
Abstract
Hormone receptor-positive breast cancers are a diverse group of tumours, with only some responding well to immunotherapy. Alternative combination strategies such as radiotherapy present an exciting opportunity to improve immunotherapy responses. We review an intriguing overlap between the impact of oestrogen signalling and radiation on multiple signalling pathways and immune cells that may be exploited for therapeutic gains in breast cancer. This is synthesised with the pre-clinical data and clinical trial landscape supporting the use of combined radiation and immunotherapy to derive insights for future neo-adjuvant trial design.
Similar content being viewed by others

Introduction
Breast cancer (BC) is the most common female malignancy. Approximately 70% are hormone receptor positive (HR+), denoted by oestrogen receptor (ER) and/or progesterone receptor (PR) positivity on immunohistochemistry. Whilst many localised HR+ tumours are associated with favourable long-term prognosis, around 10–40% experience metastatic relapse1. Many relapses occur later than in other subtypes of BC (triple-negative and HER2+), but some HR+ tumours behave aggressively with poor prognosis2.
Immune checkpoint inhibition (ICI) is an established treatment for triple-negative BC (TNBC) in combination with chemotherapy3,4,5. While ICI has not yet been approved for HR+ BC, recent results from two phase 3 trials demonstrate a doubling of pCR in this patient population when ICI are added to neo-adjuvant chemotherapy6,7. However, pCR was achieved only in ~24% of patients, indicating that patients may require additional therapies such as radiotherapy (RT), which can induce immunological stimulation to improve responses8,9. Cellular responses to RT can induce an inflammatory tumour immune microenvironment (TIME) and enhance T-cell/dendritic cell (DC) activation, migration and tumour cell recognition10. There is significant interplay between the impact of RT and oestrogen signalling on several molecular pathways and cells within the TIME, suggesting an under-explored potential immunogenic role for RT in the HR+ BC setting11,12.
Whilst RT is traditionally used in the adjuvant BC setting, neo-adjuvant RT and ICI are feasible separately3,13 and in combination14. Neo-adjuvant combined RT and ICI (RT-ICI) is an area of interest because the ability of RT to promote priming of anti-tumour T-cells requires the primary tumour in situ8. Pre-clinical evidence also suggests immunotherapy may also be more effective in the neo-adjuvant versus adjuvant BC setting15.
This review discusses the pre-clinical and clinical trial evidence supporting the concept of using RT-ICI for HR+/HER2- BC, alongside progress in HR+/HER2- BC mouse models that could advance research in this field. These are synthesised to explore future directions for neo-adjuvant RT-ICI trials in HR+ BC.
HR+/HER2- BCs are a heterogenous tumour group
Within the complex system of a primary BC tumour, cells possess differing levels of ER expression and intrinsic dependence on oestrogen signalling. Indeed, tumours can be considered ‘strongly’ ER+ if ≥11% of cells stain with strong ER intensity16. Transcriptomic profiling using PAM50 classification highlights the heterogeneity amongst HR+/HER2- tumours, with most categorised as luminal A or luminal B and smaller proportion as basal-like, normal-like or HER2-enriched17. Compared to luminal A tumours, luminal B tumours are associated with inferior prognosis and defined by higher histological grade, higher proliferation rate, lower PR positivity, lower ER expression, and lower ER signalling dependence18. They also possess increased type 1 interferon gene products, antigenicity and T-cell clonality, suggesting an increased sensitivity to immunotherapy19.
The role of the immune system in BC is complex and the BC TIME varies greatly across receptor subtypes20. Luminal tumours are traditionally viewed as particularly immune ‘cold’, with lower levels of stromal tumour infiltrating lymphocytes (sTILs), tumour cell MHC expression, and tumour mutational burden (TMB)/genomic instability resulting in reduced neo-antigen presentation21,22. However, luminal B tumours tend to possess higher sTIL levels than luminal A21, and some HR+/HER2- tumours (particularly those with lower ER positivity) harbour a TIME more similar to TNBC/HER2+ BCs23,24,25. High sTIL levels are a validated positive prognostic biomarker in triple-negative and HER2+ tumours but their prognostic role in HR+/HER2- BC is less clear; perhaps unsurprising given the diversity of tumours encompassed within this tumour group26.
Deeper sub-classification of lymphocytic infiltrates and their spatial arrangement in HR+ BCs also highlights TIME diversity and demonstrates these approaches may improve prognostic correlations than sTIL levels alone27,28. Increased ER expression also correlates with reduced immune infiltrates such as tumour-associated macrophages (TAMs) and CD4+ T-cells29. Other cell populations play an important role within the HR+ TIME and can be associated with inferior prognosis, including TAMs and cancer-associated fibroblasts (CAFs)30,31. Further exploration of the effects of neo-adjuvant therapies including chemotherapy and RT on the HR+ BC TIME is required, although data suggests they may cause a lymphodepletive effect32,33.
The heterogeneity of HR+ BC may be exploited to identify patients who respond more favourably to immunotherapy, and those who will benefit from additional combination therapies such as chemotherapy or RT, in a similar fashion to TNBC34,35. For example, post-hoc analysis of an I-SPY2 trial arm using baseline MammaPrint classification identified that the 53% of ‘MP2 ultra-high risk’ HR+ tumours exhibited enhanced responses to neo-adjuvant combined durvalumab/olaparib/chemotherapy29. Improved understanding of the molecular impacts of oestrogen signalling on tumour and immune cells could facilitate novel predictive biomarker identification.
Oestrogen signalling and radiation impact similar pathways and cells implicated in anti-tumour immune responses
The links between oestrogen signalling, inflammation and proliferation that favour BC formation and progression are well established36. ERs (principally ERα and ERβ) bound to oestrogens (principally E2; 17β-oestradiol) influence gene expression directly or indirectly via binding of coactivator/corepressor proteins and oestrogen response elements on DNA. The multifaceted nature of oestrogen signalling may explain how it can both drive tumour growth/proliferation whilst also often conferring a less aggressive phenotype. Mutations in ESR1 (encoding ERα) confer endocrine therapy resistance and are implicated in luminal BC radioresistance in vitro37.
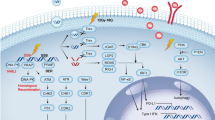
The complex nature of ER transcription with coregulatory proteins results in intricate context-dependent interactions between oestrogen signalling and signalling pathways/immune cells involved in the co-ordination of anti-tumour immune responses, summarised by previous reviews11,12,21,36,38,39,40. Several of these pathways (Fig. 1) and immune cells (Fig. 2) are also implicated in radiation responses. Multiple immune cells express ERs and RT/oestrogens can both increase PD-L1/PD-1 expression on tumour cells and immune cells39,41.

(1) Radiation and excess oestrogens induce double strand DNA (dsDNA) breaks, directly and indirectly via production of reactive oxygen species. (2) Recognition of DNA damage by ATM/ATR induces DNA damage responses including DNA repair (homologous recombination repair and non-homologous end-joining) and cell cycle checkpoint arrest. This is achieved via activation of proteins including p53 and inhibition of CDKs/cyclins/CDC25A/c-myc. Oestrogen signalling can influence multiple points in these pathways, for example increasing expression/activity of CDKs/cyclin D1/CDC25A/c-myc whilst inhibiting tumour suppressor proteins such as Rb and p53, with the overall effect of cell cycle progression which may influence radiosensitivity. (3) dsDNA breaks lead to cytoplasmic dsDNA release which triggers cGAS/STING signalling. This typically leads to an acute type 1 interferon response which activates and attracts DCs/cytotoxic T-cells, but chronic or disrupted cGAS/STING signalling can lead to pro-tumour effects via non-canonical NF-kB signalling. cGAMP can be taken up by neighbouring immune cells and activate further cGAS/STING-mediated type 1 IFN responses. However, tumour cells may upregulate CD73/ENPP1 to convert extracellular cGAMP to adenosine, with immunosuppressive consequences. Type 1 interferon/STAT1 signalling can also increase Trex1 levels as part of a self-regulatory feedback loop. (4) Type 1 interferons (e.g. IFN-β) released in response to radiation increase expression of interferon-stimulated genes via JAK/STAT signalling. Oestrogen signalling can inhibit type 1 IFN responses by inhibition of STAT2 and exerts other non-genomic effects on cells including upregulation key oncogenic pathways including EGFR/MAPK and PI3K/AKT. There is also complex context-dependent cross-talk between transcriptional activity of oestrogen receptors and NF-kB. (5) Radiation triggers release of DAMPs which can promote DC activation. Radiation can also increase MHC-I expression on cells which increases recognition by immune cells, but oestrogen signalling and radiation can both lead to PD-L1 upregulation on tumour cells which reduces T-cell activation. Finally, radiation and oestrogen signalling can both increase release of TNF-α which has further impacts on NF-kB signalling. ES oestrogen signalling, dsDNA double-strand DNA, DAMPs damage-associated molecular patterns, HRR homologous recombination repair, NHEJ non-homologous end-joining, ROS reactive oxygen species, IFNs interferons, IFN-Rs interferon receptors, DC dendritic cell, CDK cyclin-dependent kinase.

Both radiation and oestrogen signalling can also have divergent immunomodulatory effects on multiple immune cells within the HR+ TIME, with multiple immune cells expressing oestrogen receptors. Via CAFs and generation of reactive oxygen species, radiation induces TGF-β release which can increase DNA damage responses, induce Treg differentiation, promote M2 TAM polarisation, and inhibit dendritic cells which reduces cytotoxic T-cell activation. Oestrogen signalling can reduce TGF-β signalling by reducing Smad levels and TGF-β signalling from CAFs can reduce tumour cell oestrogen signalling dependence although TGF-β levels may be higher in HR+ BCs with high levels of M2 TAMs. TGF-β (predominantly released by CAFs) and oestrogen signalling have both been implicated in extracellular remodelling which could possibly promote an immune-excluded microenvironment in HR+ breast cancer. Both radiation and oestrogen signalling can increase release of various other immunosuppressive cytokines which further influence multiple immune cell populations. Oestrogen signalling can induce Treg differentiation via ERα binding to the FOXP3 promoter. Oestrogen signalling tends to steer TAMs towards M2 polarisation via IL-10/STAT3/CCL2/CCL5 signalling, whereas the effect of radiation of TAM polarisation remains unclear. Oestrogen signalling can increase the production of pro-tumoural neutrophil extracellular traps, whereas radiation may reduce neutrophil abundance. Radiation can attract CAFs to tumours via increased expression of NOTCH ligand on HR+ tumour cells but can also trigger their senescence and reduce their invasive capability. ES oestrogen signalling, DCs dendritic cells, Tregs T regulatory cells, TAMs tumour associated macrophages, ECM extracellular matrix, Neuts neutrophils, CAFs cancer-associated fibroblasts, NETs neutrophil extracellular traps, NOTCH-l NOTCH ligand.
DNA damage and repair
Radiation causes cell death by inducing double strand DNA (dsDNA) breaks. Tumour cells with deficient DNA damage repair pathways are more susceptible to cell death from irradiation. Excess oestrogens can also induce dsDNA breaks at oestrogen-responsive genes42. Oestrogens can exert influence on DNA damage response pathways, including non-homologous end joining (NHEJ) and homologous recombination repair (HRR). For example, they can downregulate ATM/ATR expression but increase DNA-PKcs, BRCA1/2 and MRN complex protein expression, which may explain a preponderance for oestrogen-dependent tumours in patients with intrinsic HRR deficiencies43. ATM is a master regulator of DNA damage responses to RT44, suggesting tumour oestrogen-dependence may alter radiosensitivity. In HR+ BC clonogenic survival assays and an immunocompromised HR+ mouse model, the addition of endocrine therapy to RT had a radiosensitising effect through NHEJ inhibition45. Both RT and oestrogens are also known to induce reactive oxygen species (ROS) that contribute to indirect DNA damage46.
Cell cycle and proliferation
Oestrogen signalling drives cell cycle progression, particularly through the G1/S checkpoint, by increasing the expression and activity of cyclin D1, c-myc, cyclin-dependent kinases (CDKs including CDK4/6) and CDC25A47,48. CDK4/6 binds to Rb, phosphorylates and inactivates it to promote S phase and release of the E2F/DP transcription factors, whilst E2 can also directly downregulate Rb expression49,50. Endocrine therapy combined with CDK4/6 inhibition can therefore elongate a G0-G1 state to inhibit tumour proliferation. Oestrogen signalling can also inhibit p53 function which can drive proliferation in hormone-dependent cancers51. The intrinsic radiosensitivity of tumour cells varies in different cell cycle phases, with late S phase the most radioresistant due to increased HRR52. Oestrogen signalling may drive increased redistribution through the cell cycle, which may influence radiosensitivity to fractionated RT regimens.
However, direct protein-protein actions of oestrogen signalling, including upregulation of pro-survival pathways such as EFGR, MAPK and PI3K/AKT, can confer radioresistance and drive tumour proliferation53,54. There is also evidence of a positive feedback loop between E2 and the pro-inflammatory cytokine TNF-α, which is implicated in tumour proliferation, epithelial-mesenchymal transition and immune evasion55,56. Cell death from RT can also induce release of immunosuppressive cytokines including TNF-α, TGF-β and IL-657.
T-cells and type 1 IFN responses
DNA damage from RT triggers dsDNA translocation to the cytosol, leading to an acute release of pro-inflammatory type 1 interferons (IFNs) including IFN-β via the cGAS/STING pathway58. cGAMP, produced upon cGAS recognition of cytosolic dsDNA, is taken up by neighbouring immune cells which induces further type 1 IFN gene expression59. RT also triggers release of damage-associated molecular patterns (DAMPs) such as calreticulin, HMGB1 and ATP, promoting cancer cell uptake by DCs and DC activation mediated by Toll-like receptors (TLRs) and P2X7R9. Alongside promoting release of tumour-associated antigens, RT increases tumour cell expression of MHC-I and other cell surface molecules that improves their recognition by CD8+ T-cells60. Together, these processes promote recruitment and activation of DCs that cross-present tumour antigens, leading to cytotoxic T-cell activation which contributes to RT’s immunostimulatory anti-tumour effects. Given that ERα signalling in HR+ BC cells can downregulate MHC-I expression and IFN-γ61, this suggests a role for RT to overcome this mechanism of immune evasion.
Conversely, RT-induced chronic/constitutive cGAS/STING signalling can have pro-tumour effects, including T-cell exhaustion and induction of immature, dysfunctional or tolerogenic DCs6,38. These effects may be exacerbated in HR+ BC62 and are mediated by increased non-canonical NF-kB signalling downstream to STING activation63. Extracellular transport and conversion of cGAMP to adenosine by overexpressed ENPP1/CD73 cell surface proteins on tumour cells also promotes immunosuppression64. Trex1, a DNA exonuclease inducible by high-dose RT, inhibits cGAS/STING signalling and is itself regulated by type 1 IFN/STAT1 signalling as part of an autocrine feedback loop65,66.
Intriguingly, ERα signalling can assist DCs to prime antigen-specific T-cells following TLR stimulation67. In vitro, E2 prevents DC growth in response to FLT3 ligand, whereas it stimulates CD11c+/MHC-II+ DC differentiation in the presence of GM-CSF68. ERα signalling in tumour cells can also inhibit type 1 IFN-stimulated gene transcription through induction of H2A.Z expression and inhibition of STAT2 activity, which prevents the formation and function of ISGF3 complexes69. Increased interferon-stimulated gene expression is seen in BC cells with endocrine and RT cross-resistance70. Complex interactions between NF-kB and oestrogen signalling including evidence of synergy and trans-repression in different contexts may drive endocrine resistance and radioresistance71.
Overall, effects of oestrogen signalling on T-cells may increase immunosuppression. Oestrogens can reduce migration of naïve T-cells to the thymus and influence T-cell selection72. ERα can bind to the FOXP3 promoter which promotes T-cell differentiation to Tregs73,74. Aromatase inhibition can decrease TIME Treg levels in BC75. ERα is also linked to downregulation of HLA-II on BC cells76. High E2 levels favour Th2 CD4+ T-cell differentiation through increased IL-4 release77.
TAMs
RT and oestrogen signalling exert complex effects on TAMs, including their maturation, migration and polarisation. TAMs polarise on a spectrum between a pro-inflammatory M1 phenotype and a pro-healing immunosuppressive M2 phenotype. The acute type 1 IFN response, DAMP release, ROS production and increased NF-KB signalling in response to RT activates M1 TAMs78,79. M2 macrophages decrease in response to RT in triple-negative mouse models80. Conversely, RT-related increases in CSF-1, IL-10, TGF-β and CCL2 levels favour M2 TAM polarisation81. Doses ≤2 Gy favour an M2 TAM phenotype whereas cumulative doses of 10 Gy favour an M1 phenotype82,83. RT also induces TAM recruitment to tumours via the CCL2-CCR2 and CCL5-CCR5 axes84.
Oestrogen signalling also increases CCL2 and CCL5 secretion by many ER+ BCs which increases M2 macrophage infiltration84. Oestrogen signalling tends to steer TAMs towards an M2 phenotype by upregulating the STAT3-IL-10 axis and suppressing NF-kB signalling, which can be prevented with oestrogen inhibition85,86. However, in some circumstances oestrogens may also lead to M1 polarisation via upregulation of STAT187. In patients with highly immune-infiltrated tumours, higher levels of TGF-β signalling are present in HR+ tumours versus TNBCs, which may derive from M2 TAMs55. Conversely, ER+ tumour cells can also decrease TGF-β signalling by increasing Smad ubiquitination88.
Neutrophils
Most evidence implies TIME neutrophils typically exert immunosuppressive pro-tumour effects. Although the impact of RT on neutrophils is not well-defined, recent evidence suggests RT reduced TIME neutrophil abundance in an HR+ BC mouse model89. Oestrogen signalling influences neutrophil production of pro-tumoural neutrophil extracellular traps90. Oestrogens also increase infiltration of neutrophils expressing lymphocyte function-associated antigen 1 and TGF-β release in the HR+ TIME91.
CAFs
CAFs are a heterogeneous radioresistant stromal cell population that exhibit pro-tumourigenic or tumour-restraining properties. RT can induce CAF senescence and reduce their invasive capabilities via increased integrin expression92,93. RT can induce NOTCH ligand expression in luminal tumour cells which increases CAF attraction89. Increased TGF-β release in response to RT may also be predominantly CAF-mediated88.
Recent pre-clinical evidence in luminal BC models also suggests that increased CAFs are linked to a TGF-β-mediated reduction in expression of ERα and reduction in oestrogen signalling dependence94. ERα36-expressing CAFs release pro-tumourigenic cytokines including CXCL5 when co-cultured with TNBC cells and may contribute to M2 macrophage polarisation95. CD146- CAFs and CAFs expressing an alternative ER (GPR30) have also been associated with tamoxifen resistance in ER+ BC96,97. Finally, oestrogen signalling can increase extracellular remodelling, which may contribute to the ‘immune excluded’ nature of many HR+ tumours40,98.
Summary
Oestrogen signalling drives tumour cell cycle progression and RT acts through DNA damage, suggesting RT may be particularly beneficial in HR+ BC (Fig. 1). In particular, many immunostimulatory effects of RT arise downstream from responses to DNA damage, so may be enhanced in HR+ tumours. Conversely, overall oestrogen signalling appears to exert immunosuppressive influences on many TIME subpopulations (Fig. 2).
Notably, many studies discussed above relied on models that cannot fully capture the complexity of the human HR+ BC TIME. The interplay between RT and oestrogen signalling and resultant impact on anti-tumour immune responses is not yet fully understood, particularly in specific pre-clinical immunocompetent HR+ BC models. Local oestrogens produced by aromatase enzymes in breast tissue are likely to exert the influences of oestrogen signalling discussed above on immune cells in the HR+ BC TIME. However, where effects are mediated through oestrogens on ER-expressing immune cells in the TIME, these may not be exclusive to HR+ BCs and may affect all cancer-bearing pre-menopausal females with higher levels of circulating oestrogens. Finally, the context-dependent influence of oestrogen signalling on ER-expressing cancer cells may have specific downstream effects on other immune/stromal cells which require further characterisation in HR+ BC.
RT-ICI in preclinical BC models
The abscopal effect, where an unirradiated lesion responds following RT, was demonstrated to be immune-mediated using a 67NR syngeneic mammary carcinoma mouse model99. Since then, multiple studies have shown that tumour irradiation promotes immune cell recruitment into tumours seeded in immunocompetent mice, including CD4+ T-cells, CD8+ T-cells and DCs100. This partly occurs due to tumour cell secretion of IFN-β following a cGAS/STING-mediated DNA damage cascade, and can be augmented by a STING agonist66,101. RT can also lead to improved DC antigen processing102 and increase presentation of RT-upregulated neoantigens, leading to improved T-cell recognition and tumour cell killing103. Together, these studies demonstrate the potentially immunogenic effects of RT and underline the potential for additional immunomodulatory agents that enhance T-cell/DC activity to maximally exploit an RT-induced immunogenic TIME.
The ability of RT to sensitise resistant tumours to anti-CTLA-4 treatment, resulting in increased control of the irradiated tumour and non-irradiated lung metastases, was first demonstrated in the 4T1 mouse model of TNBC104. These observations were subsequently confirmed in other BC models and with the combination of RT and anti-PD-1/anti-CTLA-4 treatment, including demonstration of antigen-specific clonal expansion of tumour-specific CD8+ T-cells and abscopal effects105,106,107,108.
Timing and treatment sequencing may influence RT-ICI responses. Dovedi et al. demonstrated that PD-L1 expression on tumour cells peaks 3 days after the final fraction of RT due to CD8+ T-cell production of IFNγ109. Concurrent anti-PD-L1 with RT (5x4Gy) reduced tumour burden in the 4T1 model whereas sequential ICI treatment with anti-PD-L1 7 days post-RT did not enhance survival in a colorectal model. Similarly, increased PD-L1/PD-1 expression on tumour/immune cells were observed 3 days post-RT in a HER2+ lobular BC model110. Addition of anti-PD-L1 treatment to RT increased survival and protected mice from tumour regrowth following rechallenge experiments. Wei et al. also observed primary tumour control and abscopal effects with the combination of RT (3x8Gy) followed by anti-PD1 treatment commencing on the final day of RT in the 4T1 model111. Conversely, they observed anti-PD1 treatment prior to RT prevented abscopal responses and reduced TIME CD8+ T-cell expansion in a colorectal model, as it increased CD8+ T-cell susceptibility to RT-induced apoptosis. These studies highlight the importance of targeting the PD-L1/PD-1 axis within the window of its RT-induced upregulation. Regarding dose/fractionation, fractionated RT regimens (either 3x8Gy or 5x6Gy) with anti-CTLA-4 treatment controlled growth of both a primary tumour and an unirradiated secondary tumour in a TS/A model, whereas a single 20 Gy fraction with anti-CTLA-4 treatment did not achieve an abscopal effect108.
Targets that modulate anti-tumour immune responses have been trialled alongside RT-ICI in murine pre-clinical models (Table 1)80,112,113,114,115,116,117,118,119,120,121,122,123,124. For example, TGF-β blockade enhances T-cell reactivity and extends survival when added to RT-ICI115. This study also showed upregulation of PD-1 on intratumoral T-cells limited long-term responses to RT but could be overcome by anti-PD-1 blockade.
OX-40 targeting has helped demonstrate that immune checkpoint pathways are not only non-redundant but temporally distinct in the context of RT112,113. Targeting CD137 and CD40 demonstrated that RT can enrich the TIME with cytotoxic lymphocytes and simultaneous ICI overcomes resistance that RT alone cannot114. CD40 agonism also improves RT-ICI responses by improving DC activation and enhancing priming of de novo T-cell responses, which is particularly relevant for immune-cold tumours80.
CD73 is upregulated in response to RT in 4T1 and TS/A mouse models. Addition of an anti-CD73 antibody to anti-CTLA4 treatment and RT (1x20Gy) resulted in improved local tumour control, reduced lung metastasis burden and increased DC migration in a TS/A model124. Bromodomain-containing protein 4 (BDR4) is upregulated in tumour cells and exerts pro-proliferative/immunosuppressive effects. BDR4 inhibition can enhance the effect of RT (3x8Gy) in the 4T1 model, in part by decreasing M2 macrophage TIME infiltration125. In TS/A and 4T1 mouse models, LTX-315 (an oncolytic peptide that induces immunogenic cell death) improves tumour controls by RT (3x8Gy), possibly through NK cell-mediated immunity126. Finally, in the 4T1 model, RT has also improved responses to an irradiated induced stem cell vaccine127, cancer vaccine against fibroblast activation protein alpha128, and neoantigen-based vaccines through increased immunostimulatory effects129.
Overall, pre-clinical BC studies demonstrate that RT-ICI can improve tumour control. Responses may be enhanced with additional agents targeting alternative aspects of anti-tumour immune responses. Importantly, studies have begun to address questions regarding optimal RT-ICI dose-fractionations and sequencing which are critical when considering clinical translatability. However, the success of RT-ICI in BC mouse models, including induction of abscopal responses, has not yet been reliably reproduced in clinical trials.
The requirement for immunocompetent HR+ mouse models
Current pre-clinical challenges
The paucity of immunocompetent HR+ BC murine models has been a fundamental limitation to investigating the impact of RT and immunotherapy in HR+ disease130, hence most above studies used triple-negative models. Immunodeficient HR+ BC models are not compatible with interrogation of anti-tumour immune responses to therapies131. Breast tumours in immunocompetent mice induced by carcinogens (e.g. progesterone receptor agonists) can possess luminal B features and ICI resistance, but grow slower than implanted HR+ cell lines which limits their feasibility126,132. Transgenic models are also used, such as MMTV-PyMT where mammary tumours are generated via expression of polyoma virus middle T oncoprotein by mouse mammary tumour virus LTR133. In this model, mammary cells are initially ER/PR+ but often lose their expression once tumours become established, and can express HER2 at low levels. Transgenic models are not well recognised by immunosurveillance due to a low TMB134.
Therefore, immunocompetent models with tumours derived from transplantable TNBC cell lines such as 4T1 are commonly preferred. The 4T1 model is considered poorly immunogenic in comparison to other tumour types135. It also possesses high metastatic potential, is poorly differentiated, and has relatively poor immune cell infiltration with a predominance of immunosuppressive TAMs136,137. It may not optimally capture the distinct TIME and immune responses in HR+ BC and may exhibit distinct immune signatures to RT versus HR+ models.
Advances in immunocompetent HR+ models
There is variation in the defining features of an HR+ mouse model, which can include some/all of ER/PR expression, demonstration of ER signalling, and requirement of oestrogen dependence for tumour growth in vivo. Several immunocompetent HR+ BC murine models have been developed and used in the RT-ICI setting.
The non-basal ER+TS/A mammary adenocarcinoma cell line, developed in 1983, has been used in immunocompetent mouse models to investigate RT ± immunotherapy115,126. TS/A tumours have less propensity for distant metastases than 4T1 tumours, possess low immunogenicity, and exhibit an immunosuppressive TIME enriched for M2 TAMs138. Turrell et al. used a TS/A cell line variant (TSAE1) to form epithelial mammary tumours in syngeneic immunocompetent BALB/c mice and demonstrated that increased rates of distant metastases were induced by a PDGF-C-dependent aged tumour microenvironment139.
Improved outcomes with endocrine therapy and RT-ICI in HR+ BC were recently demonstrated using an orthotopically transplanted TC11 cell line in an immunocompetent mouse model140. TC11 cells were developed from a prolactin-induced tumour in an NRL-PRL mouse model which allows spontaneous development of HR+ BCs, commonly driven by Kras mutations141. This simulates advanced endocrine therapy-resistant HR+ BC, which grows independently of oestrogen signalling but exhibits transcriptomic changes in response to anti-oestrogens.
The E0771 cell line possesses luminal B characteristics including PR and ERβ positivity, with less ERα positivity142. An immunocompetent E0771 C57BL/6J mouse model was used to show that an IL2/IL15 receptor agonist (NL-201) can enhance RT and anti-PD-1 responses by increasing cGAS/STING-mediated antigen presentation and T-cell infiltration143. Buque et al. have also developed a luminal B-like model, with mammary tumours induced by the carcinogens medroxyprogesterone acetate and 7,12-dimethylbenz[a]anthracene in C57BL/6 mice132. This model may better mimic aspects of human BC including effective immune escape following immunosurveillance and immune-editing. In this model, ablative RT (2x20Gy) achieved control of 90% of the irradiated tumours, but the improvement in overall survival (OS) was similar to non-ablative RT regimens due to increased secondary tumour development144. The addition of various immunotherapies (anti-PD1 ± FLT3 ligand, anti-CTLA4, anti-IL-1β) to fractionated RT (given at 6x8Gy, 3x10Gy and 2x20Gy) did not increase OS over RT alone, possibly partly explained by the RT dose/fractionations used and higher degree of intertumoural heterogeneity in this model.
Perez-Lanzon et al. recently developed an ICI-resistant HR+ BC cell line (B6BC) which establish tumours in syngeneic immunocompetent C57BL/6 mice145. They tend to exhibit low T-cell and high macrophage infiltration which recapitulates many typical HR+ TIMEs. Langsten et al. have genetically engineered the triple-negative 4T1.2 cell line to express ERα and produce mammary tumours in immunocompetent mice146. They found ER+ 4T1.2 tumours display a ‘colder’ TIME than triple-negative 4T1.2 tumours and display less preponderance for bone metastases. Orthotopic HR+ BC models with immunocompetent 129/SvEv mice and ER+ SSM3/SSM2 cell lines have also been established147,148.
Human ER+ cell lines such MCF-7 (luminal A-like) and 59-2-HI (HR+ epithelial) have been used in NOD-scid mice with xenografts of human CD34+ haemopoietic stem cells or BALB/C murine bone marrow to create a functioning immune system149,150,151. In this model RT (1x6Gy) ± anti-PD-L1 controlled MCF-7 tumours and TIME changes in response to treatment could be assessed152. Furthermore, Bruss et al. transplanted a HR+/HER2- patient-derived tumour xenograft within this humanised mouse model and demonstrated differential expression of immune checkpoints in immune cell populations151.
Homogenous cell line transplantation is unlikely to recapitulate the complexity of human TIMEs or heterogeneity of the broad group of HR+ BCs. Cell lines derived from late-stage tumours may not represent early BCs as they possess more mutations conferring immune evasion134. HR+ models which require exogenous oestrogens to promote metastases may confound TIME analyses, given the immune impacts of oestrogen signalling. The radiosensitivity and impact of dose/fractionation on HR+ cell lines in immunocompetent mouse models also requires further delineation.
Future pre-clinical work should recognise the difference between distinct HR+ tumour subtypes (e.g. luminal A or B) and build models which can recapitulate these, or encompass experiments using multiple models. Rat mammary gland tumours are largely ER+, which can be more representative of human BC ER signalling and its interplay with anti-tumour immune responses, particularly in immunocompetent models153. Immune-compatible models utilising humanised mice with patient-derived xenografts may also provide a more representative TIME but have not yet been used to explore the effects of RT ± ICI, which could be explored. BC organoid models can also be useful for examining cancer cell responses to RT154. Correlation of pre-clinical results with translational analyses is crucial as these samples allow the most reliable assessment of TIME changes in response to therapies.
Clinical data supporting RT-ICI in HR + BC
Combined systemic agents with ICI
The evidence of benefits from chemotherapy-ICI combinations for TNBC that has led to licensed indications for such combinations has not yet been followed by similar evidence to support routine use in HR+/HER2- BC. The modest responses to ICI monotherapy in HR+ disease broadly mirror those in TNBC. This highlights the requirement for improved identification of responding sub-populations and additional therapeutic agents to augment responses. In response to pembrolizumab, expanding T-cells from HR+ BCs expressed more genes associated with higher naivety compared to expanded T-cells from TNBC155, suggesting a potential role for RT to attract and activate functional cytotoxic T-cells within the TIME.
There have been many trials reporting mixed results with ICI for HR+ BC patients (comprehensively summarised in Supplementary Table 1). Phase 2-3 trial results have reported significant increases in pathological complete response (pCR) rates with the addition of neo-adjuvant ICI versus NACT alone6,7,29,156. Notably, the I-SPY2 trial and subsequent KEYNOTE-756 trial selected for ‘high-risk’ HR+/HER2- tumours. Maximal pCR rates were 30%, suggesting a potential role for additional therapies. Interestingly, in Checkmate 7FL7, higher sTILs were associated with improved pCR rates, raising the possibility that additional therapies (e.g. RT) to increase sTIL infiltration may further improve outcomes.
Chemotherapy-ICI responses have generally been weaker in metastatic HR+ BC. However, three phase 2 trials in metastatic BRCA-mutated and HRR-deficient tumours yielded response rates of 41-69.2%, with higher response rates for HR+ BCs versus TNBCs157,158,159. Finally, combining CDK4/6 inhibitors with ICI has produced encouraging response rates up to 55% but often alongside high rates of severe adverse events (notably hepatotoxicity) which has limited progress160,161. It could therefore be beneficial to identify a subset of patients that can benefit from a more local immunostimulatory stimulus alongside ICI, with less associated toxicity.
Lessons learnt from TNBC RT-ICI trials
Discordance exists between promising pre-clinical data for RT-ICI and divergent trial outcomes in multiple tumour types104,162,163. Efforts to improve metastatic TNBC outcomes with RT-ICI directed at metastatic lesions have also proved largely unsuccessful in phase 2 trials, with response rates between 8-17.6%164,165,166.
Possible reasons for the limited efficacy in these metastatic trials include choices of RT dose/fractionation, timing of RT in relation to ICI, and site of RT167. Upregulated neo-antigen presentation from RT to a metastatic site may have limited effects on anti-tumour immune responses to other lesions within a patient. Metastases may also harbour a more immunosuppressive TIME which may be challenging to overcome with RT compared to irradiation of a primary lesion168.
Contrastingly, in the neo-adjuvant setting, the combination of RT-ICI using 3x8Gy stereotactic body RT (SBRT) and concurrent pembrolizumab followed by NACT resulted in a response rate of 67.6%35. Translational multiomic studies assessed TIME changes induced by anti-PD-1 and RT and identified that RT can induce responses in a group who would otherwise fail to respond to immunotherapy. Similar approaches may identify RT-ICI responsive HR+ subgroups.
HR+ BC RT-ICI trials
The Neo-CheckRay trial has reported results of a safety run-in adding concurrent neo-adjuvant SBRT (3x8Gy), durvalumab and oleclumab (an anti-CD73 antibody) to NACT14. Patients have HR+/HER2- luminal B disease, defined as Ki-67 score ≥15% or grade 3 disease, plus high-risk MammaPrint status. Axillary nodes were delineated as avoidance structures during SBRT planning. 2/6 patients achieved pCR and 2/6 patients had only scattered residual disease. Following these promising preliminary results, a randomised phase 2 trial is ongoing169. Another neo-adjuvant RT-ICI trial, PEARL/IIT2017-07-HO-PembroRT (n = 66), used two doses of pembrolizumab in combination with primary tumour RT (3x8Gy) prior to neo-adjuvant chemotherapy170. This included 12 patients with high-risk HR+/HER2- BC, 4 of whom achieved pCR.
Metastatic HR+/HER2- BC RT-ICI trials have been less successful to date171. KBCRN-B002, which included 20/28 HR+/HER2- patients, combined RT (1x8Gy) to a bony metastasis with nivolumab172. The ORR was 7%, with both partial responses in HR+/HER2- patients. Barroso-Sousa et al. did not observe any clinical responses in 8 HR+/HER2- patients treated with RT (5x4Gy) to ≥1 metastatic lesion and concurrent pembrolizumab173. Trials including HR+/HER2- patients have demonstrated feasibility and intracranial control with the ICI and whole brain RT or stereotactic radiosurgery to BC brain metastases174,175. All patients in these RT-ICI studies had bony or brain metastases irradiated. The TIME of brain and bony metastases is likely distinct from the TIME of soft tissue/visceral/nodal metastases and primary tumours, for example more immunosuppressed or immune-excluded, so may not represent the optimal target for RT to trigger immunogenic effects176,177. Some of the disconnect between robust pre-clinical data and these trials may also reflect the over-representation of triple-negative mouse models which may not be representative of the HR+TIME.
There are twelve registered HR+/HER2- BC RT-ICI trials ongoing in the early and metastatic settings (Table 2). These trials build upon the pre-clinical data discussed above, including targeting additional immunomodulatory receptors such as CD73, FLT3 and CD40 that have shown promise in BC mouse models.
Three neo-adjuvant trials are evaluating the combination of pembrolizumab with RT (P-RAD, CBCV and BreastVax). In contrast to CBCV and BreastVax, P-RAD selects ‘high-risk’ HR+/HER2- patients and will deliver NACT + pembrolizumab following RT-ICI. Impacts of dose/fractionation and timing of RT in relation to ICI may start to be addressed given the variation in schedules between these trials. The BreastVax co-primary endpoint defines ‘major’ pathological response as tumours with <10% viable tumour remaining, while CBCV employs the residual cancer burden (RCB) calculator which has been developed and validated in multiple studies of neo-adjuvant chemotherapy for BC178. Given the lower pCR rates in HR+ disease, these may prove preferable endpoints to identify patients deriving clinical benefit from RT-ICI in HR+ disease, as they capture patients with good responses without pCR. The selection of nodal pCR as a co-primary endpoint in P-RAD reinforces an expectation that RT and ICI will improve systemic anti-tumour immune responses. Finally, all three trials include secondary translational endpoints such as longitudinal immune cell subpopulation mapping, which is important for evaluating disease biology and guiding future HR+/HER2- BC RT-ICI trial design.
Considerations for future neo-adjuvant HR+ RT-ICI trials
Neo-adjuvant RT is an attractive proposition in HR+ BC. Fractionated RT is safe prior to definitive surgery13,179 and neo-adjuvant RT-ICI appears feasible14. Neo-adjuvant RT may improve local recurrence rates for HR+ tumours versus adjuvant RT180. Delivery of RT with tumour in situ can potentially prime new T-cell responses and/or increase the ability of pre-existing T-cells to eliminate the cancer cells that survive RT.
In contrast, adjuvant RT is thought to work by eliminating residual microscopic disease, but there is no evidence that it can enhance ICI effectiveness. Indeed, adjuvant RT in BC may not effectively stimulate the TIME in immune-infiltrated tumours181. Upcoming randomised trials of neo-adjuvant vs adjuvant RT will include patients that have received neo-adjuvant chemo-immunotherapy and would be well placed to provide translational insights (e.g. PRADA-II; NIHR163836, in set-up).
A potential downside to neo-adjuvant RT (versus adjuvant RT) is that full pathological information is not available to clinicians for patient risk stratification and selection of RT dose/fractionation and volumes. Adjuvant tumour bed boost RT is widely used to increase local tumour control in high-risk patients. Therefore, an appealing strategy (utilised by Neo-CheckRay) transfers this to the neo-adjuvant setting with ICI in the form of a ‘primary tumour boost’, followed by adjuvant whole breast/chest wall +/− regional nodal RT based on response. Fractionated regimes may be preferable in HR+ disease due to the impact of oestrogen signalling on promotion of cell cycle progression.
Neo-adjuvant RT is associated with low toxicity, and based on experience from current trials is unlikely to cause significant delays to surgical intervention13, although theoretically possible in isolated cases. RT-ICI could be delivered concurrently with NACT or trialled in an early-phase ‘window of opportunity’ trial for high-risk HR+/HER2- patients that respond less well to NACT. Patient selection could incorporate emerging predictive biomarkers. This type of neo-adjuvant RT-ICI trial design could be used to test emerging novel immunotherapies and is well placed to deliver translationally rich outputs as biopsy sites are easily accessible for longitudinal sampling.
HR+ BC mouse data suggests fulvestrant may improve RT-ICI responses and improve TIME CD8:T-regulatory cell ratios140. This data, combined with the overall immunosuppressive effects of oestrogen signalling discussed above, raises the possibility that combining hormonal therapies with RT-ICI may improve the immunogenicity of RT and response rates. This concept merits further consideration and is undergoing testing in the CBCV trial, in which HR+ BC patients receive letrozole and neo-adjuvant RT with/without addition of FLT3 ligand and/or anti-PD-1.
Much of the existing RT evidence base in BC has been derived from trials conducted before the use of immunotherapy for non-metastatic disease. The optimal axillary management strategy, particularly following neo-adjuvant treatment, is a topic of debate with support for both treatment escalation (e.g. inclusion of the internal mammary chain) and de-escalation (e.g. omission of regional nodal RT) in certain settings. Elective regional nodal RT may be detrimental to RT-ICI responses and delaying nodal RT improves RT-ICI responses in melanoma/colorectal mouse models182,183. Furthermore, inclusion of regional nodes in RT fields influences the composition of the primary TIME including reduced CD8 T-cell levels versus tumour-only RT184. In the context of ICI, BC is a strong candidate to explore the influence of nodal RT on treatment response, given the feasibility of separating targeted neo-adjuvant RT to the primary tumour from broader adjuvant RT fields (including nodal areas if required) for eradication of microscopic disease.
Finally, consideration regarding optimal definitions of clinical benefit is required in the neo-adjuvant HR+ RT-ICI setting. A key benefit for ICI ± RT may be improved systemic disease control, but will require longer follow-up periods to prove within trials (e.g. for disease-free survival). This is particularly relevant as (a) HR+/HER2- BC is more associated with late relapses, and (b) the correlation of pCR with long-term survival is less strong for HR+/HER2- BC, so may be less reliable as a short-term surrogate outcome in this setting185. Circulating biomarkers such as cell-free circulating DNA, extracellular vesicles, non-canonical circulating tumour cells, cell-free RNAs, and specific immune cell subsets may emerge as candidates to predict likelihood of recurrence or response to therapy186.
Conclusions
Whilst most HR+ tumours do not respond to ICI (with or without adjunctive therapies such as RT), there is clearly a subgroup of HR+ tumours that do derive significant benefit from ICI. Pre-clinical/clinical data refute the traditional view of HR+ tumours always exhibiting a non-immunogenic ‘cold’ TIME and demonstrate there is scope for RT-induced TIME modulation for immunotherapy sensitisation. Summarising the above data, the following key points emerge as considerations for current and future HR+ BC RT-ICI trials:
-
1.
The rationale for neo-adjuvant RT-ICI has more evidence versus metastatic or adjuvant RT-ICI, so could be considered as a preferred treatment context for further investigation.
-
2.
A focus on identification of subgroups within higher-risk HR+/HER2- tumours may yield improved outcomes.
-
3.
Patient selection by BC biological subtype appears crucial. Studies should investigate emerging HR+/HER2- specific predictive biomarkers for ICI ± RT beyond sTILs and account for quantitative receptor analysis.
-
4.
Neo-adjuvant RT can comprise a targeted tumour-only volume with the aim of eliciting an immunostimulatory response. Adjuvant whole breast/chest wall ±nodal volumes can be delivered to tissue outside this resected target.
-
5.
Given the immunosuppressive effects of oestrogen signalling on several immune/stromal cell subpopulations, further trials of anti-oestrogens in combination with RT-ICI should be considered, especially for higher stage tumours that are strongly ER+.
-
6.
Alternative endpoints to dichotomised pCR versus non-pCR analyses, such as ordinal or continuous scores like RCB, or emerging liquid biopsy-based biomarkers, may be preferable for identifying patient subgroups benefiting from neo-adjuvant RT-ICI.
-
7.
Empirical approaches to the investigation of RT-ICI may fail to detect effective regimens in biologically defined subgroups. Patient selection and clinical trial design should continue to be underpinned by emerging pre-clinical data. In the HR+ BC setting, this should make use of the increasing availability of immunocompetent HR+ BC animal models and BC organoid models.
Higher-risk HR+ tumours broadly appear to derive more benefit from ICI combinations. These tumours tend to possess luminal B phenotype, with higher grade, higher Ki67 score or less dependence on oestrogen signalling. Earlier use of ICI combinations in a patients’ disease course appears preferable, with improved responses largely seen in neo-adjuvant trials. A shift towards testing ICI combinations in the neo-adjuvant setting perhaps reflects lessons learnt from metastatic BC trial results. Some of the best responses to date with ICI combinations for HR+ disease have resulted from integration of improved selection of high-risk HR+ tumours and early neo-adjuvant intervention in dedicated HR+/HER2- BC-specific trials.
However, improved understanding of the impact of RT on the TIME and anti-tumour immune responses is required to further refine patient selection and harness maximal outcomes with RT-ICI in HR+ BC. Advances in immunocompetent HR+ BC mouse model development are a significant step towards this. RT and oestrogen signalling both produce divergent immunomodulatory effects, so deeper understanding of the interplay between these may produce novel therapeutic targets to combine with RT. If novel endocrine therapies that inhibit the immunosuppressive effects of oestrogen signalling on immune cells whilst maintaining its influence on cell cycle progression were developed, this could be beneficial in combination with RT-ICI for HR+ BC. Given the prevalence of HR+/HER2- BC, establishing RT-ICI in the neo-adjuvant HR+/HER2- setting would be clinically significant, even if only for a selected subpopulation within this diverse group.
Data availability
No datasets were generated or analysed during the current study.
Abbreviations
- BC:
-
breast cancer
- CAF:
-
cancer-associated fibroblast
- DC:
-
dendritic cell
- Gy:
-
Gray
- HR+:
-
hormone-receptor positive
- HRR:
-
homologous recombination repair
- IFN:
-
interferon
- ICI:
-
immune checkpoint inhibition
- MDSC:
-
myeloid-derived suppressor cell
- NACT:
-
neo-adjuvant chemotherapy
- NHEJ:
-
non-homologous end-joining
- pCR:
-
pathological complete response
- RCB:
-
residual cancer burden
- RT:
-
radiotherapy
- RT-ICI:
-
radio-immunotherapy
- SBRT:
-
stereotactic body radiotherapy
- sTILs:
-
stromal tumour-infiltrating lymphocytes
- TAM:
-
tumour-associated macrophage
- TIME:
-
tumour immune microenvironment
- TMB:
-
tumour mutational burden
- TNBC:
-
triple-negative breast cancer
- Treg:
-
T regulatory cell
References
Pan, H. et al. 20-year risks of breast-cancer recurrence after stopping endocrine therapy at 5 Years. N. Engl. J. Med. 377, 1836–1846 (2017).
Rueda, O. M. et al. Dynamics of breast-cancer relapse reveal late-recurring ER-positive genomic subgroups. Nature 567, 399–404 (2019).
Schmid, P. et al. Event-free survival with pembrolizumab in early triple-negative breast cancer. N. Engl. J. Med. 386, 556–567 (2022).
Cortes, J. et al. Pembrolizumab plus chemotherapy in advanced triple-negative breast cancer. N. Engl. J. Med. 387, 217–226 (2022).
Schmid, P. et al. Atezolizumab plus nab-paclitaxel as first-line treatment for unresectable, locally advanced or metastatic triple-negative breast cancer (IMpassion130): updated efficacy results from a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol. 21, 44–59 (2020).
Cardoso, F. et al. Pembrolizumab and chemotherapy in high-risk, early-stage, ER(+)/HER2(-) breast cancer: a randomized phase 3 trial. Nat. Med. 31, 442–448 (2025).
Loi, S. et al. Neoadjuvant nivolumab and chemotherapy in early estrogen receptor-positive breast cancer: a randomized phase 3 trial. Nat. Med. 31, 433–441 (2025).
Demaria, S., Golden, E. B. & Formenti, S. C. Role of local radiation therapy in cancer immunotherapy. JAMA Oncol. 1, 1325–1332 (2015).
McLaughlin, M. et al. Inflammatory microenvironment remodelling by tumour cells after radiotherapy. Nat. Rev. Cancer 20, 203–217 (2020).
Guo, S. et al. Radiation-induced tumor immune microenvironments and potential targets for combination therapy. Signal Transduct. Target. Ther. 8, 205 (2023).
Segovia-Mendoza, M. & Morales-Montor, J. Immune tumor microenvironment in breast cancer and the participation of estrogen and its receptors in cancer physiopathology. Front. Immunol. 10, 348 (2019).
Rong, C., Meinert, E. & Hess, J. Estrogen receptor signaling in radiotherapy: from molecular mechanisms to clinical studies. Int. J. Mol. Sci. 19, 713 (2018).
Thiruchelvam, P. T. R. et al. Primary radiotherapy and deep inferior epigastric perforator flap reconstruction for patients with breast cancer (PRADA): a multicentre, prospective, non-randomised, feasibility study. Lancet Oncol. 23, 682–690 (2022).
De Caluwe, A. et al. First-in-human study of SBRT and adenosine pathway blockade to potentiate the benefit of immunochemotherapy in early-stage luminal B breast cancer: results of the safety run-in phase of the Neo-CheckRay trial. J. Immunother. Cancer 11, e007279 (2023).
Liu, J. et al. Improved efficacy of neoadjuvant compared to adjuvant immunotherapy to eradicate metastatic disease. Cancer Discov. 6, 1382–1399 (2016).
Harvey, J. M., Clark, G. M., Osborne, C. K. & Allred, D. C. Estrogen receptor status by immunohistochemistry is superior to the ligand-binding assay for predicting response to adjuvant endocrine therapy in breast cancer. J. Clin. Oncol. 17, 1474–1481 (1999).
Sorlie, T. et al. Gene expression patterns of breast carcinomas distinguish tumor subclasses with clinical implications. Proc. Natl. Acad. Sci. USA 98, 10869–10874 (2001).
Parker, J. S. et al. Supervised risk predictor of breast cancer based on intrinsic subtypes. J. Clin. Oncol. 27, 1160–1167 (2009).
Hammerl, D. et al. Clonality, antigen recognition, and suppression of CD8(+) T cells differentially affect prognosis of breast cancer subtypes. Clin. Cancer Res. 26, 505–517 (2020).
Wu, S. Z. et al. A single-cell and spatially resolved atlas of human breast cancers. Nat. Genet. 53, 1334–1347 (2021).
Dieci, M. V., Griguolo, G., Miglietta, F. & Guarneri, V. The immune system and hormone-receptor positive breast cancer: is it really a dead end?. Cancer Treat. Rev. 46, 9–19 (2016).
Lee, H. J. et al. Differential expression of major histocompatibility complex class I in subtypes of breast cancer is associated with estrogen receptor and interferon signaling. Oncotarget 7, 30119–30132 (2016).
Criscitiello, C. et al. Tumor-infiltrating lymphocytes (TILs) in ER+/HER2- breast cancer. Breast Cancer Res. Treat. 183, 347–354 (2020).
Voorwerk, L. et al. Immune landscape of breast tumors with low and intermediate estrogen receptor expression. NPJ Breast Cancer 9, 39 (2023).
Curtis, C. et al. The genomic and transcriptomic architecture of 2,000 breast tumours reveals novel subgroups. Nature 486, 346–352 (2012).
El Bairi, K. et al. The tale of TILs in breast cancer: a report from the international immuno-oncology biomarker working group. NPJ Breast Cancer 7, 150 (2021).
Chung, Y. R., Kim, H. J., Jang, M. H. & Park, S. Y. Prognostic value of tumor infiltrating lymphocyte subsets in breast cancer depends on hormone receptor status. Breast Cancer Res. Treat. 161, 409–420 (2017).
Amgad, M. et al. A population-level digital histologic biomarker for enhanced prognosis of invasive breast cancer. Nat. Med. 30, 85–97 (2024).
Pusztai, L. et al. Durvalumab with olaparib and paclitaxel for high-risk HER2-negative stage II/III breast cancer: Results from the adaptively randomized I-SPY2 trial. Cancer Cell 39, 989–998.e5 (2021).
Harris, M. A. et al. Towards targeting the breast cancer immune microenvironment. Nat. Rev. Cancer 24, 554–577 (2024).
Ali, H. R., Chlon, L., Pharoah, P. D., Markowetz, F. & Caldas, C. Patterns of immune infiltration in breast cancer and their clinical implications: a gene-expression-based retrospective study. PLoS Med. 13, e1002194 (2016).
Waks, A. G. et al. The immune microenvironment in hormone receptor-positive breast cancer before and after preoperative chemotherapy. Clin. Cancer Res. 25, 4644–4655 (2019).
Yoneyama, M. et al. Longitudinal assessment of tumor-infiltrating lymphocytes in primary breast cancer following neoadjuvant radiation therapy. Int. J. Radiat. Oncol. Biol. Phys. 120, 862–874 (2024).
Wolf, D. M. et al. Redefining breast cancer subtypes to guide treatment prioritization and maximize response: predictive biomarkers across 10 cancer therapies. Cancer Cell 40, 609–623.e6 (2022).
Shiao, S. L. et al. Single-cell and spatial profiling identify three response trajectories to pembrolizumab and radiation therapy in triple negative breast cancer. Cancer Cell 42, 70–84.e8 (2024).
Clusan, L., Ferriere, F., Flouriot, G. & Pakdel, F. A basic review on estrogen receptor signaling pathways in breast cancer. Int. J. Mol. Sci. 24, 6834 (2023).
Alluri, N. Abstract P1-09-01: ESR1 mutations confer radiation resistance in breast cancer. Cancer Res. 80, P1–09-1 (2020).
Chakraborty, B. et al. Estrogen receptor signaling in the immune system. Endocr. Rev. 44, 117–141 (2023).
Nicolini, A., Rossi, G. & Ferrari, P. Experimental and clinical evidence in favour of an effective immune stimulation in ER-positive, endocrine-dependent metastatic breast cancer. Front Immunol. 14, 1225175 (2023).
Schuler, L. A. & Murdoch, F. E. Endogenous and therapeutic estrogens: maestro conductors of the microenvironment of ER+ breast cancers. Cancers 13, 3725 (2021).
Wang, N. H. et al. Radiation-induced PD-L1 expression in tumor and its microenvironment facilitates cancer-immune escape: a narrative review. Ann. Transl. Med. 10, 1406 (2022).
Williamson, L. M. & Lees-Miller, S. P. Estrogen receptor alpha-mediated transcription induces cell cycle-dependent DNA double-strand breaks. Carcinogenesis 32, 279–285 (2011).
Yedidia-Aryeh, L. & Goldberg, M. The interplay between the cellular response to DNA double-strand breaks and estrogen. Cells 11, 3097 (2022).
Shiloh, Y. & Ziv, Y. The ATM protein kinase: regulating the cellular response to genotoxic stress, and more. Nat. Rev. Mol. Cell Biol. 14, 197–210 (2013).
Michmerhuizen, A. R. et al. Estrogen receptor inhibition mediates radiosensitization of ER-positive breast cancer models. NPJ Breast Cancer 8, 31 (2022).
Okoh, V., Deoraj, A. & Roy, D. Estrogen-induced reactive oxygen species-mediated signalings contribute to breast cancer. Biochim. Biophys. Acta 1815, 115–133 (2011).
Foster, J. S., Henley, D. C., Bukovsky, A., Seth, P. & Wimalasena, J. Multifaceted regulation of cell cycle progression by estrogen: regulation of Cdk inhibitors and Cdc25A independent of cyclin D1-Cdk4 function. Mol. Cell Biol. 21, 794–810 (2001).
Moghadam, S. J., Hanks, A. M. & Keyomarsi, K. Breaking the cycle: an insight into the role of ERalpha in eukaryotic cell cycles. J. Carcinog. 10, 25 (2011).
Gottardis, M. M. et al. Regulation of retinoblastoma gene expression in hormone-dependent breast cancer. Endocrinology 136, 5659–5665 (1995).
Topacio, B. R. et al. Cyclin D-Cdk4,6 drives cell-cycle progression via the retinoblastoma protein’s C-terminal helix. Mol. Cell 74, 758–70.e4 (2019).
Liu, W. et al. Estrogen receptor-alpha binds p53 tumor suppressor protein directly and represses its function. J. Biol. Chem. 281, 9837–9840 (2006).
Pawlik, T. M. & Keyomarsi, K. Role of cell cycle in mediating sensitivity to radiotherapy. Int. J. Radiat. Oncol. Biol. Phys. 59, 928–942 (2004).
Zhao, X. Z. et al. Role of estrogen in lung cancer based on the estrogen receptor-epithelial mesenchymal transduction signaling pathways. Onco Targets Ther. 8, 2849–2863 (2015).
Britton, D. J. et al. Bidirectional cross talk between ERalpha and EGFR signalling pathways regulates tamoxifen-resistant growth. Breast Cancer Res. Treat. 96, 131–146 (2006).
To, S. Q., Cheung, V., Lazarus, K. A., Knower, K. C. & Clyne, C. D. Estradiol regulates tumor necrosis factor-alpha expression and secretion in estrogen receptor positive breast cancer cells. Mol. Cell. Endocrinol. 394, 21–28 (2014).
Mercogliano, M. F., Bruni, S., Elizalde, P. V. & Schillaci, R. Tumor necrosis factor alpha blockade: an opportunity to tackle breast cancer. Front Oncol. 10, 584 (2020).
McKelvey, K. J., Hudson, A. L., Back, M., Eade, T. & Diakos, C. I. Radiation, inflammation and the immune response in cancer. Mamm. Genome 29, 843–865 (2018).
Wilkins, A. C., Patin, E. C., Harrington, K. J. & Melcher, A. A. The immunological consequences of radiation-induced DNA damage. J. Pathol. 247, 606–614 (2019).
Carozza, J. A. et al. Extracellular cGAMP is a cancer cell-produced immunotransmitter involved in radiation-induced anti-cancer immunity. Nat. Cancer 1, 184–196 (2020).
Reits, E. A. et al. Radiation modulates the peptide repertoire, enhances MHC class I expression, and induces successful antitumor immunotherapy. J. Exp. Med. 203, 1259–1271 (2006).
Song, I. H. et al. The association of estrogen receptor activity, interferon signaling, and MHC Class I expression in breast cancer. Cancer Res. Treat. 54, 1111–1120 (2022).
Fu, X., De Angelis, C. & Schiff, R. Interferon signaling in estrogen receptor-positive breast cancer: a revitalized topic. Endocrinology 163, bqab235 (2022).
Storozynsky, Q. & Hitt, M. M. The impact of radiation-induced DNA damage on cGAS-STING-mediated immune responses to cancer. Int. J. Mol. Sci. 21, 8877 (2020).
Al-Rawi, D. H., Lettera, E., Li, J., DiBona, M. & Bakhoum, S. F. Targeting chromosomal instability in patients with cancer. Nat. Rev. Clin. Oncol. 21, 645–659 (2024).
Tani, T. et al. TREX1 inactivation unleashes cancer cell STING-interferon signaling and promotes antitumor immunity. Cancer Discov. 14, 752–765 (2024).
Vanpouille-Box, C. et al. DNA exonuclease Trex1 regulates radiotherapy-induced tumour immunogenicity. Nat. Commun. 8, 15618 (2017).
Douin-Echinard, V. et al. Estrogen receptor alpha, but not beta, is required for optimal dendritic cell differentiation and [corrected] CD40-induced cytokine production. J. Immunol. 180, 3661–3669 (2008).
Carreras, E. et al. Estradiol acts directly on bone marrow myeloid progenitors to differentially regulate GM-CSF or Flt3 ligand-mediated dendritic cell differentiation. J. Immunol. 180, 727–738 (2008).
Cao, L. B. et al. Estrogen receptor alpha-mediated signaling inhibits type I interferon response to promote breast carcinogenesis. J. Mol. Cell Biol. 15, mjad047 (2024).
Post, A. E. M. et al. Interferon-stimulated genes are involved in cross-resistance to radiotherapy in tamoxifen-resistant breast cancer. Clin. Cancer Res. 24, 3397–3408 (2018).
Sas, L. et al. The interaction between ER and NFkappaB in resistance to endocrine therapy. Breast Cancer Res 14, 212 (2012).
Zoller, A. L. & Kersh, G. J. Estrogen induces thymic atrophy by eliminating early thymic progenitors and inhibiting proliferation of beta-selected thymocytes. J. Immunol. 176, 7371–7378 (2006).
Adurthi, S. et al. Oestrogen Receptor-alpha binds the FOXP3 promoter and modulates regulatory T-cell function in human cervical cancer. Sci. Rep. 7, 17289 (2017).
Prieto, G. A. & Rosenstein, Y. Oestradiol potentiates the suppressive function of human CD4 CD25 regulatory T cells by promoting their proliferation. Immunology 118, 58–65 (2006).
Generali, D. et al. Immunomodulation of FOXP3+ regulatory T cells by the aromatase inhibitor letrozole in breast cancer patients. Clin. Cancer Res. 15, 1046–1051 (2009).
Mostafa, A. A. et al. Activation of ERalpha signaling differentially modulates IFN-gamma induced HLA-class II expression in breast cancer cells. PLoS ONE 9, e87377 (2014).
Harding, A. T. & Heaton, N. S. The impact of estrogens and their receptors on immunity and inflammation during infection. Cancers 14, 909 (2022).
Beach, C., MacLean, D., Majorova, D., Arnold, J. N. & Olcina, M. M. The effects of radiation therapy on the macrophage response in cancer. Front. Oncol. 12, 1020606 (2022).
Zhu, L. et al. Macrophage contributes to radiation-induced anti-tumor abscopal effect on transplanted breast cancer by HMGB1/TNF-alpha signaling factors. Int. J. Biol. Sci. 17, 926–941 (2021).
Rudqvist, N. P. et al. Immunotherapy targeting different immune compartments in combination with radiation therapy induces regression of resistant tumors. Nat. Commun. 14, 5146 (2023).
Wennerberg, E. et al. Barriers to radiation-induced in situ tumor vaccination. Front. Immunol. 8, 229 (2017).
Teresa Pinto, A. et al. Ionizing radiation modulates human macrophages towards a pro-inflammatory phenotype preserving their pro-invasive and pro-angiogenic capacities. Sci. Rep. 6, 18765 (2016).
Wunderlich, R. et al. Low and moderate doses of ionizing radiation up to 2 Gy modulate transmigration and chemotaxis of activated macrophages, provoke an anti-inflammatory cytokine milieu, but do not impact upon viability and phagocytic function. Clin. Exp. Immunol. 179, 50–61 (2015).
Svensson, S. et al. CCL2 and CCL5 are novel therapeutic targets for estrogen-dependent breast cancer. Clin. Cancer Res. 21, 3794–3805 (2015).
Villa, A., Rizzi, N., Vegeto, E., Ciana, P. & Maggi, A. Estrogen accelerates the resolution of inflammation in macrophagic cells. Sci. Rep. 5, 15224 (2015).
Chakraborty, B. et al. Inhibition of estrogen signaling in myeloid cells increases tumor immunity in melanoma. J. Clin. Investig. 131, e151347 (2021).
Dai, R., Phillips, R. A., Karpuzoglu, E., Khan, D. & Ahmed, S. A. Estrogen regulates transcription factors STAT-1 and NF-kappaB to promote inducible nitric oxide synthase and inflammatory responses. J. Immunol. 183, 6998–7005 (2009).
Ito, I. et al. Estrogen inhibits transforming growth factor beta signaling by promoting Smad2/3 degradation. J. Biol. Chem. 285, 14747–14755 (2010).
Reichardt, C. M. et al. Neutrophils seeking new neighbors: radiotherapy affects the cellular framework and the spatial organization in a murine breast cancer model. Cancer Immunol. Immunother. 73, 67 (2024).
Yasuda, H. et al. 17-beta-estradiol enhances neutrophil extracellular trap formation by interaction with estrogen membrane receptor. Arch. Biochem. Biophys. 663, 64–70 (2019).
Vazquez Rodriguez, G., Abrahamsson, A., Jensen, L. D. & Dabrosin, C. Estradiol promotes breast cancer cell migration via recruitment and activation of neutrophils. Cancer Immunol. Res. 5, 234–247 (2017).
Aboussekhra, A., Alraouji, N. N., Al-Mohanna, F. H. & Al-Khalaf, H. Ionizing radiation normalizes the features of active breast cancer stromal fibroblasts and suppresses their paracrine pro-carcinogenic effects. Transl. Oncol. 37, 101780 (2023).
Hellevik, T. et al. Cancer-associated fibroblasts from human NSCLC survive ablative doses of radiation but their invasive capacity is reduced. Radiat. Oncol. 7, 59 (2012).
Reid, S. E. et al. Cancer-associated fibroblasts rewire the estrogen receptor response in luminal breast cancer, enabling estrogen independence. Oncogene 43, 1113–1126 (2024).
Nagel, A. et al. ERalpha36-high cancer-associated fibroblasts as an unfavorable factor in triple-negative breast cancer. Cancers 14, 2005 (2022).
Brechbuhl, H. M. et al. Fibroblast subtypes regulate responsiveness of luminal breast cancer to estrogen. Clin. Cancer Res 23, 1710–1721 (2017).
Liu, L. et al. GPR30-mediated HMGB1 upregulation in CAFs induces autophagy and tamoxifen resistance in ERalpha-positive breast cancer cells. Aging (Albany NY) 13, 16178–16197 (2021).
Bou-Dargham, M. J. et al. TCGA RNA-Seq and tumor-infiltrating lymphocyte imaging data reveal cold tumor signatures of invasive ductal carcinomas and estrogen receptor-positive human breast tumors. Int. J. Mol. Sci. 24, 9355 (2023).
Demaria, S. et al. Ionizing radiation inhibition of distant untreated tumors (abscopal effect) is immune mediated. Int. J. Radiat. Oncol. Biol. Phys. 58, 862–870 (2004).
Matsumura, S. et al. Radiation-induced CXCL16 release by breast cancer cells attracts effector T cells. J. Immunol. 181, 3099–3107 (2008).
Ridnour, L. A. et al. Adjuvant COX inhibition augments STING signaling and cytolytic T cell infiltration in irradiated 4T1 tumors. JCI Insight 9, e165356 (2024).
Apetoh, L. et al. Toll-like receptor 4-dependent contribution of the immune system to anticancer chemotherapy and radiotherapy. Nat. Med. 13, 1050–1059 (2007).
Lhuillier, C. et al. Radiotherapy-exposed CD8+ and CD4+ neoantigens enhance tumor control. J. Clin. Investig. 131, e138740 (2021).
Demaria, S. et al. Immune-mediated inhibition of metastases after treatment with local radiation and CTLA-4 blockade in a mouse model of breast cancer. Clin. Cancer Res. 11, 728–734 (2005).
Sharabi, A. B. et al. Stereotactic radiation therapy augments antigen-specific PD-1-mediated antitumor immune responses via cross-presentation of tumor antigen. Cancer Immunol. Res. 3, 345–355 (2015).
Song, H. N. et al. Abscopal effect of radiotherapy enhanced with immune checkpoint inhibitors of triple negative breast cancer in 4T1 mammary carcinoma model. Int. J. Mol. Sci. 22, 10476 (2021).
Rudqvist, N. P. et al. Radiotherapy and CTLA-4 blockade shape the TCR repertoire of tumor-infiltrating T cells. Cancer Immunol. Res. 6, 139–150 (2018).
Dewan, M. Z. et al. Fractionated but not single-dose radiotherapy induces an immune-mediated abscopal effect when combined with anti-CTLA-4 antibody. Clin. Cancer Res. 15, 5379–5388 (2009).
Dovedi, S. J. et al. Acquired resistance to fractionated radiotherapy can be overcome by concurrent PD-L1 blockade. Cancer Res 74, 5458–5468 (2014).
Deng, L. et al. Irradiation and anti-PD-L1 treatment synergistically promote antitumor immunity in mice. J. Clin. Investig. 124, 687–695 (2014).
Wei, J. et al. Sequence of alphaPD-1 relative to local tumor irradiation determines the induction of abscopal antitumor immune responses. Sci. Immunol. 6, eabg0117 (2021).
Young, K. H. et al. Optimizing timing of immunotherapy improves control of tumors by hypofractionated radiation therapy. PLoS ONE 11, e0157164 (2016).
Han, M. G. et al. Combination of OX40 co-stimulation, radiotherapy, and PD-1 inhibition in a syngeneic murine triple-negative breast cancer model. Cancers 14, 2692 (2022).
Verbrugge, I. et al. Radiotherapy increases the permissiveness of established mammary tumors to rejection by immunomodulatory antibodies. Cancer Res. 72, 3163–3174 (2012).
Vanpouille-Box, C. et al. TGFbeta is a master regulator of radiation therapy-induced antitumor immunity. Cancer Res. 75, 2232–2242 (2015).
Liu et al. Potentiating antitumor efficacy through radiation and sustained intratumoral delivery of anti-CD40 and anti-PDL1. Int. J. Radiat. Oncol. Biol. Phys. 110, 492–506 (2021).
Aguilera, T. A. et al. Induced tumor heterogeneity reveals factors informing radiation and immunotherapy combinations. Clin. Cancer Res. 26, 2972–2985 (2020).
Pilones, K. A. et al. Converging focal radiation and immunotherapy in a preclinical model of triple negative breast cancer: contribution of VISTA blockade. Oncoimmunology 9, 1830524 (2020).
Song, J. Y. et al. Combination of local radiotherapy and anti-glucocorticoid-induced tumor necrosis factor receptor (GITR) therapy augments PD-L1 blockade-mediated anti-tumor effects in murine breast cancer model. Radiother. Oncol. 190, 109981 (2024).
Chang, W. I. et al. PI3Kαδ inhibitor combined with radiation enhances the antitumor immune effect of anti-PD1 in a syngeneic murine triple-negative breast cancer model. Int. J. Radiat. Oncol. Biol. Phys. 110, 845–858 (2021).
Han, M. G., Jang, B. S., Kang, M. H., Na, D. & Kim, I. A. PI3Kγδ inhibitor plus radiation enhances the antitumour immune effect of PD-1 blockade in syngenic murine breast cancer and humanised patient-derived xenograft model. Eur. J. Cancer 157, 450–463 (2021).
Pieper, A. A. et al. Combination of radiation therapy, bempegaldesleukin, and checkpoint blockade eradicates advanced solid tumors and metastases in mice. J. Immunother. Cancer 9, e002715 (2021).
Wennerberg, E. et al. Exercise reduces immune suppression and breast cancer progression in a preclinical model. Oncotarget 11, 452–461 (2020).
Wennerberg, E. et al. CD73 blockade promotes dendritic cell infiltration of irradiated tumors and tumor rejection. Cancer Immunol. Res. 8, 465–478 (2020).
Kim, S., Jeon, S. H., Han, M. G., Kang, M. H. & Kim, I. A. BRD4 inhibition enhances the antitumor effects of radiation therapy in a murine breast cancer model. Int. J. Mol. Sci. 24, 13062 (2023).
Yamazaki, T. et al. LTX-315-enabled, radiotherapy-boosted immunotherapeutic control of breast cancer by NK cells. Oncoimmunology 10, 1962592 (2021).
Huang, K. C. et al. Neoantigen-augmented iPSC cancer vaccine combined with radiotherapy promotes antitumor immunity in poorly immunogenic cancers. NPJ Vaccines 9, 95 (2024).
Chen, M. et al. Stereotactic ablative radiotherapy and FAPalpha-based cancer vaccine suppresses metastatic tumor growth in 4T1 mouse breast cancer. Radiother. Oncol. 189, 109946 (2023).
Huang, K. C. et al. A novel engineered AAV-based neoantigen vaccine in combination with radiotherapy eradicates tumors. Cancer Immunol. Res. 11, 123–136 (2023).
Ozdemir, B. C., Sflomos, G. & Brisken, C. The challenges of modeling hormone receptor-positive breast cancer in mice. Endocr. Relat. Cancer 25, R319–R330 (2018).
Kumar, M. J. et al. A mouse model for luminal epithelial like ER positive subtype of human breast cancer. BMC Cancer 7, 180 (2007).
Buque, A. et al. Immunoprophylactic and immunotherapeutic control of hormone receptor-positive breast cancer. Nat. Commun. 11, 3819 (2020).
Christenson, J. L. et al. MMTV-PyMT and derived Met-1 mouse mammary tumor cells as models for studying the role of the androgen receptor in triple-negative breast cancer progression. Horm. Cancer 8, 69–77 (2017).
Buque, A. & Galluzzi, L. Modeling tumor immunology and immunotherapy in mice. Trends Cancer 4, 599–601 (2018).
Schrors, B. et al. Multi-omics characterization of the 4T1 murine mammary gland tumor model. Front. Oncol. 10, 1195 (2020).
Allen, B. M. et al. Systemic dysfunction and plasticity of the immune macroenvironment in cancer models. Nat. Med. 26, 1125–1134 (2020).
Snipstad, S., Bremnes, F., Dehli Haugum, M. & Sulheim, E. Characterization of immune cell populations in syngeneic murine tumor models. Cancer Med. 12, 11589–11601 (2023).
De Giovanni, C. et al. Bioprofiling TS/A murine mammary cancer for a functional precision experimental model. Cancers 11, 1889 (2019).
Turrell, F. K. et al. Age-associated microenvironmental changes highlight the role of PDGF-C in ER(+) breast cancer metastatic relapse. Nat. Cancer 4, 468–484 (2023).
O’Leary, K. A. et al. Estrogen receptor blockade and radiation therapy cooperate to enhance the response of immunologically cold ER+ breast cancer to immunotherapy. Breast Cancer Res. 25, 68 (2023).
Campbell, K. M. et al. A spontaneous aggressive eralpha+ mammary tumor model is driven by Kras activation. Cell Rep. 28, 1526–1537.e4 (2019).
Le Naour, A. et al. EO771, the first luminal B mammary cancer cell line from C57BL/6 mice. Cancer Cell Int. 20, 328 (2020).
Li, X. et al. Radiation synergizes with IL2/IL15 stimulation to enhance innate immune activation and antitumor immunity. Mol. Cancer Ther. 23, 330–342 (2024).
Buque, A. et al. Impact of radiotherapy dose, fractionation and immunotherapeutic partner in a mouse model of HR+ mammary carcinogenesis. J. Natl. Cancer Inst. 117, 934–947 (2024).
Perez-Lanzon, M. et al. New hormone receptor-positive breast cancer mouse cell line mimicking the immune microenvironment of anti-PD-1 resistant mammary carcinoma. J Immunother. Cancer 11, e007117 (2023).
Langsten, K. L. et al. A novel metastatic estrogen receptor-expressing breast cancer model with antiestrogen responsiveness. Cancers 15, 5773 (2023).
Hazlett, J. et al. Oestrogen deprivation induces chemokine production and immune cell recruitment in in vitro and in vivo models of oestrogen receptor-positive breast cancer. Breast Cancer Res 23, 95 (2021).
Capietto, A. H. et al. Novel ERalpha positive breast cancer model with estrogen independent growth in the bone microenvironment. Oncotarget 7, 49751–49764 (2016).
Sequeira, G. R. et al. Enhanced antitumor immunity via endocrine therapy prevents mammary tumor relapse and increases immune checkpoint blockade sensitivity. Cancer Res. 81, 1375–1387 (2021).
Wege, A. K. et al. Humanized tumor mice-a new model to study and manipulate the immune response in advanced cancer therapy. Int J. Cancer 129, 2194–2206 (2011).
Bruss, C. et al. Immune checkpoint profiling in humanized breast cancer mice revealed cell-specific LAG-3/PD-1/TIM-3 Co-expression and elevated PD-1/TIM-3 secretion. Cancers 15, 2615 (2023).
Bruss, C. et al. Neoadjuvant radiotherapy in ER(+), HER2(+), and triple-negative -specific breast cancer based humanized tumor mice enhances anti-PD-L1 treatment efficacy. Front Immunol. 15, 1355130 (2024).
Nicotra, R., Lutz, C., Messal, H. A. & Jonkers, J. Rat models of hormone receptor-positive breast cancer. J. Mammary Gland Biol. Neoplasia 29, 12 (2024).
Hacker, B. C. & Rafat, M. Organoids as complex in vitro models for studying radiation-induced cell recruitment. Cell Mol. Bioeng. 13, 341–357 (2020).
Bassez, A. et al. A single-cell map of intratumoral changes during anti-PD1 treatment of patients with breast cancer. Nat. Med. 27, 820–832 (2021).
Nanda, R. et al. Effect of pembrolizumab plus neoadjuvant chemotherapy on pathologic complete response in women with early-stage breast cancer: an analysis of the ongoing phase 2 adaptively randomized I-SPY2 trial. JAMA Oncol. 6, 676–684 (2020).
Domchek, S. M. et al. Olaparib and durvalumab in patients with germline BRCA-mutated metastatic breast cancer (MEDIOLA): an open-label, multicentre, phase 1/2, basket study. Lancet Oncol. 21, 1155–1164 (2020).
Cortesi, L. et al. A phase II study of pembrolizumab plus carboplatin in BRCA-related metastatic breast cancer (PEMBRACA). ESMO Open 8, 101207 (2023).
Guiu, S. et al. 179O Combination of olaparib, durvalumab and fulvestrant in patients with advanced ER-positive, HER2-negative breast cancer harboring homologous recombination repair (HRR) deficiency or microsatellite instability (MSI): results of the international phase II DOLAF trial. ESMO Open 9(Suppl-4), 103201 (2024).
Masuda, J. et al. Efficacy, safety, and biomarker analysis of nivolumab in combination with abemaciclib plus endocrine therapy in patients with HR-positive HER2-negative metastatic breast cancer: a phase II study (WJOG11418B NEWFLAME trial). J. Immunother. Cancer 11, e007126 (2023).
Yuan, Y. et al. Phase I/II trial of palbociclib, pembrolizumab and letrozole in patients with hormone receptor-positive metastatic breast cancer. Eur. J. Cancer 154, 11–20 (2021).
Antonia, S. J. et al. Overall survival with durvalumab after chemoradiotherapy in stage III NSCLC. N. Engl. J. Med 379, 2342–2350 (2018).
Lee, N. Y. et al. Avelumab plus standard-of-care chemoradiotherapy versus chemoradiotherapy alone in patients with locally advanced squamous cell carcinoma of the head and neck: a randomised, double-blind, placebo-controlled, multicentre, phase 3 trial. Lancet Oncol. 22, 450–462 (2021).
Voorwerk, L. et al. Immune induction strategies in metastatic triple-negative breast cancer to enhance the sensitivity to PD-1 blockade: the TONIC trial. Nat. Med 25, 920–928 (2019).
Ho, A. Y. et al. A phase 2 clinical trial assessing the efficacy and safety of pembrolizumab and radiotherapy in patients with metastatic triple-negative breast cancer. Cancer 126, 850–860 (2020).
David, S. et al. Abstract PD10-02: A randomised phase II trial of single fraction or multi-fraction SABR (stereotactic ablative body radiotherapy) with atezolizumab in patients with advanced triple negative breast cancer (AZTEC trial). Cancer Res. 82, PD10-02 (2022).
Demaria, S., Romano, E., Brackstone, M. & Formenti, S. C. Immune induction strategies to enhance responses to PD-1 blockade: lessons from the TONIC trial. J. Immunother. Cancer 7, 318 (2019).
Szekely, B. et al. Immunological differences between primary and metastatic breast cancer. Ann. Oncol. 29, 2232–2239 (2018).
De Caluwé, A. et al. Neo-CheckRay: radiation therapy and adenosine pathway blockade to increase benefit of immuno-chemotherapy in early stage luminal B breast cancer, a randomized phase II trial. BMC Cancer 21, 899 (2021).
Ho, A. Y. et al. PEARL: a phase Ib/II biomarker study of adding radiation therapy to pembrolizumab before neoadjuvant chemotherapy in human epidermal growth factor receptor 2-negative breast cancer. J. Clin. Oncol. 42, 4282–4293 (2024).
Verma, S. et al. Immunotherapy and radiation therapy sequencing in breast cancer: a systematic review. Int. J. Radiat. Oncol. Biol. Phys. 118, 1422–1434 (2024).
Takada, M. et al. Phase Ib/II study of nivolumab combined with palliative radiation therapy for bone metastasis in patients with HER2-negative metastatic breast cancer. Sci. Rep. 12, 22397 (2022).
Barroso-Sousa, R. et al. A phase II study of pembrolizumab in combination with palliative radiotherapy for hormone receptor-positive metastatic breast cancer. Clin. Breast Cancer 20, 238–245 (2020).
Ahmed, K. A. et al. Nivolumab and stereotactic radiosurgery for patients with breast cancer brain metastases: a nonrandomized, open-label phase 1b study. Adv. Radiat. Oncol. 6, 100798 (2021).
Page, D. B. et al. Brain radiotherapy, tremelimumab-mediated CTLA-4-directed blockade +/− trastuzumab in patients with breast cancer brain metastases. NPJ Breast Cancer 8, 50 (2022).
McGee, H. M. et al. Stereotactic ablative radiation therapy induces systemic differences in peripheral blood immunophenotype dependent on irradiated site. Int. J. Radiat. Oncol. Biol. Phys. 101, 1259–1270 (2018).
Sampson, J. H., Gunn, M. D., Fecci, P. E. & Ashley, D. M. Brain immunology and immunotherapy in brain tumours. Nat. Rev. Cancer 20, 12–25 (2020).
Symmans, W. F. et al. Assessment of residual cancer burden and event-free survival in neoadjuvant treatment for high-risk breast cancer: an analysis of data from the I-SPY2 randomized clinical trial. JAMA Oncol. 7, 1654–1663 (2021).
Lightowlers, S. V. et al. Neoadjuvant radiotherapy and endocrine therapy for oestrogen receptor positive breast cancers: the neo-RT feasibility study. Clin. Oncol. 37, 103669 (2024).
Poleszczuk, J. et al. Neoadjuvant radiotherapy of early-stage breast cancer and long-term disease-free survival. Breast Cancer Res 19, 75 (2017).
Stenmark Tullberg, A. et al. Immune infiltrate in the primary tumor predicts effect of adjuvant radiotherapy in breast cancer; results from the randomized SweBCG91RT trial. Clin. Cancer Res. 27, 749–758 (2021).
Darragh, L. B. et al. Elective nodal irradiation mitigates local and systemic immunity generated by combination radiation and immunotherapy in head and neck tumors. Nat. Commun. 13, 7015 (2022).
Telarovic, I. et al. Delayed tumor-draining lymph node irradiation preserves the efficacy of combined radiotherapy and immune checkpoint blockade in models of metastatic disease. Nat. Commun. 15, 5500 (2024).
Marciscano, A. E. et al. Elective nodal irradiation attenuates the combinatorial efficacy of stereotactic radiation therapy and immunotherapy. Clin. Cancer Res. 24, 5058–5071 (2018).
Spring, L. M. et al. Pathologic complete response after neoadjuvant chemotherapy and impact on breast cancer recurrence and survival: a comprehensive meta-analysis. Clin. Cancer Res. 26, 2838–2848 (2020).
Nicolo, E. et al. Circulating tumor cells et al.: towards a comprehensive liquid biopsy approach in breast cancer. Transl. Breast Cancer Res. 5, 10 (2024).
Acknowledgements
A.M., E.W. and N.S. are supported by a Cancer Research UK Radiation Research Centre of Excellence at The Institute of Cancer Research and The Royal Marsden NHS Foundation Trust (grant ref: A28724). M.F. is supported by clinical PhD research fellowship funding from Cancer Research UK. A.T. and A.M. are supported by Breast Cancer Now through programmatic funding to The Breast Cancer Now Toby Robins Research Centre at the Institute of Cancer Research. A.T. receives grant funding as a Breast Cancer Research Foundation (BCRF) Investigator. S.D. is supported by grants from NIH (2R01CA201246), US DOD (W81XWH-19-1-0142), and Breast Cancer Research Foundation (BCRF-24-053).
Author information
Authors and Affiliations
Contributions
N.S., M.F. and M.Y. conceptualised the review manuscript. M.F. wrote the manuscript and prepared figures and tables. M.Y. contributed to manuscript writing. N.S. provided supervision and review of initial drafts. All authors critically reviewed and approved the manuscript.
Corresponding author
Ethics declarations
Competing interests
A.T. is/has been a consultant for AstraZeneca, Merck KGaA, Artios, Pfizer and Gilead and has received grant/research support from AstraZeneca, Myriad, Medivation and Merck KGaA; is a stockholder in Inbiomotion; is also a named inventor on patents describing the use of DNA repair inhibitors and stands to gain from their development and use as part of the ICR ‘Rewards to Inventors’ scheme and also reports benefits from this scheme associated with patents for PARP inhibitors paid to A.T. research accounts at the Institute of Cancer Research. S.D.—Research funding to institution from Lytix Biopharma and Boehringer-Ingelheim, and compensation for consultant/advisory services from Lytix Biopharma, Genentech, and Johnson & Johnson Enterprise Innovation Inc. All other authors declare no financial or non-financial competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Fenton, M., Yoneyama, M., Wennerberg, E. et al. The untapped potential of radiation and immunotherapy for hormone receptor-positive breast cancer. npj Breast Cancer 11, 77 (2025). https://doi.org/10.1038/s41523-025-00796-x
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41523-025-00796-x
