation, using the top 39 PCs to calculate the top 250 nearest neighbors (NNs). Meanwhile, the GRNs were reconstructed for all clusters using ‘get_links’ function, incorporating cluster information. The edges exhibiting low significance and weak scores were discarded for stringent network construction. For the simulation, we combined the active TFs from both lineages, resulting in 219 TFs to be simulated. The simulation analysis produced a negative perturbation score (PS), calculated as the inner-product value between the original differentiation vector and TF KO simulation vector. To compare the perturbation between two lineages, the negative PS scores were normalized by the number of grid points in each lineage.
Experimental autoimmune encephalomyelitis
EAE was induced in 9-week-old female Cre− control and NR3C1 cKO mice by subcutaneously injecting 200 μl of MOG35-55 in complete Freund’s adjuvant (Hooke Laboratories). This was followed by the administration of 110 ng of pertussis toxin in PBS on the first day of immunization and again the following day. Mice were monitored until the end of the experiment. Clinical scores and weight were measured daily for 30 days post injection. EAE clinical scores were defined as follows: 0, no signs; 1, limp tail; 2, weakness of hind legs; 3, paralysis of hind legs; 4, partial front leg paralysis; 5, moribund. All mice that reached a clinical score of 5 were considered as having a clinical score of 5 from the day they reached this score until the end of the scoring period.
Immunohistochemistry (IHC) and image analysis
Mice were anesthetized with avertin (0.2 g/ml, Sigma-Aldrich) by intraperitoneal (i.p.) injection. The mice were perfused with DPBS followed by 4% paraformaldehyde (PFA, Wako). The spinal cords and brains were isolated and postfixed in 4% PFA overnight at 4 °C and then transferred to 30% sucrose (Sigma-Aldrich)/DPBS for 48 hours. The brains were embedded in optimal cutting temperature (OCT) compound (Leica Microsystems) and quick-frozen on dry ice. Four 30-μm brain sections were obtained with a cryostat (Leica MICROSYSTEMS) and placed in DPBS in one well of a 48-well plate. For the spinal cords, 15 μm sections of lumbar regions were obtained by cryostat (Leica MICROSYSTEMS) on a slide glass (Matsunami) and stored in −20 °C. Sections were washed in DPBS to remove OCT compounds. Using ImmEdge Hydrophobic Barrier pen (Vector Laboratories), the barrier was drawn outside of spinal cord sections. The sections were blocked with blocking solution for 1 hour at RT and incubated with primary antibodies for 48 hours at 4 °C with gentle agitation. The blocking solution consisted of 4% bovine serum albumin (BSA, Goldbio), 0.3∼1% Triton X-100 (Sigma), and DPBS. The following primary antibodies were used: rabbit anti-S100β (Abcam, ab52642, 1:500), chicken anti-GFAP (Aves Labs, GFAP, 1:500), rabbit anti-GR (Cell signaling Technology, 12041S, 1:500), rabbit anti-IBA1 (WAKO, 019-19741, 1:500), chicken anti-GFP (Aves Labs, GFP-1020, 1:1000), rat anti-CD45 (BD, 550539, 1:500), and rat anti-CD4 (Biolegend, 100506, 1:100). The brain sections were washed in DPBST (DPBS containing 0.1% Tween-20) and treated with Alexa fluorophore-conjugated secondary antibodies (Alexa Fluor 488, Alexa Fluor 594, Alexa Fluor 405, Alexa Fluor 647, Abcam, Jackson Laboratory, and Invitrogen) in blocking solution for 2 hours at RT with gentle agitation. The sections were washed five times with DPBST and then mounted on a slide glass (Matsunami) with a coverglass and Vectashield with or without DAPI (Vector Lab).
Images were acquired with a Nikon upright microscope and Zeiss LSM 780 inverted and Zeiss LSM 880 upright confocal microscopes. Confocal imaging analyses were performed with two different brain slices per mouse. For each brain slice, two sets of confocal z-stacks containing multiple single-plane confocal images were obtained from the regions of interest (ROIs). Imaging analysis was performed using individual single-plane confocal images, which were averaged to produce one data point per z-stack, thus producing four data points per mouse. All images and representative z-stack images were processed using Fiji (ImageJ).
Isolation of a single cell suspension from the adult CNS
At the peak day of EAE induction, brain and spinal cord cells were isolated. Briefly, mice were anesthetized with avertin (0.2 g/ml, Sigma-Aldrich) and perfused with DPBS. Brains and spinal cords were separately digested in an enzyme solution containing 90 units of Papain suspension (Worthington, LS003126) for 45 minutes. After enzymatic digestion, the tissues were mechanically dissociated through serial trituration using low ovomucoid diluted in an inhibitor solution. The inhibitor solution consisted of 0.46% D(+)-glucose and 26 mM NaHCO3 in an EBSS base. The low ovomucoid solution contained 3 g BSA (Sigma, A8806) and 3 g trypsin inhibitor dissolved in 150 ml DPBS. After dissociation, the cell suspension was filtered through a 70 μm cell strainer (SPL) and centrifuged at 150 × g for 10 minutes. The resulting cell pellets were suspended in 30% Percoll solution (Sigma-Aldrich) and centrifuged at 500 × g for 25 minutes at RT. CNS cell pellets were collected for further applications.
T cell stimulation
Brain and spinal cord cells were stimulated with 50 ng/ml phorbol 12-myristate 13-acetate (PMA, Sigma-Aldrich, P8139), 1 μM Ionomycin calcium salt (Sigma-Aldrich, I3909), GolgiStop (BD, 554724), and GolgiPlug (BD, 555029) diluted in RPMI medium (Gibco) supplemented with 10% FBS, 1% penicillin/streptomycin, and 50 μM 2-mercaptoethanol (Gibco, 21985023) for 4 hours in a cell culture dish. After stimulation, cell suspensions were collected and centrifuged at 220 × g for 5 minutes for further staining.
Flow cytometry
Cells were stained with Zombie Aqua (BioLegend, 423101) for live/dead staining for 10 minutes. Cells were then washed with FACS buffer consisting of 0.5% BSA and 2 mM EDTA in DPBS. To block nonspecific antibody binding, cells were incubated with anti-mouse CD16/32 (BioLegend, 101302) for 10 minutes, followed by surface staining with antibodies for 30 minutes in the dark. The following surface antibodies were used: FITC anti-mouse CD45 (BD, 553079, 1:100), PE anti-mouse CD11b (BD, 553311, 1:100), PerCP/Cyanine5.5 anti-mouse Ly-6G (BioLegend, 127615, 1:100), APC/Cyanine7 anti-mouse CD11c (BioLegend, 117323, 1:100), APC/Cyanine7 anti-mouse CD4 (BioLegend, 100525, 1:100), PE/Cyanine7 anti-mouse CD8a (BioLegend, 100721, 1:100). After staining, cells were washed with FACS buffer and prepared for flow cytometry analysis. For intracellular staining, after surface staining, cells were washed with FACS buffer, then fixed and permeabilized using Foxp3/TF Staining Buffer Set (eBioscience, 00-5523-00). After fixation and permeabilization, cells were incubated with intracellular antibodies. The following antibodies were used: PE anti-mouse Foxp3 (BioLegend, 126403, 1:100), BV510 anti-mouse IFN-γ (BioLegend, 505841, 1:100), and BV421 anti-mouse IL17A (BD, 566286, 1:100). After staining, cells were washed with FACS buffer and prepared for flow cytometry analysis. Compensation was performed with single-stained UltraComp eBeads (Thermo, 01-2222-42) and unstained CNS cells. Flow cytometry was performed on a BD FACSCanto 2 (BD) and analyzed using FlowJo software.
Single-nucleus RNA-seq of EAE mouse cortices
To obtain samples for snRNA-seq, EAE-induced female mice were euthanized by CO2 inhalation and cerebral cortices were dissected and enzymatically digested in papain solution (Worthingtons). After, the cells were dissociated into single cells through mechanical trituration. Nuclei were then extracted by treating them with lysis buffer (0.2% Triton X-100, Roche protease inhibitor, 1 mM DTT, 0.2 U/μl RNAsin, and PBS containing 2% BSA) for 5 min at 4 °C. Next, the nuclei were filtered using a 40μm cell strainer to prevent aggregation, and their count was measured to ensure the appropriate amount of nuclei was loaded onto the microfluidic devices. snRNA-seq libraries were generated using the Chromium Single Cell 3′ Library & Gel Bead Kit v3.1 (10× Genomics, Pleasanton, CA) following the manufacturer’s instructions. The nuclei were loaded to target 15,000 nuclei of the output. The libraries were constructed and sequenced to a depth of ~30,000 reads per cell using the MGI DNBSEQ-G400 platform.
The sequenced libraries were aligned to the mouse reference genome (10× Cell Ranger reference mm10 v2.1.0), which generated gene count matrices utilizing cellranger count. Subsequently, all matrices were merged using cellranger aggr using default parameters. The merged feature-barcode matrix underwent processing using the Seurat R package v3.1.558. To filter out low-quality nuclei data, we remove nuclei that expressed fewer than 500 genes, had more than 10,000 unique molecules, or had more than 2.5% mitochondrial genes of their total gene expression. Clusters of doublets were excluded based on predicted scores using scDblFinder in Seurat’s implementation. Gene expression counts were normalized based on the total gene count and then log-transformed within each cell. The top 2000 highly variable genes were selected by the vst method in Seurat R package v3.1.5. The normalized matrix was scaled, and PCA was conducted. Finally, the nuclei were clustered via unsupervised clustering (resolution 0.7) and visualized by t-SNE, using the top 8 PCs.
Subpopulation analysis of EAE snRNA-seq data
To investigate cellular dynamics during EAE, astrocytes were subjected to subclustering analysis. Briefly, excluding mitochondrial and ribosomal genes, the gene count matrices were normalized. Using the top 2000 variable genes, the normalized matrices were scaled, and PCA was performed. To minimize the experimental biases at the subpopulation level, nuclei from different samples were aligned using harmony method59 in Seurat R package v3.1.5. The aligned nuclei were clustered via unsupervised clustering (with resolution 0.15) and visualized by t-SNE, using the top 8 PCs.
Glucocorticoid receptor stimulation of primary astrocyte
Primary astrocytes were isolated from postnatal day 0 (P0) mouse brain following anesthesia, by subsequently placing the animals on ice for 10 minutes (n = 10, comprising both male and female mice). Briefly, the cerebral cortices were dissected from P0 mice and enzymatically digested in trypsin-EDTA (Gibco) for 5 minutes at 37 °C. Then, the cortices were mechanically dissociated through trituration in DMEM (Welgene) supplemented with 10% FBS (Welgene) and 1% penicillin/streptomycin (Gibco). After dissociation, the cell suspension was filtered through a 70μm cell strainer (SPL). The single-cell suspension was plated in T175 culture flask (SPL). After maintaining for 1 week, the flasks were shaken at 240 rpm for 10 hours to detach microglia and OPCs. The supernatant was removed, and the adherent astrocytes were detached using trypsin, and replated in culture plates for recovery. After one additional week of astrocyte culture, the glucocorticoid receptor was stimulated with 5 μM corticosterone (Sigma-Aldrich, 27840) for 24 hours and collected for further sequencing analysis. The astrocyte culture media consisted of 45% neurobasal medium (Gibco), 45% DMEM (Welgene), 10% FBS (Welgene), 1% penicillin/streptomycin (Gibco), 1 mM sodium pyruvate (Gibco), 292 μg/ml L-glutamine (Gibco), 1× SATO, and 5 ng/ml HBEGF (Sigma-Aldrich).
TF ChIP-seq and data analysis
We collected 3 million astrocytes to perform a ChIP-seq experiment for each TF. The collected cells were crosslinked twice: first with 2 mM disuccinimidyl glutarate for 35 min, followed by 1% formaldehyde for 10 minutes, both at room temperature. The crosslinking was quenched using 1.25 M glycine, and then cells were washed twice with PBS. The cells were then lysed in nucleus buffer (50 mM Tris-HCl pH 8.1, 10 mM EDTA, 1% SDS, and protease inhibitors), followed by sonication using a Covaris M220 (70 amplitude, 30-minute process time, alternating 30 seconds ON and 30 seconds OFF). Debris was removed by centrifugation, and DNA fragments were extracted and diluted ten times with dilution buffer (1% Triton X-100, 2 mM EDTA, 150 mM NaCl, 20 mM pH 8.1 Tris-HCl, and protease inhibitor). To conjugate TF-targeting antibody and protein A Sepharose (ThermoFisher), we added 2 μg of TF antibody into 10 μl beads (for NR3C1, ThermoFisher, PA1-511A; for c-JUN, Cell Signaling Technology, #9165). The antibody-conjugated protein A Sepharose was added to the diluted sample for overnight immunoprecipitations at 4 °C. Then, the beads were washed with TSE I buffer (0.1% SDS, 1% Triton X-100, 2 mM EDTA, 20 mM Tris-HCl (pH 8.1) and 150 mM NaCl), TSE II buffer (0.1% SDS, 1% Triton X-100, 2 mM EDTA, 20 mM Tris-HCl (pH 8.1) and 500 mM NaCl), buffer III (0.25 M LiCl, 1% NP-40, 1% deoxycholate, 10 mM Tris-HCl (pH 8.1) and 1 mM EDTA), three times with TE buffer (10 mM pH 8.0 Tris-HCl and 1 mM EDTA), and the eluted in elution buffer (1% SDS and 0.1 M NaHCO3). DNA was reverse-crosslinked by incubation at 65 °C overnight. After RNase and Proteinase K treatment, DNA was purified and prepared for next-generation sequencing using the NEBNext Ultra II DNA Library Prep Kit (NEB). The ChIP-seq library was sequenced using the DNBSEQ-G400 platform.
For the data processing, sequencing reads were aligned to the mouse reference genome (GRCm38.p6) using BWA-mem. Low-quality alignments (MAPQ < 10) were removed, and PCR duplicates were eliminated using Picard (v2.6.0). The signal-enriched peaks were called using MACS2 v2.2.7.1 (-q 1e-2).
Enrichment analysis using Enrichr and GSEA
To predict the disease association of developmental TFs, we performed gene set enrichment analysis by fgsea R package. TF’s regulatory activity for each gene was calculated by the sum of the ABC scores of linked cCREs harboring each TF motif sequence. The regulatory activity scores were subjected to enrichment analysis by comparing to brain disease-associated gene sets from DisGeNET26.
We used the web-based software for GSEA, Enrichr60, for identifying enrichment of MSigDB Hallmark pathway61 in DEGs from NR3C1 cKO astrocytes with or without EAE stimulation.
TF Motif enrichment analysis
The enrichment analysis for general TF motif sequences was performed by using HOMER62 with default parameters, with the exception of size=given. To compare motif enrichment between cCREs of upregulated and downregulated EAE-responsive genes, we used the cCREs of each group as the background for the other, applying the ‘bg’ option.
We used FIMO to calculate the enrichment score of a TF motif sequence in cCREs of upregulated and downregulated EAE-responsive genes with options ‘--bfile --uniform--’. The significance of motif enrichment was measured by the hypergeometric test.
Statistical analyses
We conducted the KS test to compare the distributions of two groups without assuming that the distributions follow normality. Welch’s t test was conducted to compare the two distributions when the distributions follow normality. The Mann-Whitney test was performed to compare the means of two groups. Two-way ANOVA was used for comparisons among multiple groups. P < 0.05 was considered statistically significant. Asterisks indicate P values (*P < 0.05, **P < 0.01, ***P < 0.001).
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
All raw and processed data generated in this study have been deposited in the GEO database under accession number GSE158329. The single-cell transcriptome data of developing mouse cortices were available at the Sequence Read Archive (https://www.ncbi.nlm.nih.gov/sra) under accession PRJNA637987 or at http://mousebrain.org/development/. Source data are provided with this paper.
References
Lee, H. G., Wheeler, M. A. & Quintana, F. J. Function and therapeutic value of astrocytes in neurological diseases. Nat. Rev. Drug Discov. 21, 339–358 (2022).
Kenny, E. M. et al. Excess of rare novel loss-of-function variants in synaptic genes in schizophrenia and autism spectrum disorders. Mol. Psychiatry 19, 872–879 (2014).
Kirov, G. et al. De novo CNV analysis implicates specific abnormalities of postsynaptic signalling complexes in the pathogenesis of schizophrenia. Mol. Psychiatry 17, 142–153 (2012).
Kraft, A. W. et al. Attenuating astrocyte activation accelerates plaque pathogenesis in APP/PS1 mice. FASEB J. 27, 187–198 (2013).
Knott, C., Wilkin, G. P. & Stern, G. Astrocytes and microglia in the substantia nigra and caudate-putamen in Parkinson’s disease. Parkinsonism Relat. Disord. 5, 115–122 (1999).
Krumbholz, M. et al. BAFF is produced by astrocytes and up-regulated in multiple sclerosis lesions and primary central nervous system lymphoma. J. Exp. Med. 201, 195–200 (2005).
Molofsky, A. V. & Deneen, B. Astrocyte development: a guide for the perplexed. Glia 63, 1320–1329 (2015).
Kang, P. et al. Sox9 and NFIA coordinate a transcriptional regulatory cascade during the initiation of gliogenesis. Neuron 74, 79–94 (2012).
Bonni, A. et al. Regulation of gliogenesis in the central nervous system by the JAK-STAT signaling pathway. Science 278, 477–483 (1997).
Nakashima, K. et al. Synergistic signaling in fetal brain by STAT3-Smad1 complex bridged by p300. Science 284, 479–482 (1999).
Tiwari, N. et al. Stage-specific transcription factors drive astrogliogenesis by remodeling gene regulatory landscapes. Cell Stem Cell 23, 557–571.e558 (2018).
Lattke, M. et al. Extensive transcriptional and chromatin changes underlie astrocyte maturation in vivo and in culture. Nat. Commun. 12, 4335 (2021).
Ge, W. P., Miyawaki, A., Gage, F. H., Jan, Y. N. & Jan, L. Y. Local generation of glia is a major astrocyte source in postnatal cortex. Nature 484, 376–380 (2012).
Tan, L. et al. Changes in genome architecture and transcriptional dynamics progress independently of sensory experience during post-natal brain development. Cell 184, 741–758.e717 (2021).
Sun, Y. et al. Spatiotemporal 3D chromatin organization across multiple brain regions during human fetal development. Cell Discov. 11, 50 (2025).
Wei, X. et al. HiCAR is a robust and sensitive method to analyze open-chromatin-associated genome organization. Mol. Cell 82, 1225–1238.e1226 (2022).
Cahoy, J. D. et al. A transcriptome database for astrocytes, neurons, and oligodendrocytes: a new resource for understanding brain development and function. J. Neurosci. 28, 264–278 (2008).
Li, X. et al. Decoding cortical glial cell development. Neurosci. Bull. 37, 440–460 (2021).
Gorina, Y. V. et al. Astrocyte activation markers. Biochemistry87, 851–870 (2022).
Kim, K. & Jung, I. covNorm: an R package for coverage based normalization of Hi-C and capture Hi-C data. Comput. Struct. Biotechnol. J. 19, 3149–3159 (2021).
Fulco, C. P. et al. Activity-by-contact model of enhancer-promoter regulation from thousands of CRISPR perturbations. Nat. Genet. 51, 1664–1669 (2019).
Zhang, K., Wang, M., Zhao, Y. & Wang, W. Taiji: System-level identification of key transcription factors reveals transcriptional waves in mouse embryonic development. Sci. Adv. 5, eaav3262 (2019).
La Manno, G. et al. Molecular architecture of the developing mouse brain. Nature 596, 92–96 (2021).
Kamimoto, K. et al. Dissecting cell identity via network inference and in silico gene perturbation. Nature 614, 742–751 (2023).
Gorkin, D. U. et al. An atlas of dynamic chromatin landscapes in mouse fetal development. Nature 583, 744–751 (2020).
Pinero, J. et al. DisGeNET: a comprehensive platform integrating information on human disease-associated genes and variants. Nucleic Acids Res. 45, D833–D839 (2017).
Park, S. W., Seo, M. K., Webster, M. J., Lee, J. G. & Kim, S. Differential expression of gene co-expression networks related to the mTOR signaling pathway in bipolar disorder. Transl. Psychiatry 12, 184 (2022).
Leng, K. et al. CRISPRi screens in human iPSC-derived astrocytes elucidate regulators of distinct inflammatory reactive states. Nat. Neurosci. 25, 1528–1542 (2022).
Wolf, F. A., Angerer, P. & Theis, F. J. SCANPY: large-scale single-cell gene expression data analysis. Genome Biol. 19, 15 (2018).
Schirmer, L. et al. Neuronal vulnerability and multilineage diversity in multiple sclerosis. Nature 573, 75–82 (2019).
Macnair, W. et al. snRNA-seq stratifies multiple sclerosis patients into distinct white matter glial responses. Neuron 113, 396–410.e399 (2025).
Cain, D. W. & Cidlowski, J. A. Immune regulation by glucocorticoids. Nat. Rev. Immunol. 17, 233–247 (2017).
Escoter-Torres, L., Greulich, F., Quagliarini, F., Wierer, M. & Uhlenhaut, N. H. Anti-inflammatory functions of the glucocorticoid receptor require DNA binding. Nucleic Acids Res. 48, 8393–8407 (2020).
Cruz-Topete, D. & Cidlowski, J. A. One hormone, two actions: anti- and pro-inflammatory effects of glucocorticoids. Neuroimmunomodulation 22, 20–32 (2015).
Hiltunen, J., Helminen, L. & Paakinaho, V. Glucocorticoid receptor action in prostate cancer: the role of transcription factor crosstalk. Front. Endocrinol.15, 1437179 (2024).
Voskuhl, R. R. et al. Gene expression in oligodendrocytes during remyelination reveals cholesterol homeostasis as a therapeutic target in multiple sclerosis. Proc. Natl. Acad. Sci. USA 116, 10130–10139 (2019).
Lee, H. G. et al. Disease-associated astrocyte epigenetic memory promotes CNS pathology. Nature 627, 865–872 (2024).
Coutinho, A. E. & Chapman, K. E. The anti-inflammatory and immunosuppressive effects of glucocorticoids, recent developments and mechanistic insights. Mol. Cell Endocrinol. 335, 2–13 (2011).
van der Knaap, L. J. et al. Glucocorticoid receptor gene (NR3C1) methylation following stressful events between birth and adolescence. The TRAILS study. Transl. Psychiatry 4, e381 (2014).
Palma-Gudiel, H., Cordova-Palomera, A., Leza, J. C. & Fananas, L. Glucocorticoid receptor gene (NR3C1) methylation processes as mediators of early adversity in stress-related disorders causality: a critical review. Neurosci. Biobehav Rev. 55, 520–535 (2015).
Bechmann, L. et al. Corticosteroid receptor expression and in vivo glucocorticoid sensitivity in multiple sclerosis. J. Neuroimmunol. 276, 159–165 (2014).
Wiechmann, T. et al. Identification of dynamic glucocorticoid-induced methylation changes at the FKBP5 locus. Clin. Epigenet. 11, 83 (2019).
Binder, E. B. et al. Association of FKBP5 polymorphisms and childhood abuse with risk of posttraumatic stress disorder symptoms in adults. JAMA 299, 1291–1305 (2008).
Souren, N. Y. et al. DNA methylation signatures of monozygotic twins clinically discordant for multiple sclerosis. Nat. Commun. 10, 2094 (2019).
Sojka, C. et al. Mapping the developmental trajectory of human astrocytes reveals divergence in glioblastoma. Nat. Cell Biol. 27, 347–359 (2025).
Clark, I. C. et al. Identification of astrocyte regulators by nucleic acid cytometry. Nature 614, 326–333 (2023).
Foo, L. C. et al. Development of a method for the purification and culture of rodent astrocytes. Neuron 71, 799–811 (2011).
Kim, D. et al. TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 14, R36 (2013).
Pertea, M. et al. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 33, 290–295 (2015).
Robinson, M. D., McCarthy, D. J. & Smyth, G. K. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26, 139–140 (2010).
Langmead, B. & Salzberg, S. L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359 (2012).
Zhang, Y. et al. Model-based analysis of ChIP-Seq (MACS). Genome Biol. 9, R137 (2008).
Liao, Y., Smyth, G. K. & Shi, W. featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30, 923–930 (2014).
Li, H. & Durbin, R. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics 26, 589–595 (2010).
Grant, C. E., Bailey, T. L. & Noble, W. S. FIMO: scanning for occurrences of a given motif. Bioinformatics 27, 1017–1018 (2011).
Polanski, K. et al. BBKNN: fast batch alignment of single cell transcriptomes. Bioinformatics 36, 964–965 (2020).
Cusanovich, D. A. et al. A single-cell atlas of in vivo mammalian chromatin accessibility. Cell 174, 1309–1324.e1318 (2018).
Hao, Y. et al. Integrated analysis of multimodal single-cell data. Cell 184, 3573–3587.e3529 (2021).
Korsunsky, I. et al. Fast, sensitive and accurate integration of single-cell data with Harmony. Nat. Methods 16, 1289–1296 (2019).
Kuleshov, M. V. et al. Enrichr: a comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res. 44, W90–W97 (2016).
Liberzon, A. et al. The Molecular Signatures Database (MSigDB) hallmark gene set collection. Cell Syst. 1, 417–425 (2015).
Heinz, S. et al. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol. Cell 38, 576–589 (2010).
Acknowledgements
We thank the members of the Jung and Chung laboratory, who provided support and suggestions throughout the course of this work. This study was supported by the Ministry of Science and ICT through the National Research Foundation in the Republic of Korea (2023R1A2C3002773, 2022R1A5A1026413, RS-2023-00262527, and RS-2023-00223069 to IJ), and the SUHF Fellowship (to I.J.). This study was also supported by the Ministry of Science and ICT through the National Research Foundation in the Republic of Korea (2021R1A2C3005704, to W.-S.C.), and the Institute for Basic Science (IBS-R025-A1, to W.-S.C.).
Author information
Authors and Affiliations
Contributions
S.P., H.P., W.-S.C. and I.J. designed and conceived the study. S.P. led the sequencing library preparation and bioinformatics analyses with assistance from X.W., J.E., J.J., A.J.L. and Y.D. H.P. led the mouse experiments and imaging analyses with assistance from Y.G.B. S.P. prepared the manuscript with assistance from H.P., W.-S.C. and I.J. All authors read and approved the final manuscript. Correspondence and requests for materials should be addressed to I.J. (ijung@kaist.ac.kr) or W.-S.C. (wonsuk.chung@kaist.ac.kr).
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Park, S., Park, H., Byun, Y.G. et al. NR3C1-mediated epigenetic regulation suppresses astrocytic immune responses in mice. Nat Commun 16, 8330 (2025). https://doi.org/10.1038/s41467-025-64088-5
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-64088-5
This article is cited by
-
NR3C1 limits the imprinting of astrocyte epigenetic inflammatory memory early in life
Nature Communications (2025)
