
Abstract
Laryngeal squamous cell carcinoma (LSCC) presents significant treatment challenges, especially regarding recurrence after larynx-preservation therapy. We identified distinct microbial community structures between recurrence and non-recurrence groups, particularly highlighting the genera abundance of Fusobacterium and Serratia. However, larynx-preserving therapy did not significantly alter microbial diversity in recurrent patients. Survival analysis identified Fusobacterium and Serratia as independent prognostic factors for recurrence, leading to the development of a Serratia-Fusobacterium (SF) prognostic scoring model. The SF model achieved an AUC of 81.37% for predicting recurrence, outperforming the TNM staging system. LSCC patients classified as high-risk by the SF model exhibited significantly shorter disease-free survival (DFS) compared to low-risk patients in the LSCC cohort. Furthermore, the SF model demonstrated an AUC of 78.48% in the multi-center cohort for predicting recurrence. In conclusion, the Serratia-Fusobacterium prognostic scoring model can predict LSCC recurrence after larynx-preserving therapy and provide valuable insights to inform recommendations for LSCC surveillance.

Similar content being viewed by others


Introduction
Laryngeal squamous cell carcinoma (LSCC) remains a serious issue for global public health, with an estimated 184,615 new cases and 99,840 deaths worldwide in 20201. LSCC, in addition to potentially increasing mortality, can also adversely affect the quality of life, including phonation, deglutition, and respiratory function. In LSCC treatment, preserving laryngeal function as much as possible is a complex and challenging task. The current treatment strategy for larynx preservation requires a multidisciplinary approach, involving partial laryngectomy, radiotherapy, and/or chemotherapy alone or in combination2. Despite significant improvements in survival rates for most carcinomas over the past several decades, advancements in survival for LSCC have been limited3. Therefore, there is an urgent need to improve outcomes by identifying accurate and efficient prognostic factors and recognizing LSCC patients at high risk of recurrence.
The larynx is a mucosal organ located within the aerodigestive tract, covered by a complex microbial community4,5,6. Extensive research has demonstrated the significant contribution of the microbiota to the initiation and development of alimentary and respiratory carcinomas7,8. Our previous studies have revealed significant differences in microbial diversity and composition between LSCC and non-cancerous tissue samples, including adjacent normal tissues and vocal fold polyp tissues9,10. However, research to date has not yet determined the actual relationship between microbiota and LSCC recurrence. Several aspects should be considered in this regard: (1) Head and neck squamous cell carcinoma (HNSCC) comprises malignant tumor types with different anatomical structures, which host a diverse range of microbiota compositions11,12; (2) it remains unclear whether the same microbiota community composition and diversity persists from primary LSCC to recurrence; (3) most studies have been single-center studies with small sample sizes. Therefore, addressing these questions can help identify the key microbiota in LSCC recurrence, which may serve as potential prognostic factors for improve survival and quality of life.
This study endeavors to comprehensively profile the microbiota composition and diversity in LSCC recurrence. We enrolled LSCC patients who underwent larynx-preserving therapy at two other hospitals to minimize single-center sampling bias. In addition to elucidating the microbial factors associated with LSCC recurrence, we aim to establish and validate a prognostic prediction model. This microbial model not only enhances our understanding of the microbial factors associated with recurrence, but also has the potential to guide personalized therapeutic strategies.
Results
Recurrent LSCC characteristics and microbial community composition
We characterized the microbial communities of formalin-fixed paraffin-embedded (FFPE) tissue samples from 123 patients with LSCC in the LSCC cohort. Overall, a total of 180 LSCC samples were categorized into three distinct groups: the non-recurrence group (NR), the recurrence group (RC), and the postoperative recurrence group (PRC). To mitigate potential data bias and confounding variables, we ensured that there were no statistically significant differences in demographics and clinical characteristics between the NR group and RC group (P > 0.05, Supplementary Table S1).
Amplicon sequence variants (ASVs) served as the foundation for microbiota analysis, with classification of 16S rRNA gene amplicon sequencing achieved through exact matching between ASVs and reference sequences in the SILVA database, which provides 16S rRNA sequences for taxonomic classification and phylogenetic analysis13. Consequently, a total of 40 phyla, 98 classes, 242 orders, 421 families, and 943 genera were annotated in the LSCC cohort.
Distinct microbial profiles in LSCC recurrence
We first observed that Proteobacteria comprised the largest proportion among the 27 phyla, with Vulcaniibacterium showing the highest relative abundance (6.94%) and prevalence (79.03%) at the genus level in the RC group (Fig. 1A). Additionally, Proteobacteria represented the largest proportion among the 39 phyla, with Raoultella showing the highest relative abundance (5.87%) and prevalence (83.61%) at the genus level in the NR group (Fig. 1B).

Schematic phylogenetic tree depicting the representative bacterial phylum (inner circle) and genus (middle circle) abundance of the recurrence group (RC group, A) and the non-recurrence group (NR group, B). The circle size corresponds to the relative abundance at the genus level, while the height of the shadow (outer circle) indicates the prevalence level. C Alpha diversity, determined using richness indices and diversity indices, is depicted for the RC and NR groups. Beta diversity, as illustrated in PCoA plots (D) and NMDS plots (E), is assessed using unweighted UniFrac, weighted UniFrac, and Jaccard distances for the RC and NR groups.
To explore microbial community dynamics and map microbial communities in LSCC recurrence, we summarized the microbial diversity of each sample in the RC and NR groups based on alpha diversity metrics. Microbial richness and diversity indices revealed significant variations between the RC and NR groups (P < 0.01, Fig. 1C). To depict between-group diversity, beta diversity was assessed using principal coordinate analysis (PCoA) and nonmetric multidimensional scaling (NMDS). Statistical significance between the RC and NR groups was evaluated using permutational multivariate analysis of variance (PERMANOVA) tested by Adonis (P = 0.001, Fig. 1D). NMDS was generated based on Jaccard distance, and the statistical significance of similarity was assessed using ANOSIM (P = 0.001, Fig. 1E). Consequently, we identified notable microbial heterogeneity in community composition and diversity in LSCC recurrence.
Microbial functional pathway alterations in LSCC recurrence
The pathway abundances for microbial functions in LSCC recurrence were inferred from ASVs using phylogenetic investigation of communities by reconstruction of unobserved states 2 (PICRUSt2)-predicted functional profiles, and were annotated with the KEGG and MetaCyc metabolic pathway databases. The Wnt signaling pathway was closely associated with the microbiota in the RC group (P < 0.05, Supplementary Fig. S1A). Additionally, the metabolic pathways of L-tryptophan degradation XII (PWY-6505) and 2-aminophenol degradation (PWY-6210) exhibited statistically significant increases in the RC group (P < 0.001, Supplementary Fig. S1B). These results suggest that the microbiota may contribute to LSCC recurrence after larynx-preserving therapy by influencing transcriptional and metabolic factors in LSCC.
Larynx-preserving therapy failed to alter the microbial profiles of LSCC recurrence
Proteobacteria accounted for the greatest proportion among the 32 phyla, with Prauserella occupying the highest relative abundance (5.71%) and prevalence (91.23%) at the genus level in the PRC group.
To investigate whether larynx-preserving therapy affects microbial community composition and diversity in LSCC patients with recurrence, we compared alpha diversity and beta diversity between the RC and PRC groups. In terms of alpha diversity, the Chao1, observed species, Simpson, and Faith’s phylogenetic diversity (PD) metrics did not exhibit significant differences between the RC and PRC groups (P > 0.05, Fig. 2B). Additionally, LSCC microbial diversity was similar between the RC and PRC groups according to Adonis (P > 0.05, Fig. 2C) and ANOSIM (P > 0.05, Fig. 2D). Consequently, LSCC patients who underwent larynx-preserving therapy did not experience significant changes in the composition or diversity of the microbial communities.

A Schematic phylogenetic tree depicting the representative bacterial phylum (inner circle) and genus (middle circle) abundance of the postoperative recurrence group (PRC group). The circle size corresponds to the relative abundance at the genus level, while the height of the shadow (outer circle) indicates the prevalence level. B Alpha diversity, determined using richness indices and diversity indices, is depicted for the RC and PRC groups. Beta diversity, as illustrated in PCoA plots (C) and NMDS plots (D) is assessed using unweighted UniFrac, weighted UniFrac, and Jaccard distances for the RC and PRC groups.
Identification of key microbial genera associated with LSCC recurrence
To further investigate the differences in microbial community composition between LSCC patients with and without recurrence, we found that the RC group had 2603 unique ASVs after filtering out contaminants (Fig. 3A). The rank abundance visualization using linear discriminant analysis (LDA) with a threshold greater than 3.5 highlighted differences in the microbial composition of LSCC patients with and without recurrence. The RC group had a significantly higher abundance of the Fusobacterium taxonomic group, including phylum Fusobacteria, class Fusobacteriia, order Fusobacteriales, family Fusobacteriaceae, and genus Fusobacterium, relative to the NR group (P < 0.001, Fig. 3B). Furthermore, the RC group had significantly higher abundances of theVulcaniibacterium taxonomic group, including order Xanthomonadales, family Xanthomonadaceae, and genus Vulcaniibacterium, relative to the NR group (P < 0.0001, Fig. 3B). Conversely, the RC group showed a lower abundances of the Raoultella taxonomic group, including order Enterobacterales, family Enterobacteriaceae, and genus Raoultella, relative to the NR group (P < 0.0001, Fig. 3B). Additionally, the RC group showed a lower abundance of the Serratia taxonomic group, including order Enterobacterales, family Yersiniaceae, and genus Serratia, relative to the NR group (P < 0.0001, Fig. 3B). Similarly, there were significant differences in the abundances of Fusobacterium and Serratia between these two groups, as determined by the Mann–Whitney test (P < 0.001, Fig. 3C). Consequently, the abundances of Fusobacterium, Vulcaniibacterium, Raoultella, and Serratia were closely associated with LSCC recurrence.

A The Venn diagram showed the overlap of observed ASVs for the NR and RC groups. B Linear discriminant analysis (LDA) score effect size (LEfSe) analysis was coupled with the effect size measurements in the RC and NR groups. Only taxa with LDA scores greater than the threshold of 3.5 are presented. C Statistical analysis of the dominant genera.
Identification of the microbial predictive model associated with LSCC recurrence
Based on their discriminative power at specific genus levels associated with LSCC recurrence, we constructed stratified 10-fold cross-validated random forest (RF) models to assess their ability to accurately predict LSCC recurrence. The abundances of Raoultella and Serratia were significantly higher in the NR group than in the RC group (P < 0.05, Fig. 4A). Conversely, the abundances of Vulcaniibacterium and Fusobacterium showed a marked decrease in the RC group compared to the NR group (P < 0.05, Fig. 4A).

A Microbial biomarkers were identified to construct a random forest (RF) model for discriminating LSCC recurrence. B Area under the curve (AUC) transfer validation classifiers were evaluated using different sets of genera, both alone and in combination. C Receiver operating characteristic (ROC) curve analysis of the optimized models constructed with genus level alone and in combination for discriminating LSCC recurrence. D Statistical analysis was conducted in the LSCC cohort based on the Microbial model, and recurrence rate was calculated using Chi-square test. E Disease-free survival (DFS) was determined in patients with LSCC from the LSCC cohort who were stratified by the status of the Microbial model as assessed by the log-rank test.
To optimize microbial classifiers for LSCC recurrence based on the abundance levels of all 186 genera, we investigated whether combining genera abundances could enhance the prognostic and predictive utility for LSCC. This was assessed using the area under the curve (AUC) from receiver operating characteristic (ROC) curve analysis with 10-fold stratified cross-validation (CV). Our analysis revealed that the combination of four genera (Raoultella, Serratia, Vulcaniibacterium, and Fusobacterium) maintained an AUC of over 98%, demonstrating strong predictive capabilities (Fig. 4B). Given their promise as prognostic parameters, we further validated our findings to develop a practical and effective microbial model for predicting recurrence in LSCC patients. The validation results confirmed the abundance of these four genera as key components of the microbial model, whose AUC was superior to that of any specific genus (P < 0.0001, Fig. 4C). The microbial model had a sensitivity of 95.16% and a specificity of 90.16% for LSCC recurrence, indicating strong predictive ability (P < 0.0001, Fig. 4D). Furthermore, the high-risk patients had significantly shorter disease-free survival (DFS) than the low-risk patients stratified by the microbial model (P < 0.0001, Fig. 4E). Consequently, these results suggest that the microbial model is pathologically and clinically associated with LSCC recurrence.
Establishment and validation of the Serratia-Fusobacterium prognostic scoring model for LSCC recurrence
To validate the sequencing results of bacterial 16S rRNA genes and enhance clinical usefulness and practicality, we utilized a quantitative real-time polymerase chain reaction (qPCR) approach with specific bacterial primers to quantify the amounts of the genera Vulcaniibacterium, Serratia, Raoultella, and Fusobacterium. We observed that the RC group exhibited a significant decrease in the amount of Serratia, and a significant increase in the amount of Fusobacterium, in the LSCC cohort (P < 0.0001, Fig. 5A). Furthermore, these qPCR results for Serratia and Fusobacterium amounts showed significant concordance with our findings from bacterial 16S rRNA gene sequencing (P < 0.0001, Fig. 5B). However, the amount of Raoultella was too low to detect, and the amount of Vulcaniibacterium did not differ between the RC and NR groups (P > 0.05). To determine the prognostic value of specific genera amounts, LSCC patients were classified into high-risk and low-risk groups using the optimal cutoff value determined by X-tile in the LSCC cohort (Supplementary Fig. S2). Consistent with this result, low amounts of Serratia group (−ΔCt < −8.3950) were associated with shorter DFS than high amounts of Serratia group, contrasting with the shorter DFS observed in the LSCC cohort with high amounts of Fusobacterium (−ΔCt > −7.6800, P < 0.0001, Fig. 5C). To eliminate potential confounding factors influencing observed correlations between clinicopathological characteristics and specific genus abundances, we conducted univariate and multivariate analyses for DFS. We found that Serratia and Fusobacterium amounts were identified as independent risk factors for DFS (P = 0.0001, Fig. 5D). Consequently, we established the Serratia-Fusobacterium prognostic scoring model (SF model) to predict LSCC recurrence (Fig. 5E).

A Statistical analysis of the amounts of Serratia and Fusobacterium between the NR and RC groups in the LSCC cohort was calculated using the Mann–Whitney test. B The correlation between the amounts of Serratia and Fusobacterium, as determined by quantitative real-time polymerase chain reaction (qPCR) and the abundance as measured by 16S rRNA sequencing in the LSCC cohort. C DFS was determined in patients with LSCC from the LSCC cohort who were stratified by the cut-off value of the amounts of Serratia and Fusobacterium, as assessed by the log-rank test. D Multivariate analyses of the level of Serratia and Fusobacterium for DFS were conducted in the LSCC cohort. The bars represent 95% confidence intervals. E Schematic illustration of the generation of microbial prognostic scores in the Serratia-Fusobacterium prognostic scoring model (SF model). F ROC curve analysis was performed based on the levels of Serratia and Fusobacterium alone, SF model, and the TNM staging system to discriminate LSCC recurrence in the LSCC cohort. G DFS was determined in patients with LSCC from the LSCC cohort who were stratified by the status of the SF model, as assessed by the log-rank test. H ROC curve analysis was performed based on the level of Serratia and Fusobacterium alone, SF model, and the TNM staging system, for discriminating LSCC recurrence in the Multi-center LSCC cohort. I DFS was determined in patients with LSCC from the Multi-center LSCC cohort who were stratified by the status of the SF model, as assessed by the log-rank test.
To test the robustness and generalizability of the SF model in LSCC prognosis, the SF model was trained in the LSCC cohort (discovery cohort) and validated in the Multi-center LSCC cohort (validation cohort). We predicted LSCC recurrence using the SF model, Fusobacterium amounts, Serratia amounts, and the 8th edition of the American Joint Committee on Cancer (AJCC) cancer staging manual for LSCC staging through ROC curve analysis. The AUC value of the SF model was significantly greater than that of individual genus amounts alone and the TNM staging system in the LSCC cohort (P < 0.0001, Fig. 5F). Survival analysis demonstrated that the high-risk group had worse DFS than the low-risk group in the LSCC cohort (P < 0.0001, Fig. 5G). To validate and assess the generalizability of the SF model, we collected 60 FFPE samples for external validation in the Multi-center LSCC cohort. The predictive ability of the SF model (P = 0.0018) persisted and remained superior to that of the TNM staging system (P = 0.4511, Fig. 5H). Furthermore, similar results were observed for DFS in the Multi-center LSCC cohort (P = 0.0003, Fig. 5I). Consequently, these results support the use of the SF model for predicting LSCC recurrence.
Discussion
With advances in larynx-preservation treatment, LSCC patients are increasingly concerned about maintaining their quality of life rather than simply prolonging their lives. However, larynx-preserving therapy has not been entirely successful in preventing LSCC recurrence. Despite the close association between specific microbiota and the prognosis of LSCC patients in our previous studies, there is a paucity of high-quality and reliable research providing a comprehensive understanding of the association between microbiota composition and LSCC recurrence.
Dysbiosis of intratumor microbiota is closely associated with the prognosis of HNSCC patients, and is an important factor in regulating signaling pathways and host metabolism14. Fusobacterium nucleatum (F. nucleatum) activates the Wnt signaling pathway by binding its virulence factor FadA to E-cadherin, a process that plays a crucial role in the progression of oral squamous cell carcinoma (OSCC)15. Furthermore, intratumor microbiota-induced L-tryptophan degradation may influence patients’ prognosis by modulating the kynurenine pathway, which affects immune responses and contributes to tumor progression16.
Total laryngectomy, due to permanent alterations in the respiratory tract, may reduce the risk of LSCC recurrence by significantly lowering the abundance of LSCC-promoting microbiota17. Fusobacterium is considered a component of the core microbiota of the laryngeal cavity, with its abundance strongly associated with the poor prognosis in LSCC patients in our previous studies18,19. Within the Fusobacterium genus, F. nucleatum is a major determinant of ethanol metabolic decompensation in LSCC, and interactions involving F. nucleatum collectively promote carcinoma invasion and metastasis, thus affecting LSCC prognosis18. F. nucleatum influences epigenetic alterations of DNA mismatch repair (MMR) and microsatellite instability in LSCC by suppressing MMR-related gene expression via the TLR4/MYD88/miR-205-5p signaling pathway19. Additionally, F. nucleatum promotes LSCC progression by restricting purine metabolism20. However, our results indicate that larynx-preserving therapy does not reduce Fusobacterium abundance, which may serve as a key factor contributing to LSCC recurrence.
Recent studies have identified microbiota abundances as prognostic factors for patients with various cancers, providing potential references for assessing therapeutic efficacy21,22. Fusobacterium abundance has served as a predictive biomarker in patients with metastatic colorectal cancer undergoing treatment with regorafenib plus toripalimab in a phase Ib/II clinical trial23. Furthermore, F. nucleatum has been found to impair therapeutic efficacy during radiotherapy in colorectal cancer24. In HNSCC, F. nucleatum enhances the progression and chemoresistance of esophageal squamous cell carcinoma (ESCC) by promoting the secretion of chemotherapy-induced senescence-associated secretory phenotype through activation of the DNA damage response pathway25. F. nucleatum also predicts therapeutic response to neoadjuvant chemotherapy in ESCC26. Furthermore, Fusobacterium species can increase both PD-L1 mRNA levels and surface PD-L1 protein expression on HNSCC cell lines27. Therefore, identifying novel cancer-promoting microbiota for predicting cancer prognosis and monitoring outcomes holds significant importance in cancer research.
While many microbiota are implicated in promoting carcinogenesis, it is noteworthy that several microbiota demonstrate anti-cancer immunity and therapeutic responses28. Serratia, a Gram-negative genus within the Enterobacteriaceae family, tends to colonize the respiratory and gastrointestinal tracts29,30. Prodigiosin (PG, PubChem CID: 5377753) is predominantly synthesized by Serratia marcescens, and its synthetic derivatives, including undecylprodigiosin and cycloprodigiosin hydrochloride, exhibit various anticancer effects31,32. In HNSCC, PG demonstrates anticancer efficacy comparable to that of 5-FU in nasopharyngeal cancer, particularly in inhibiting carcinoma proliferation, migration, and invasion, while also exhibiting lower toxicity than conventional chemotherapeutic drugs33. Additionally, PG effectively induces cell death via LC3-mediated autophagy and causes cell-cycle arrest by suppressing cyclin D1 expression in OSCC34. Therefore, the presence of low Serratia and high Fusobacterium levels plays a crucial role in the adverse prognosis of LSCC.
To our knowledge, this is the first study to comprehensively assess alterations in the microbial communities among LSCC patients with recurrence. Our results indicate that larynx-preserving therapy does not alter the composition and diversity of the laryngeal microbiota in LSCC patients with recurrence. When LSCC patients undergo larynx-preserving therapy, we suggest that employing various intervention methods, such as probiotics, antibiotics, and phages, may reduce the abundance of LSCC-promoting microbiota, thereby minimizing the risk of LSCC recurrence35. Furthermore, we have established a prognostic model for predicting LSCC recurrence by utilizing specific microbiota levels. This model can identify patients at risk of early recurrence and provide valuable insights to inform recommendations on LSCC surveillance. Although this multicenter study included LSCC patients from three hospitals in China, the prognostic prediction model may not be generalizable to cohorts from other countries. Thus, further prospective trials are necessary to validate these promising results.
In this study, our results indicate that larynx-preserving therapy does not alter the composition and diversity of the laryngeal microbiota in LSCC patients with recurrence and highlight the significance of specific genera as independent prognostic factors for LSCC. Furthermore, we have established a prognostic model for predicting LSCC recurrence by utilizing specific microbiota levels. The Serratia-Fusobacterium prognostic scoring model can identify patients at risk of early recurrence and provide valuable insights to inform recommendations for LSCC surveillance.
Methods
Human subject enrollment
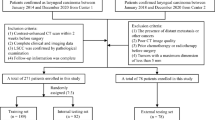
The study comprised three cohorts and involved the collection of 240 FFPE samples from 183 LSCC patients who underwent larynx-preserving therapy between 2011 and 2020. The LSCC cohort from the Eye & ENT Hospital of Fudan University included 123 patients and was divided into the RC, NR, and PRC groups, with a total of 180 LSCC tissue samples. The RC group, consisting of 62 patients, included 62 primary samples and 57 paired recurrence samples (PRC group) collected within five years after larynx-preserving therapy. The NR group included 61 LSCC tissue samples from patients who remained recurrence-free after larynx-preserving therapy, among whom 59 patients were followed for at least five years, while two patients were censored at 48 and 57 months due to causes unrelated to LSCC recurrence. The LSCC cohort had a follow-up time of 2 to 71 months (median: 48 months), with tissue samples from the NR and RC groups collected between 2015 and 2020, and those from the PRC group collected between 2016 and 2021 (Supplementary Table S1).
The Multi-center LSCC cohort included 60 LSCC tissue samples from 60 LSCC patients, including 28 LSCC tissue samples (22 primary samples and six recurrence samples) from The First Affiliated Hospital of Nanjing Medical University between 2015 and 2020, and 32 LSCC tissue samples (25 primary samples and seven recurrence samples) from Renji Hospital, Shanghai Jiao Tong University. The Multi-center LSCC cohort had a follow-up time of 2–75 months (median: 47 months), with samples collected between 2011 and 2020 (Supplementary Table S2).
The inclusion criteria were as follows: (i) written informed consent was obtained from LSCC patients; (ii) LSCC staging was classified according to the 8th edition of the AJCC cancer staging manual by experienced pathologists36; and (iii) all patients successfully underwent the protocol treatment of larynx-preserving therapy. The exclusion criteria were as follows: (i) clinical evidence of active bacterial or viral infection and antibiotic therapy prior to larynx-preserving therapy; (ii) presence of residual tumor after larynx-preserving therapy; and (iii) lack of complete clinical information or complete follow-up. The clinicopathological characteristics were obtained from archived records (Supplementary Tables S1 and S2).
Ethics approval
Our study protocol was reviewed and approved by the Ethics Committee of the Eye & ENT Hospital, Fudan University (2022076), the Institutional Review Board of the First Affiliated Hospital with Nanjing Medical University (2021SR-391), and the Ethics Committee of Renji Hospital, Shanghai Jiaotong University School of Medicine (KY2021-062-B), all in accordance with the Declaration of Helsinki. The requirement for written informed consent was waived, as this waiver did not adversely affect the rights or health of the participants.
DNA extraction and 16S rRNA sequencing
Total genomic DNA was extracted from LSCC tissue samples using the GeneRead DNA FFPE Kit (180134, Qiagen, Germany) and stored at −20 °C until further analysis. PCR amplification of the V3-V4 regions of bacterial 16S rRNA genes was performed using the forward primer 338F (5’-ACTCCTACGGGAGGCAGCA-3’) and the reverse primer 806R (5’-GGACTACHVGGGTWTCTAAT-3’). The PCR amplicons were purified using VAHTSTM DNA Clean Beads (N411, Vazyme, China), quantified using the Quant-iT PicoGreen dsDNA Assay Kit (P7589, Invitrogen, USA), and sequenced on the NovaSeq platform using the NovaSeq 6000 SP Reagent Kit (20029137, Illumina, USA) at Genekinder Medicaltech Shanghai Co., Ltd. (China).
Microbiome analysis
Microbiome bioinformatics analyses were primarily performed using QIIME2 (v.2020.11, USA) and R packages (v.3.2.0, USA)37. ASVs were assigned taxonomy using the feature-classifier plugin’s classify-sklearn naive Bayes taxonomy classifier compared to the SILVA database (v.132). ASV-level alpha diversity indices, which measure within-individual diversity, were calculated and visualized as box plots. These included Chao1, observed species, Simpson, and Faith’s PD metrics. Beta diversity analysis was performed to investigate the diversity of microbial communities using PCoA and NMDS among the NR, RC, and PRC groups. To visualize the results, PCoA plots were generated based on unweighted and weighted UniFrac distances, while NMDS plots were generated based on Jaccard distance. Microbial functions were predicted by PICRUSt2 based on the KEGG and MetaCyc databases38,39.
Establishment and validation of a microbial prognosis model for LSCC recurrence
Based on the 16S rRNA sequencing data, all genera with an occurrence frequency less than 20% were excluded before constructing the RF model for the RC and NR groups. The RF model learning was conducted using the 10-fold stratified CV for automated hyperparameter optimization and sample prediction, facilitating the ranking of different prediction models based on microbial genera40. The final microbial prediction model was selected based on the highest AUC value and the lowest number of specific microbial predictors41.
qPCR with SYBR Green fluorescence, synthesized by Jierui Biotechnology (China), was utilized to quantify the amounts of Serratia, Fusobacterium19, Vulcaniibacterium, and Raoultella. The primer sequence for the common primer (all bacterial 16S rRNA) was described previously17. Bacterial primers were designed and validated to detect these specific genera using Primer-BLAST from the NCBI (https://www.ncbi.nlm.nih.gov/tools/primer-blast/). The primers for all candidate genes are listed (Supplementary Table S3). The cycle threshold (Ct) values obtained from LSCC samples were compared using the -ΔCt method.
To confirm and validate the clinical significance of Serratia and Fusobacterium amounts, we used X-tile software (v.3.6.1, USA) to determine the cutoff values. Subsequently, patients were categorized into two groups, and a score was assigned to each specific genus42. A score of one was assigned to each genus when a sample had low amounts of Serratia (−ΔCt < −8.3950) and high amounts of Fusobacterium (−ΔCt > −7.6800), which were identified as independent factors according to the multivariate Cox regression model. LSCC patients were then assigned to either the high-risk group (low Serratia and high Fusobacterium amounts) or the low-risk group (Fig. 5E).
Statistical analysis
The relative abundance and quantification of significant genera were assessed using the nonparametric Mann–Whitney U test. The correlation between genus abundance and genus amount was investigated using two-tailed nonparametric Spearman correlation. ROC analysis was conducted to identify the sensitivity and specificity of genera levels in predicting LSCC recurrence. Survival of LSCC patients was calculated using the Kaplan-Meier method and compared using the log-rank test. Univariate and multivariate regression analyses were conducted using Cox proportional hazards models to determine associations between independent clinicopathologic parameters. All P values were two-tailed, and P values < 0.05 were considered statistically significant. All statistical analyses were performed using GraphPad Prism software (v.10.0.3, USA) and IBM SPSS Statistics software (v.20.0, USA).
Data availability
All raw sequence files for all samples used in this study have been deposited in the National Genomics Data Center (NGDC) under accession number CRA014541.
References
Sung, H. et al. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. Ca. Cancer J. Clin. 71, 209–249 (2021).
Steuer, C. E., El-Deiry, M., Parks, J. R., Higgins, K. A. & Saba, N. F. An update on larynx cancer: an update on larynx cancer. Ca. Cancer J. Clin. 67, 31–50 (2017).
Pulte, D. & Brenner, H. Changes in survival in head and neck cancers in the late 20th and early 21st century: a period analysis. Oncologist 15, 994–1001 (2010).
Gong, H. L. et al. The composition of microbiome in larynx and the throat biodiversity between laryngeal squamous cell carcinoma patients and control population. PLOS One 8, e66476 (2013).
Gong, H. et al. Alterations of microbiota structure in the larynx relevant to laryngeal carcinoma. Sci. Rep. 7, 5507 (2017).
Mao, X. et al. Complex of intratumoral mycobiome and bacteriome predicts the recurrence of laryngeal squamous cell carcinoma. Appl. Environ. Microbiol. e01954-24 https://doi.org/10.1128/aem.01954-24 (2025).
Wong-Rolle, A., Wei, H. K., Zhao, C. & Jin, C. Unexpected guests in the tumor microenvironment: microbiome in cancer. Protein Cell 12, 426–435 (2021).
Brennan, C. A. & Garrett, W. S. Fusobacterium nucleatum — symbiont, opportunist and oncobacterium. Nat. Rev. Microbiol. 17, 156–166 (2019).
Gong, H. et al. Microbiota in the throat and risk factors for laryngeal carcinoma. Appl. Environ. Microbiol. 80, 7356–7363 (2014).
Gong, H. et al. Composition and abundance of microbiota in the pharynx in patients with laryngeal carcinoma and vocal cord polyps. J. Microbiol. 55, 648–654 (2017).
Lau, H.-C. et al. Oropharynx microbiota transitions in hypopharyngeal carcinoma treatment of induced chemotherapy followed by surgery. BMC Microbiol. 21, 310 (2021).
Lau, H.-C. et al. Cross-comparison of microbiota in the oropharynx, hypopharyngeal squamous cell carcinoma and their adjacent tissues through quantitative microbiome profiling. J. Oral. Microbiol. 14, 2073860 (2022).
Quast, C. et al. The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res. 41, D590–D596 (2012).
Dorobisz, K., Dorobisz, T. & Zatoński, T. The microbiome’s influence on head and neck cancers. Curr. Oncol. Rep. 25, 163–171 (2023).
Cai, L. et al. Integrative analysis reveals associations between oral microbiota dysbiosis and host genetic and epigenetic aberrations in oral cavity squamous cell carcinoma. npj Biofilms Microbiomes 10, 39 (2024).
Platten, M., Nollen, E. A. A., Röhrig, U. F., Fallarino, F. & Opitz, C. A. Tryptophan metabolism as a common therapeutic target in cancer, neurodegeneration and beyond. Nat. Rev. Drug Discov. 18, 379–401 (2019).
Hsueh, C.-Y. et al. Throat microbial community structure and functional changes in postsurgery laryngeal carcinoma patients. Appl. Environ. Microbiol. 86, e01849–20 (2020).
Hsueh, C.-Y. et al. A positive feed-forward loop between fusobacterium nucleatum and ethanol metabolism reprogramming drives laryngeal cancer progression and metastasis. iScience 25, 103829 (2022).
Hsueh, C.-Y. et al. Fusobacterium nucleatum impairs DNA mismatch repair and stability in patients with squamous cell carcinoma of the head and neck. Cancer 128, 3170–3184 (2022).
Li, F. et al. Fusobacterium nucleatum-triggered purine metabolic reprogramming drives tumorigenesis in head and neck carcinoma. Discov. Oncol. 14, 120 (2023).
Sepich-Poore, G. D. et al. The microbiome and human cancer. Science 371, eabc4552 (2021).
Yang, L., Li, A., Wang, Y. & Zhang, Y. Intratumoral microbiota: roles in cancer initiation, development and therapeutic efficacy. Sig. Transduct. Target. Ther. 8, 35 (2023).
Wang, F. et al. Regorafenib plus toripalimab in patients with metastatic colorectal cancer: a phase ib/II clinical trial and gut microbiome analysis. Cell Rep. Med. 2, 100383 (2021).
Dong, J. et al. Oral microbiota affects the efficacy and prognosis of radiotherapy for colorectal cancer in mouse models. Cell Rep. 37, 109886 (2021).
Zhang, J.-W. et al. fusobacterium nucleatum promotes esophageal squamous cell carcinoma progression and chemoresistance by enhancing the secretion of chemotherapy-induced senescence-associated secretory phenotype via activation of DNA damage response pathway. Gut Microbes 15, 2197836 (2023).
Yamamura, K. et al. Intratumoral Fusobacterium Nucleatum levels predict therapeutic response to neoadjuvant chemotherapy in esophageal squamous cell carcinoma. Clin. Cancer Res. 25, 6170–6179 (2019).
Michikawa, C. et al. Fusobacterium is enriched in oral cancer and promotes induction of programmed death-ligand 1 (PD-L1). Neoplasia. 31, 100813 (2022).
Montalban-Arques, A. et al. Commensal clostridiales strains mediate effective anti-cancer immune response against solid tumors. Cell Host Microbe 29, 1573–1588.e7 (2021).
Appaneal, H. J. et al. Trends in collection of microbiological cultures across veterans affairs community living centers in the united states over 8 years. J. Am. Med. Dir. Assoc. 21, 115–120 (2020).
Harris, V. et al. Rotavirus vaccine response correlates with the infant gut microbiota composition in pakistan. Gut Microbes 9, 93–101 (2018).
Mohan, C. D. et al. Bacteria as a treasure house of secondary metabolites with anticancer potential. Semin. Cancer Biol. 86, 998–1013 (2022).
Montaner, B. & Pérez-Tomás, R. The prodigiosins: a new family of anticancer drugs. Curr. Cancer Drug Targets 3, 57–65 (2003).
Liu, Y. et al. Prodigiosin inhibits proliferation, migration, and invasion of nasopharyngeal cancer cells. Cell. Physiol. Biochem. 48, 1556–1562 (2018).
Cheng, M.-F. et al. Inhibitory growth of oral squamous cell carcinoma cancer via bacterial prodigiosin. Mar. Drugs 15, 224 (2017).
Raman, V. et al. Build-a-bug workshop: Using microbial-host interactions and synthetic biology tools to create cancer therapies. Cell Host Microbe 31, 1574–1592 (2023).
Amin, M. B. et al. The Eighth Edition AJCC Cancer Staging Manual: continuing to build a bridge from a population-based to a more “personalized” approach to cancer staging. CA Cancer J. Clin. 67, 93–99 (2017).
Rai, S. N. et al. Microbiome data analysis with applications to pre-clinical studies using QIIME2: statistical considerations. Genes Dis. 8, 215–223 (2021).
Douglas, G. M. et al. PICRUSt2 for prediction of metagenome functions. Nat. Biotechnol. 38, 685–688 (2020).
Caspi, R. et al. The MetaCyc database of metabolic pathways and enzymes - a 2019 update. Nucleic Acids Res. 48, D445–D453 (2020).
Zhang, M. et al. Radiomic signatures of posterior fossa ependymoma: molecular subgroups and risk profiles. Neuro Oncol. 24, 986–994 (2022).
Sze, M. A., Baxter, N. T., Ruffin, M. T., Rogers, M. A. M. & Schloss, P. D. Normalization of the microbiota in patients after treatment for colonic lesions. Microbiome 5, 150 (2017).
Tian, M.-X. et al. Histopathology-based immunoscore predicts recurrence for intrahepatic cholangiocarcinoma after hepatectomy. Cancer Immunol. Immunother. 68, 1369–1378 (2019).
Acknowledgements
This study was supported by the National Natural Science Foundation of China (82203727 to C.Y.H., 82273097 to M.Z., and 81502343 to H.L.G.), the Clinical Research Plan of SHDC (SHDC2020CR6011 to L.T.), the Science and Technology Commission of Shanghai Municipality (23ZR1409700 to M.Z., 21Y11900100 to H.L.G. and 19411961300 to H.L.G.), Shanghai “Rising Stars of Medical Talent” Youth Development Program-Specialist Program (2019-72 to H.L.G.), and Shanghai Sailing Program (22YF1405700 to C.Y.H.). We thank for the sequencing and analysis service provided by Genekinder Medicaltech (Shanghai) Co., Ltd, China.
Author information
Authors and Affiliations
Contributions
Study concept and design: C.Y.H., X.H.Y., H.Y.H., M.Z., J.S. and H.L.G. Clinical specimen and data collection: X.H.Y., J.S., W.D.D., D.H.L., H.Y.H., Y.J.S., Q.H., X.H.M. and S.M.L. Methodology: C.Y.H., X.H.Y., J.S. and H.L.G. Data acquisition and interpretation: All authors. Data curation, visualization, software, and formal analysis: C.Y.H., X.H.Y., H.Y.H., J.S., Y.J.S. and H.L.G. Manuscript drafting: C.Y.H. and X.H.Y. Critical comments on the manuscript and important intellectual content: J.S., L.T., M.Z. and H.L.G. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Hsueh, CY., Yuan, X., Huang, H. et al. Evaluating the prognostic value of microbial communities in predicting recurrence of laryngeal carcinoma: a multicenter case-control study. npj Biofilms Microbiomes 11, 159 (2025). https://doi.org/10.1038/s41522-025-00789-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41522-025-00789-5
