
Abstract
Breast cancer etiology traditionally emphasizes genetic mutations, hormonal dynamics, and tissue aging. However, recent decades have seen a steady rise in breast cancer with a growing proportion of these tumors exhibiting estrogen receptor–positive (ER + ) phenotypes along with an alarming rise in early-onset breast cancer occurring in individuals without a family history of the disease. While increased screening and lifestyle changes explain part of this trend, they do not fully account for the rising incidence, particularly among specific racial and geographic subgroups. We hypothesize that this rising trend in hormone-sensitive and early-onset cancers is a manifestation of chronic, cumulative environmental exposures, particularly to endocrine-disrupting chemicals (EDCs), that profoundly alter breast tissue biology. Since EDCs alter estrogen receptor signaling, the epigenetic landscape, and disrupt immune surveillance, we hypothesize that this may underpin the rising incidence of hormone-sensitive early-onset breast cancers through a mechanism that affects field cancerization and hormone-mediated aging.
Similar content being viewed by others

Introduction
Historical perspective and current trends
Since the 1970s, breast cancer incidence trends have been understood through genetic predispositions, cumulative hormone exposure, and increased mammography screening. Lifestyle changes including earlier menarche, delayed childbirth, obesity, and hormone replacement therapy usage have also contributed to the rise of breast cancers1,2,3,4,5,6,7. However, these factors alone cannot explain epidemiological studies showing that each new generation carries a slightly higher risk of breast cancer than the prior one2,8,9,10,11. Moreover, these factors cannot explain the alarming increases specifically in estrogen receptor-positive (ER + ) breast cancers, but also in early-onset breast cancers (<50 years old), or the shift in breast cancer subtypes in younger women particularly among Hispanic American, Asian American, and Pacific Islander populations2,10.
Traditionally, early-onset breast cancers were linked with exposure to radiation around the time of menarche12, or linked with inherited mutations in cancer causing genes (eg. BRCA1/2, TP53, ATM, PALB etc)13,14,15,16,17. Early-onset breast cancers were also predominantly characterized by estrogen receptor-negative (ER-) profiles18,19,20. However, recent trends have shown a shift in the presentation of these cancers with an increasing number of early-onset breast cancers identified as ER+ and the vast majority with no family history21. This change underscores a possible evolution in the biological mechanisms underlying the formation of these cancers. Most notably, early-onset breast cancer incidence has been shown to be enriched in the Northeast, Midwest, and Southeast United States22.
In this perspective, we argue that the rising trends in hormone-sensitive and early-onset cancers, is a manifestation of chronic and cumulative environmental exposures that profoundly alter breast tissue biology. Specifically, the continuous exposure to endocrine disrupting chemicals (EDCs) permeates highly plastic and susceptible stages of breast development, overwhelming intrinsic tumor‑suppressive programs. In utero, post-natal, peri-pubertal, and pubertal exposures to EDCs remodel the breast epigenome, accelerate tissue‑specific aging, and impair immunosurveillance, leading to increased cancer development and especially early-onset breast cancers.
Geographic clustering and environmental exposure
In a landmark study that followed a cohort of 203,691 twins (80,309 identical and 123,382 same-sex fraternal twins) for 32 years and recorded cancer incidence, researchers found that the majority of cancer risk (69%) comes from individual lifestyle choices (e.g. reproductive health choices, diet, alcohol consumption, physical activity, etc.) but more importantly, shared environmental exposures (e.g. pollutants, chemicals, or radiation)23,24. Environmental exposures such as DNA damaging carcinogens are ubiquitous in the environment25. These include per- and polyfluoroalkyl substances (PFAS), polycyclic aromatic hydrocarbons (PAHs), and certain pesticides26,27,28. Endocrine disrupting chemicals (EDCs), which are also highly prevalent in the environment, have been shown to exhibit hormone binding activity via activation of hormone receptors (e.g. ERs, PRs, ARs or TRs)29,30,31,32. These EDCs include PFAS, PAH, and pesticides but also bisphenols (BPXs), phthalates, dioxins, polychlorinated biphenyls (PCBs), organophosphate esters (OPEs), and brominated flame retardants (BFRs)33,34,35,36,37,38. EDCs initiate or exacerbate cancer risks by disrupting hormone signaling and causing DNA damage39.
A recent study has linked rising exposure to environmental factors with the uptrend in breast cancer cases in developed nations. Kehm et al. highlighted significant geographic disparities in early-onset breast cancer incidence across the United States22. Utilizing data from the U.S. Cancer Statistics database, researchers calculated age-adjusted incidence rates and assessed trends in women aged 25 to 39 from 2001 to 2020. They found the Northeast region of the U.S. exhibited the highest absolute incidence rates, with a significant upward trend over time, while the Western region experienced the highest annual increase in early-onset breast cancer rates, despite having the lowest absolute incidence rates (Fig. 122). Interestingly, the geographic distribution of high-incidence early-onset breast cancer regions shows a striking overlap with states with a legacy of industrial pollution, PFAS contamination, and urban living (Fig. 140). Geospatial distribution of EPA Superfund sites, which represent remedial long-term, comprehensive sites contaminated by PCBs, dioxins, and heavy metals, mirrors that of the geographical incidence of early-onset breast cancers40. While this geographic correlation suggests a potential link between environmental pollutants and rising breast cancer rates among young women, establishing a direct causal relationship and identifying specific environmental factors influencing early-onset breast cancer incidence requires further research.

A Map of EPA-designated Superfund sites in the continental United States, representing areas with confirmed hazardous contamination using data obtained from EnviroAtlas Data and B state-level incidence rates of early-onset breast cancer (diagnosed before age 50), highlighting regional variation using data obtained from20. Notably, areas with high Superfund site density overlap with states showing higher early-onset breast cancer incidence.
Observations in wildlife does offer strong sentinel geographic evidence however, that environmental exposures to EDCs are biologically active, persistent, and capable of inducing hormone-related pathologies (Table 1)41,42,43,44,45. For example, the Hudson River, Cape Cod, and the Great Lakes have all documented elevated concentrations of EDCs including PCBs, and PFAS which bioaccumulate through the food chain, concentrating in apex predators such as osprey, river otters, and bald eagles44,46,47,48,49. These species exhibit biological phenotypes including reproductive failure, immune dysfunction, and altered thyroid and sex hormone levels. This raises concerns about human dietary and other exposures in these regions. Mammals and birds living in urbanized or industrialized areas exhibit elevated tissue levels of EDCs, correlating with disrupted hormone levels, abnormal sexual development, and reduced fertility50,51. This raises concerns about similar biological effects in humans since they are exposed to the same air, water, and food systems and hence share similar physiological disruptions to reproductive and endocrine health. In amphibians and reptiles, species such as frogs, turtles, and alligators living in watersheds contaminated by EDCs including atrazine, DDT derivatives, and bisphenol A (BPA), exhibit intersex characteristics, testicular oocytes, and abnormal sex ratios41 Similarly, white-tailed deer sampled near wastewater treatment sites or pesticide application zones have shown detectable concentrations of phthalates and PCBs, alongside histological signs of endocrine disruption52,53. Taken together, these consistent cross-species effects across diverse ecosystems and geographic areas underscore the biological potency of EDCs in real-world settings and heighten concern that chronic, low-dose exposures in humans, particularly in similarly contaminated environments, may contribute to rising rates of breast cancer, especially early-onset cancers.
Environmental exposures, particularly to certain EDCs, have been shown to contribute to cancer development. These include persistent EDCs such as PCBs, DTT, PBDEs, organochlorine pesticides, and dioxins, which are not easily metabolized and therefore bioaccumulate in lipophilic tissues like the breast fat depots. There they can remain in the body for years, sometimes even decades slowly released over time54,55,56. Although PCBs and DDT were banned in the 1970s due to experiments directly linking their exposure with DNA damage and cellular transformation56,57, they still persist in the environment today and remain in adipose tissues for up to 20−50 years after exposure. Moreover, despite these bans, persistent organochlorine pesticides are being replaced with chemicals that exhibit similar chemical stability, leading to continued concerns about the evolving bioaccumulation and potential interactions with hormone receptors, or enhancing hormonal effects27.
Bioaccumulation of other EDCs varies significantly among different EDCs. Polybrominated diphenyl ethers (PBDEs), which are commonly used as flame retardants, can mimic progesterone activity, alter hormone levels, and affect the menstrual cycle and reproductive outcomes in humans and animals58,59,60. Depending on the specific variant, they can persist in fat tissue for 2−12 years. Dioxins, which are present in bleached paper products such as disposable diapers, paper towels, and tampons exhibit half-lives in human fat tissue ranging from 7−11 years55. Because they are highly lipophilic, these chemicals preferentially reside within adipocytes, where they can remain for years and be released when those fat stores are mobilized, making their presence nearly constant once they have entered the body55. The prolonged residence of these EDCs in adipose depots, especially in the breast, implies that even banned substances, such as PCBs and DDT, will continue to exert their effects decades after their use has ceased. The ongoing substitution of these compounds with similarly stable chemicals only prolongs and adds to the cumulative dose exposure of the breast to hormones.
Non-persistent EDCs (e.g., phthalates, parabens, bisphenols, etc.) in contrast, are rapidly metabolized and excreted60. Although they do not accumulate in fat depots, given their ubiquitous prevalence in the environment, they are readily measured in the blood and urine of nearly all children and adults60. Silicone wristbands used to collect exposures to chemicals in pregnant women and office workers has revealed detectable levels of EDCs in nearly all samples across various classes with organophosphate esters (OPEs) and phthalates present in 100% of samples, followed by PAHs, BFRs PCBs and BPXs61,62. In fact, every wristband extracted in these studies were found to be hormonally active in human hormone receptor cell assays, disrupting estrogen, androgen, and thyroid hormone receptors61,62. Thus, the chronic and daily exposure of EDCs leads to a nearly continuous total hormonal load that exceeds the natural range63. In addition, the interaction of EDCs with endogenous estrogens rapidly results in a significantly higher cumulative exposure to hormones in breast tissue especially at a much younger age. As we describe below, this elevated and chronic exposure has substantial impact on tissue aging, field cancerization and the immune microenvironment that may explain the rising incidence of hormone-sensitive breast cancers especially among younger women.
EDCs as accelerators of hormone-mediated tissue aging
The breast (and other hormone sensitive tissues) exhibits a unique aging trajectory that is dictated not by chronological age, but by cumulative hormonal exposure, particularly to estrogen and progesterone, over a woman’s reproductive lifespan. Each menstrual cycle, pregnancy, lactation period, and the timing of menopause increase the aggregate of hormone driven cell proliferation, genomic stress, and epigenetic drift, together which accelerate tissue-level aging processes and influence cancer risk. This concept of “hormonal aging” was epidemiologically introduced by Pike et al. in 1983 where “breast tissue age” was used to explain the atypical age-incidence curve of breast cancer compared with that of other cancers64. This work demonstrated that breast tissue ages rapidly from menarche until the first full-term pregnancy (FFTP) (explaining the short‑term risk increase with late first birth), at which point aging rate markedly decelerates after childbirth, but rises again, though less sharply, with subsequent pregnancies, and drops once more at menopause. If a woman never becomes pregnant or delays FFTP beyond age 30, the rate of breast tissue aging remains high until perimenopause64,65.
Hormonal aging has now been corroborated at the molecular level. Studies have shown that normal breast tissue in healthy women accumulates significantly more somatic mutations and DNA methylation changes than matched blood, and at a faster rate64,66. The mutational accumulation rate in breast tissue is higher in nulliparous women and increases significantly with age but becomes less pronounced after the FFTP67, consistent with a deceleration rate of aging upon FFTP64. However, this deceleration is only observed in young first-time mothers since older first-time mothers showed a significantly higher probability of accumulating oncogenic events compared to nulliparous women. Likewise, the rate of epigenetic aging as gauged by DNA methylation is also more rapid in breast tissue compared to matched blood64,68,69,70. The rapid accumulation starts around menarche and decelerates as women approach menopause. This implies that unlike other tissues, the rate of cellular and molecular aging and the accumulation of DNA damage in breast tissue is heavily influenced by reproductive milestones and hormonal exposure.
The mechanism behind this aging involves the repeated cycles of estrogen- and progesterone-driven epithelial proliferation. Each menstrual cycle induces a rapid ~2-fold wave of cell proliferation, E + P driven by estrogen and progesterone during the mid‑luteal phase71,72, increasing opportunities for replication-associated mutations71,73,74,75. Estrogen metabolites can also form DNA adducts, directly damaging the genome76,77. Concentrations as low as 10 nM 4-OHE₂, which is within the physiologic range of breast tissue estrogen, can generate measurable, mutagenic DNA adducts if exposure is chronic or detox pathways are compromised77,78,79. In women, adduct signal is detectable in urine and rises several-fold in people who later develop, or already have, hormone-sensitive cancers76,80,81. In parallel, inflammatory and oxidative stress, especially during periods of active proliferation, contribute to DNA damage via reactive oxygen species (ROS) and additional adduct formation82,83. Thus, when there is an overlap between the presence of estrogen (or chemicals that mimic them), and epithelial cell proliferation, the risk for DNA mutation increases and consequently so does the risk of malignant transformation84.
Both high dose radiation exposure (eg. Hodgkin’s Disease, atomic bomb)85,86,87 and even low-level, chronic DNA damage from replicative stress necessitates sustained repair88,89,90,91. However, the breast epithelium, especially prior to pregnancy, exhibits less effective DNA damage response, with reduced activation of key protective pathways like p53/p21 and diminished apoptosis compared to tissues like skin or lymphocytes86,92. However, following pregnancy, efficiency of these pathways improves, potentially reducing transformation risk93,94,95.
In addition to mutational burden, epigenetic reprogramming also tracks with hormonal aging. DNA methylation patterns in breast tissue show accelerated changes compared to peripheral blood, especially in early reproductive life64,66,68,69,70. DNA methylation in the breast is higher compared to that seen in the blood of the same individuals64,66. The rapid accumulation starts around menarche and decelerates as women approach menopause. This implies that unlike other tissues, the rate of cellular and molecular aging and the accumulation of DNA damage in breast tissue is heavily influenced by reproductive milestones and hormonal exposure.
Several lines of evidence implicate epimutations as contributors to breast cancer development and are affected by environmental exposures that can shape tumor subtype. While methylation of BRCA1 or RAD51C are well established somatic and constitutional epimutations more commonly found in ER- breast cancers, other genes do show frequent methylation patterns, especially in ER+ tumors. RASSF1A promoter hypermethylation is detectable in 40% of DCIS and 65% of matched invasive tumors96,97,98. Promoter methylation of PTEN is found in ~30% of primary tumors and predicts worse 10 year outcome in HR‑positive early breast cancer99,100. APC methylation is higher in cancer vs. normal breast, and higher in luminal breast cancers compared to TNBCs101,102. Lobular carcinomas exhibit promoter methylation of CDH1 (E‑cadherin) which tends to be ER+ cancers103,104, and hypermethylation of GSTP1 is mostly observed in ER‑positive tumors and increases in cancer vs. hyperplasia105.
Genome wide methylation profiling has revealed distinct luminal A and luminal B epigenotypes with recurrent methylation at ESR1, PGR, APC, GSTP1, CDH1and others, underscoring that luminal cancers accumulate a constellation of epimutations rather than a single driver106. Associations between EDCs exposure-related DNA methylation changes at specific CpG sites in different breast cancer subtypes has also been reported107, as well as unique epigenetic signatures among twins to identify environmentally mediated breast cancer risk108.
Phenolic EDCs, parabens, and polyaromatic hydrocarbons have all been shown to increase proliferation of human breast epithelial cells109,110,111,112. PCBs, bisphenols (e.g., BPA), phthalates, and flame retardants also directly induce DNA damage and interfere with DNA repair pathways, even at low-level, chronic exposure92,113. EDC exposure also leads to profound epigenetic changes, disrupting DNA methylation, histone modification, and chromatin remodeling that persist long after exposure has ceased114,115, even over multiple generations, altering gene expression patterns essential for normal cell function116. EDCs induce epigenetic changes that modify methylation kinetics and alter gene expression which can lead to unexpected abnormalities resulting in a predisposition to cancer. They can impact various mechanisms of epigenetic modifications to DNA and chromatin by modulating the availability of methyl donor, S-adenosylmethionine (SAM), for both DNA methyltransferase (DNMT) and histone methyltransferase (HMT) and by affecting expression of TET2 for DNA hydroxymethylation117,118. Therefore, whether lipophilic, persistent, or chronic exposure, EDCs will add to the already high baseline level of DNA damage sustained in breast epithelial cells as well as increase DNA methylation, thereby accelerating key cellular aging processes.
Recent studies have shown that women with Luminal breast cancer exhibit significant epigenetic age acceleration in histologically normal adjacent tissue92. Moreover, breast tumors, as well as normal adjacent tissue in very young women (<35) exhibited accelerated DNA methylation age compared to those in older women66,69. This suggests that long before a malignancy is detected, breast tissue in younger women has already undergone an epigenetic transformation aligned with premature or dysregulated aging, possibly initiated or sustained by chronic exposure to EDCs.
Together, these findings support a model in which chronic EDC exposure mimics and intensifies hormonal cellular aging, effectively accelerating genomic instability and DNA methylation. This is especially concerning given the trends in earlier menarche, delayed childbirth, and prolonged reproductive windows, which already demonstrated rapid aging in the absence of pregnancy. These demographic shifts, when combined with ubiquitous EDC exposure, likely synergize to promote earlier, more aggressive onset of hormone-sensitive breast cancers in young women.
EDCs as amplifiers of ER+ field cancerization
Field cancerization refers to the emergence of genetically and epigenetically altered yet histologically normal cells that expand clonally and increase cancer risk. Rather than a single oncogenic transformation event, tumorigenesis in this model occurs within a “primed field”, a patch of epithelium harboring subtle but accumulating alterations that confer a proliferative or survival advantage119. In breast tissue, this concept is especially relevant due to the gland’s cyclical remodeling, hormonal sensitivity, and expansive ductal-lobular architecture.
During developmental windows such as puberty and pregnancy, breast tissue undergoes rapid proliferation and morphogenesis, making it uniquely susceptible to genomic and epigenetic perturbation. As described above, normal hormonal cycling drives repeated rounds of epithelial expansion, increasing the risk of DNA replication errors and mutation accumulation. Concurrently, the tissue is exposed to oxidative stress from reactive oxygen species and inflammation, further challenging genomic stability.
These dynamics create a fertile environment for the emergence of cancer-primed cells. The combination of hormonal proliferation, DNA damage, and imperfect repair allows for the persistence and expansion of cells with advantageous traits, an early step toward field cancerization.
The architecture and geometry of the mammary ductal epithelium inadvertently promotes the rapid expansion of a few mutated cells that can then spread over large areas120. These extensive patches of pre-cancerous cells are the regions from which subsequent tumors form within the tissue120. While observing real-time field cancerization has not been possible in humans, similar to mice, somatic mutations, chromosomal alterations, and copy number variations as well as DNA methylation and histone modification patterns have been reported in ostensibly normal tissues adjacent to and surrounding breast cancers121,122. Interestingly, the presence of a breast cancer molecular field effect that extends beyond the adjacent normal breast tissue and includes the entire mammary gland has recently been reported123. A cancerized field may present a dual nature: it can appear morphologically abnormal, displaying characteristics of dysplastic tissue, or it may look morphologically normal while still harboring significant genetic or epigenetic alterations such as the recent study that included mammary tissue far beyond the epithelium surrounding a tumor124. This dichotomy between morphological appearance and underlying molecular alterations complicates detection and highlights the need for molecular-level assessment of breast tissue.
EDCs disrupt cellular homeostasis by binding hormone receptors and dysregulating downstream transcriptional programs. In vitro, phenolic EDCs, parabens, and polyaromatic hydrocarbons enhance proliferation in both normal and malignant breast cells, including ER+ and ER- phenotypes, demonstrating broad carcinogenic potential109,110,111,112,125. In addition to disruption of normal proliferation programs, EDCs can also cause cells to fail to differentiate properly, leading to a buildup of immature, potentially cancerous cells. Several epidemiological studies have followed pubertal-aged adolescents after incidental exposure to either polychlorinated aromatic hydrocarbons or polybrominated biphenyls and found significant delays in breast development and earlier onset of menarche, respectively126,127. These anecdotal epidemiologic data are supported by extensive studies in mice and rats confirming that prenatal exposure to EDCs disrupts normal mammary gland development through subtle morphological changes in both the epithelial and stromal fractions128,129. These changes indicated a more developmentally-advanced mammary gland in the EDC-exposure conditions, including increased numbers of terminal ducts and terminal end buds at earlier developmental time points130. Similar studies reported transcriptomic changes in both the stromal and epithelial fractions after EDC exposure that underlie the morphological phenotype of prenatal exposure, including dysregulation of cell differentiation and extracellular matrix genes128. The effect of these EDCs on both epithelial and stromal fractions of the mammary gland is significant given that interaction between these compartments is vital to normal development, and disruption of this interaction has been implicated in the development of breast cancer131,132. When rats that were prenatally exposed to BPA matured, they had an increased number of hyperplastic lesions in the ducts of the mammary glands, as well as desmoplasia of the stromal tissue133. In human breast cancer cells, it has been shown that exposure to BPA significantly decreases expression of KRT14, a defining differentiation marker of breast epithelial cells, in addition to increasing proliferation and migration134.
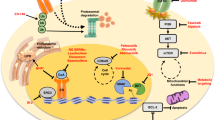
By sustaining a proliferative, plastic epithelial state, EDCs may reprogram hormone-sensitive ER+ luminal cells from a quiescent, differentiated identity into a growth-permissive progenitor-like state, a key feature of Luminal cancer biology. Thus, it could be likely that chronic and cumulative exposure to EDCs initiates and sustains the formation of cancerized fields in the breast composed of ER+ epithelial cells that retain histological normal but are genetically and epigenetically primed for transformation. These fields originate in breast lobules, where chronic hormonal mimicry by EDCs drives DNA damage, epigenetic plasticity, dedifferentiation, and loss of growth arrest. The resulting cells are more proliferative, progenitor-like, and poised for malignant transformation and create a fertile ground for ER+ Luminal breast cancer emergence (Fig. 2).

A Schematic depicting how chronic exposure to EDCs reprograms estrogen receptor-positive (ER⁺) luminal epithelial cells in the mammary gland. In normal tissue, non-proliferative ER⁺ cells integrate hormonal signals to regulate proliferation of ER - cells. EDC exposure induces epigenetic changes, including altered DNA methylation and increased chromatin accessibility, promoting a more proliferative, plastic phenotype of ER⁺ luminal cells. Over time, this results in the emergence of a “cancerized field” of altered ER⁺ cells that are primed for transformation into aggressive luminal tumors. B Developmental timing of exposure shapes the extent of field cancerization. Illustrations show three critical windows of susceptibility—in utero, puberty, and pregnancy—during which environmental exposures may differentially impact mammary epithelial cell fate. Earlier exposures are hypothesized to result in more extensive field cancerization, as depicted by the gradient and corresponding cellular diagrams below.
Although histological and methylation profiling of tumor-adjacent breast tissue has identified focal epigenetic alterations consistent with field effects, robust, large-scale human data remain scarce. Most insights derive from small surgical series or autopsy samples that may not fully capture population heterogeneity. Consequently, the concept of EDC-driven field cancerization in human breast tissue, while highly plausible, needs to be further validated.
EDCs as modifiers of the stromal and immune microenvironments
In addition to accelerating the molecular and functional aging of hormone-sensitive luminal epithelial cells and amplifying field cancerization, chronic exposure to EDCs may also reshape the stromal and immune microenvironment of the breast in ways that suppress anti-tumor immunity, sustain chronic inflammation, and promote tumor escape. These effects are particularly relevant in hormonally responsive tissues like the breast, where immune-epithelial crosstalk governs both homeostasis and transformation risk.
The immune system plays a critical role in maintaining homeostasis by detecting and eliminating nascent tumor cells through mechanisms involving natural killer (NK) cells135 and cytotoxic T lymphocytes (CTLs)136,137,138. Under normal conditions, NK cells and CTLs recognize and eliminate transformed cells prior to the establishment of malignancies. EDC exposure compromises immune surveillance critical for maintaining homeostasis, thereby allowing cancer cells more opportunities to escape detection and targeted killing. EDCs have been shown to suppress the function of immune cells, potentially impairing immunosurveillance and immunoediting139,140,141. Bisphenols and phthalates have been shown to reduce NK cell cytotoxicity and disrupt T cell differentiation142,143, which could lead to an overall weaker anti-tumor response. These immunosuppressive effects may be particularly relevant in younger women, where early-life and cumulative exposure to EDCs could compromise immune resilience before age related immune decline occurs144,145,146.
While acute inflammation is a necessary component of immune defense, chronic low-grade inflammation contributes to tumorigenesis by sustaining a microenvironment rich in pro-inflammatory cytokines, growth factors, and reactive oxygen species147. Endocrine disruptors have been implicated in promoting chronic inflammation in the brain of both mice and rats, where microglia are negatively impacted by the presence of bisphenols, including BPA, phenols, and phthalates148,149,150. Inflammatory cytokines are also upregulated, increasing the production of TNF-α, IL-6, and IL-1β. While these studies clearly link EDCs with microglial activation and increased pro-inflammatory cytokine production in the brain; similarly, systemic inflammation driven by EDC exposure could extend to peripheral tissues, including the breast, where a persistent inflammatory environment may enhance epithelial cell proliferation and support immune evasion mechanisms that allow pre-malignant cells to persist.
Macrophages play a central role in maintaining tissue homeostasis, clearing senescent cells, and initiating immune responses151. In the presence of phthalates, however, macrophages can undergo phenotypic shifts that favor tumor growth, as well as secrete increased inflammatory cytokines152,153. Tumor associated macrophages (TAMs), which arise from dysregulated immune signaling, play a complex role in tumor biology, and have both pro-tumor and anti-tumor effects154. They promote angiogenesis, suppress cytotoxic immune responses, and remodel the extracellular matrix to promote tumor invasion. Studies in mice have also found a decrease in macrophages when exposed to nonylphenol (NP), and when pre-challenged with NP prior to injection of melanoma, the rates of tumor formation and relative tumor growth increased155. These studies suggest that endocrine disrupting chemicals cause dysfunction and dysregulation across multiple immune cell populations, and have the capacity to increase tumorigenicity in vivo.
The link between immune dysfunction and breast cancer extends beyond direct immune suppression and chronic inflammation. Emerging evidence shows that EDC exposure can exacerbate allergy and asthma incidence143,156,157,158,159, shifting immune responses toward a T-helper 2 (Th2)-dominant phenotype. Th2 cytokines, particularly IL-4 and IL-13, not only drive allergic inflammation but also inhibit effective anti-tumor immunity160,161. This shift towards a Th2-skewed immune environment has been observed in multiple cancer types, including breast cancer, where increased IL-4 signaling is associated with enhanced tumor growth and metastasis162,163. Given that allergies and asthma have been linked to EDC exposure, it is possible that EDC-modulated immune responses contribute to increased breast cancer risk.
Beyond individual immune cell dysfunction, EDCs contribute to broader changes in the tumor microenvironment. Exposure to these chemicals has been shown to increase populations of regulatory T cells (Tregs) and myeloid-derived suppressor cells (MDSCs), which inhibit effective anti-tumor immunity and can promote immune evasion164. These immune shifts mirror those seen in aggressive breast cancers, where an immunosuppressive microenvironment allows tumor cells to persist and expand unchecked.
In addition to the effect of EDCs on immune cells, they induce metabolic dysregulation and promote obesity165, creating a vicious cycle of increased estrogen biosynthesis in adipose tissue and enhanced breast cancer risk. In recent years, there has been significant research showing that endocrine-disrupting chemicals affect both cellular and organ metabolism166,167,168,169,170,171, and this body of research has led to a new categorization of certain EDCs as metabolism-disrupting chemicals. Chronic exposure to these chemicals has been shown to increase risk of disorders such as obesity, which are known to contribute to cancer risk166. Studies in mice have shown an increase in overall body weight and breast weight when exposed to BPA133. Human clinical data indicates that there is an exposure-related association of prenatal exposure to low doses of polychlorinated biphenyls (PCBs) to pubertal-age girls having increased body size167. Further work has shown that EDCs affect the differentiation and proliferation of adipocytes168,169, as well as the metabolic processes of pancreatic cells that regulate systemic glucose and lipid levels170. These studies highlight mechanisms by which exposure to EDCs may contribute to increasing body weight. Additionally, fat cells contain aromatase and can store steroid hormones, such as estrogen, as well as lipophilic endocrine disruptors171, thus further creating a positive feedback loop that increases the effects of hormone pathways detailed above.
EDCs as field primers to the luminal breast cancer
Evidence already indicates the association between breast cancer risk and estradiol levels172,173. The connection between EDC exposure and cancer is also well-established; from studies of women who were exposed to diethylstilbestrol (DES) during pregnancy, to animal studies demonstrating in utero exposure to BPA, TCDD, phthalates, PFOA, and even certain phytoestrogens results in altered mammary development, and increased risk of neoplasms later in life39. Follow-up studies in mice as well as the ongoing NCI DES Third Generation Study have suggested that effects of DES exposure persist through multiple generations, thus supporting the mechanistic hypothesis of an epigenetic imprint of exposure174,175.
Given today’s exposure to the vast number of EDCs within our environment, substantially more research is needed to identify which EDCs alter in utero development and a deeper understanding of their long-term effects after birth. Based on the evidence outlined above, we propose that early-life EDC exposure may create even more extensive cancer-primed fields within breast tissue. Early-life EDC exposure encompasses both prenatal (in utero) and postnatal exposures, including during infancy, childhood, and puberty: critical developmental windows during which breast tissue is particularly sensitive to hormonal disruption. We posit that chronic and cumulative EDC exposure likely serves as an environmental trigger to initiate Luminal tumorigenesis by promoting DNA damage, enhancing epigenetic plasticity, inducing dedifferentiation of ER+ cells, accelerating breast tissue aging, and fostering immunosuppression (Fig. 2). EDC exposure alters the epigenetic landscape of luminal cells by inducing methylation changes, leading to increased chromatin accessibility and altered gene expression. This epigenetic reprogramming disrupts ER-mediated growth arrest in hormone sensing ER+ luminal cells, which normally are differentiated and post-mitotic. The changes induced upon chronic EDC exposure pushes luminal cells into a more plastic, proliferative, progenitor-like state. These cellular and molecular alterations culminate in the cancerization of hormone-independent ER+ fields, marking a pivotal step toward Luminal tumor development.
We further posit that the epigenetic plasticity induced by chronic hormonal disruption transitions ER+ luminal cells from a differentiated, growth-arrested state to a proliferative progenitor-like phenotype. Such widespread fields of genetically and epigenetically altered cells become fertile grounds for Luminal breast cancer emergence, that combined with suppressed immune surveillance, characterizes the rapid and early onset of aggressive and hormone responsive breast cancer. This model, where EDCs promote field cancerization not only at the cellular and molecular level but also by disabling immune surveillance and reprogramming the tissue microenvironment, may explain the rising incidence of hormone-sensitive breast cancers among younger women. Understanding these immunologic vulnerabilities opens new avenues for prevention and therapeutic intervention, including strategies to restore immune function in high-risk, environmentally exposed populations. Furthermore, epigenetic changes are therapeutically targetable. Therefore, learning about the contributions of EDCs to carcinogenic dysregulation of the epigenetic landscape to create therapeutic strategies to ameliorate risk or treat EDC-driven cancers.
Conclusion
Current epidemiological shifts in breast cancer incidence underscore the urgency of understanding environmental contributions to carcinogenesis. However, it has been challenging to study environmental exposures and their potential role in health trends due to a variety of reasons. First, there has been, and continues to be, limited methods to accurately measure exposure in humans. EDCs with a short half‑life in vivo (eg. phthalates, phenols and parabens) are cleared within hours; thus a single urine or plasma sample could misclassify chronic exposure. More persistent EDCs (eg. PCBs, PFAS and organochlorine pesticides) are typically simultaneously present making it difficult to isolate any single agent. Furthermore, dose exposure and responses in the real world fluctuate with low‑dose effects having differing effects from high‑dose effects. In addition, both the types, nature, and formulations of environmental chemicals continue to shift over time. The types and concentrations of legacyEDCs (e.g. DTT, some PCBs, certain phthalates etc.) have fallen since the early 2000s, while structurally replacement chemicals now dominate exposure profiles (eg. BPA vs. BPS, BPF)61. Therefore, specimens collected from earlier cohorts will likely lack chemicals that are currently relevant, while specimens collected in recent cohorts may dilute exposure signals limiting the direct comparability of longitudinal data. All of these various complexities make it difficult to identify and interpret exposure data. Overcoming complications in capturing real‑world exposures may require repeated sampling designs and pooling biospecimens or the use of wearable passive samplers (e.g. silicone wristbands) to complement biofluid collection, especially for measuring exposure in children62,63. High‑resolution, simultaneous detection of known and unknown chemical metabolites in serum, plasma, urine, saliva or even lymph, could allow for reconstruction of past and present exposures that together could provide “chemical exposomics” signatures. This could be accomplished using ultrasensitive mass spectrometry to small volumes of plasma, lymph fluid, or urine to identify and quantify chemical metabolites. Additionally, identifying molecular fingerprinting of exposure based on DNA methylation, DNA adduct formation signature patterns, and/or RNA expression could complement or substitute for direct chemical exposure measurements.
Second, breast tissue is most sensitive to hormones and exposures during specific developmental and reproductive stages of life; yet most cohort biospecimens are typically collected in mid‑life, decades after the etiologic window of susceptibility. Longitudinal studies using cohorts starting at birth, through adolescence and pregnancy as well as using nested case-control analyses could help overcome the historical limitations needed to study exposure. Generation R and the Breast Cancer and the Environment Research Program (BCERP) are two such long-term initiatives launched in 2002 and 2003 respectively, aimed at studying environmental and genetic factors focusing on exposures during sensitive periods by following cohorts from before birth into young adulthood to identify early factors influencing health176,177.
Third, the long latency period between exposure and the onset of breast cancer (10-40 yrs)12 combined with the modest risk of any single chemical exposure makes it difficult for traditional epidemiologic study designs to isolate the effect of that risk while fully adjusting for confounders such as parity, obesity, and alcohol consumption. Employing complementary approaches using prospective birth and adolescent cohorts, and geographic information that incorporates EPA-superfund geospatial information, as well as wildlife-based environmental indicators could improve any causal claims regarding EDCs and their role in breast cancer risk.
Finally, biological studies on EDCs have historically relied on 2D cell cultures that are almost exclusively on ER+ breast cancer cell lines and l stromal, immune and adipose compartments. 3D human mammary organoid co‑cultures that are hormone sensitive and recreate ductal-lobular architecture and resident immune cells178,179, represents the new frontier for studying chemical exposure on breast tissue development. This methodological advancement enables low‑dose and multi-chemical exposure, as well as the study of developmental stage specific exposures, which could generate far more physiologically relevant and clinically translatable insights into how EDCs could be driving breast tissue transformation.
Advances in tools and technologies make it possible to finally close the knowledge gap and address rising breast cancer risk from environmental exposures. Additional funding that integrates research with real‑world exposure assessment and tissue‑specific functional read‑outs will not only clarify which chemical mixtures accelerate breast‑tumor initiation but will also generate biomarkers for early detection and targets for primary prevention. Together, these advances can position the field to move beyond correlation toward mechanistic causation.
References
Valla, M. et al. Molecular subtypes of breast cancer: long-term incidence trends and prognostic differences. Cancer Epidemiol. Biomark. Prev. 25, 1625–1634 (2016).
Li, N. H. Y. & Li, C. I. Incidence rate trends of breast cancer overall and by molecular subtype by race and ethnicity and age. JAMA Netw. Open 8, e2456142 (2025).
Momenimovahed, Z. & Salehiniya, H. Epidemiological characteristics of and risk factors for breast cancer in the world. Breast Cancer Targets Ther. 11, 151–164 (2019).
McPherson, K., Steel, C. M. & Dixon, J. M. ABC of breast diseases. Breast Cancer Epidemiol. Risk factors, Genet. BMJ 321, 624–628 (2000).
Dall, G. V. & Britt, K. L. Estrogen effects on the mammary gland in early and late life and breast cancer risk. Front. Oncol. 7, 110 (2017).
Anderson, K. N., Schwab, R. B. & Martinez, M. E. Reproductive risk factors and breast cancer subtypes: a review of the literature. Breast Cancer Res. Treat. 144, 1–10 (2014).
Bhardwaj, P. et al. Estrogens and breast cancer: mechanisms involved in obesity-related development, growth and progression. J. Steroid Biochem. Mol. Biol. 189, 161–170 (2019).
Sung, H. et al. Differences in cancer rates among adults born between 1920 and 1990 in the USA: an analysis of population-based cancer registry data. Lancet Public Health 9, e583–e593 (2024).
Rosenberg, P. S. & Miranda-Filho, A. Cancer incidence trends in successive social generations in the US. JAMA Netw. Open 7, e2415731 (2024).
Xu, S., Murtagh, S., Han, Y., Wan, F. & Toriola, A. T. Breast cancer incidence among US women aged 20 to 49 years by race, stage, and hormone receptor status. JAMA Netw. Open 7, e2353331 (2024).
Cai, Y., Dai, F., Ye, Y. & Qian, J. The global burden of breast cancer among women of reproductive age: a comprehensive analysis. Sci. Rep. 15, 9347 (2025).
Tokunaga, M. et al. Incidence of female breast cancer among atomic bomb survivors, 1950-1985. Radiat. Res. 138, 209–223 (1994).
Moskowitz, C. S. et al. Radiation-associated breast cancer and gonadal hormone exposure: a report from the childhood cancer survivor study. Br. J. Cancer 117, 290–299 (2017).
Inskip, P. D. et al. Radiation dose and breast cancer risk in the childhood cancer survivor study. J. Clin. Oncol. J. Am. Soc. Clin. Oncol. 27, 3901–3907 (2009).
Apostolou, P. et al. Genetic testing of breast cancer patients with very early-onset breast cancer (≤30 years) yields a high rate of germline pathogenic variants, mainly in the BRCA1, TP53, and BRCA2 genes. Cancers 16, 2368 (2024).
De Silva, S., Tennekoon, K. H. & Karunanayake, E. H. Overview of the genetic basis toward early detection of breast cancer. Breast Cancer Targets Ther. 11, 71–80 (2019).
Pal, M., Das, D. & Pandey, M. Understanding genetic variations associated with familial breast cancer. World J. Surg. Oncol. 22, 271 (2024).
Bertrand, K. A., Bethea, T. N., Adams-Campbell, L. L., Rosenberg, L. & Palmer, J. R. Differential patterns of risk factors for early-onset breast cancer by er status in african american women. Cancer Epidemiol. Biomark. Prev. Publ. Am. Assoc. Cancer Res. Cosponsored Am. Soc. Prev. Oncol. 26, 270–277 (2017).
Ganz, P. A., Greendale, G. A., Petersen, L., Kahn, B. & Bower, J. E. Breast cancer in younger women: reproductive and late health effects of treatment. J. Clin. Oncol. J. Am. Soc. Clin. Oncol. 21, 4184–4193 (2003).
Turkman, Y. E., Sakibia Opong, A., Harris, L. N. & Knobf, M. T. Biologic, demographic, and social factors affecting triple negative breast cancer outcomes. Clin. J. Oncol. Nurs. 19, 62–67 (2015).
Acheampong, T., Kehm, R. D., Terry, M. B., Argov, E. L. & Tehranifar, P. Incidence trends of breast cancer molecular subtypes by age and race/ethnicity in the US from 2010 to 2016. JAMA Netw. Open 3, e2013226 (2020).
Kehm, R. D., Daaboul, J. M., Tehranifar, P. & Terry, M. B. Geographic differences in early-onset breast cancer incidence trends in the USA, 2001-2020, is it time for a geographic risk score? Cancer Causes Control CCC 36, 707−717 (2025).
Mucci, L. A. et al. Familial risk and heritability of cancer among twins in nordic countries. JAMA 315, 68–76 (2016).
Ogbenna, B. T. et al. Healthy lifestyle index and breast cancer risk among postmenopausal women: the multiethnic cohort study. Cancer Epidemiol. Biomark. Prev. 34, 875–884 (2025).
Barnes, J. L., Zubair, M., John, K., Poirier, M. C. & Martin, F. L. Carcinogens and DNA damage. Biochem. Soc. Trans. 46, 1213–1224 (2018).
Patterson Jr et al. Levels in the US Population of those persistent organic pollutants (2003 − 2004) included in the stockholm convention or in other long-range transboundary air pollution agreements. Environ. Sci. Technol. 43, 1211–1218 (2009)..
Thornton, J. W., McCally, M. & Houlihan, J. Biomonitoring of industrial pollutants: health and policy implications of the chemical body burden. Public Health Reports® 117, 315–323 (2002).
CDC. National Exposure Report Home Page. National Report on Human Exposure to Environmental Chemicals https://www.cdc.gov/environmental-exposure-report/index.html (2025).
Combarnous, Y. & Nguyen, T. M. D. Comparative overview of the mechanisms of action of hormones and endocrine disruptor compounds. Toxics 7, 5 (2019).
Shanle, E. K. & Xu, W. Endocrine disrupting chemicals targeting estrogen receptor signaling: identification and mechanisms of action. Chem. Res. Toxicol. 24, 6–19 (2011).
Tan, H. et al. Structures of endocrine-disrupting chemicals determine binding to and activation of the estrogen receptor α and androgen receptor. Environ. Sci. Technol. 54, 11424–11433 (2020).
Toporova, L. & Balaguer, P. Nuclear receptors are the major targets of endocrine disrupting chemicals. Mol. Cell. Endocrinol. 502, 110665 (2020).
Fischer, F. C. et al. Binding of per- and polyfluoroalkyl substances (PFAS) to serum proteins: implications for toxicokinetics in humans. Environ. Sci. Technol. 58, 1055–1063 (2024).
Perera, L. et al. Binding of bisphenol A, bisphenol AF, and bisphenol S on the androgen receptor: coregulator recruitment and stimulation of potential interaction sites. Toxicol. Vitro 44, 287–302 (2017).
McKinney, J. D. & Waller, C. L. Polychlorinated biphenyls as hormonally active structural analogues. Environ. Health Perspect. 102, 290–297 (1994).
Perminova, I. V., Grechishcheva, N., Yu & Petrosyan, V. S. Relationships between structure and binding affinity of humic substances for polycyclic aromatic hydrocarbons: relevance of molecular descriptors. Environ. Sci. Technol. 33, 3781–3787 (1999).
Zhang, H. et al. Relationships between structure and binding affinity of humic substances for polycyclic aromatic hydrocarbons: relevance of molecular descriptors. Chem. Res. Toxicol. 28, 1196–1204 (2015).
Cao, L.-Y., Ren, X.-M., Li, C.-H. & Guo, L.-H. Organophosphate esters bind to and inhibit estrogen-related receptor γ in cells. Environ. Sci. Technol. Lett. 5, 68–73 (2018).
Macon, M. B. & Fenton, S. E. Endocrine disruptors and the breast: early life effects and later life disease. J. Mammary Gland Biol. Neoplasia 18, 43–61 (2013).
US EPA, O. EnviroAtlas Interactive Map. https://www.epa.gov/enviroatlas/enviroatlas-interactive-map (2015).
Marlatt, V. L. et al. Impacts of endocrine disrupting chemicals on reproduction in wildlife and humans. Environ. Res. 208, 112584 (2022).
Rannaud-Bartaire, P., Demeneix, B. A. & Fini, J.-B. Pressures of the urban environment on the endocrine system: Adverse effects and adaptation. Mol. Cell. Endocrinol. 583, 112125 (2024).
Ullah, S. et al. A review of the endocrine disrupting effects of micro and nano plastic and their associated chemicals in mammals. Front. Endocrinol. 13, 1084236 (2023).
Giesy, J.P. et al. Perfluorinated compounds in the great lakes. In Persistent Organic Pollutants in the Great Lakes. The Handbook of Environmental Chemistry, (ed. Hites, R.A.) (Springer, 2005)
Matthiessen, P., Wheeler, J. R. & Weltje, L. A review of the evidence for endocrine disrupting effects of current-use chemicals on wildlife populations. Crit. Rev. Toxicol. 48, 195–216 (2018).
Yang, Y. et al. Per- and polyfluoroalkyl substances (PFAS) in consumer products: an overview of the occurrence, migration, and exposure assessment. Molecules 30, 994 (2025).
ProQuest. Mercury and PCB Residues in Massachusetts River Otters: Comparisons on a Watershed basis. https://www.proquest.com/openview/d4dffbbebbdfaacf48e64dba6970df7f/1?pq-origsite=gscholar&cbl=18750&diss=y.
US EPA, O. 2020 Great Lakes Human Health Fish Fillet Tissue Study. https://www.epa.gov/choose-fish-and-shellfish-wisely/2020-great-lakes-human-health-fish-fillet-tissue-study (2024).
Ruyle, B. J. et al. Isolating the AFFF signature in coastal watersheds using oxidizable pfas precursors and unexplained organofluorine. Environ. Sci. Technol. 55, 3686–3695 (2021).
Rolland, R. M. A review of chemically-induced alterations in thyroid and vitamin A status from field studies of wildlife and fish. J. Wildl. Dis. 36, 615–635 (2000).
Wainstein, M. et al. Highly contaminated river otters (Lontra canadensis) are effective biomonitors of environmental pollutant exposure. Environ. Monit. Assess. 194, 670 (2022).
U.S. Geological Survey. A National Assessment of Pesticide, PFAS, Microplastic, and Antibiotic Resistance Gene Exposures in White-Tailed Deer. https://www.usgs.gov/centers/columbia-environmental-research-center/science/a-national-assessment-pesticide-pfas (2023).
Berheim, E. H. et al. Effects of neonicotinoid insecticides on physiology and reproductive characteristics of captive female and fawn white-tailed deer. Sci. Rep. 9, 4534 (2019).
Longnecker, M. P., Rogan, W. J. & Lucier, G. The human health effects of ddt (dichlorodiphenyltrichloroethane) and pcbs (polychlorinated biphenyls) and an overview of organochlorines in public health1. Annu. Rev. Public Health 18, 211–244 (1997).
Institute of Medicine (US) Committee on the Implications of Dioxin in the Food Supply. Dioxins and Dioxin-like Compounds in the Food Supply: Strategies to Decrease Exposure. (National Academies Press (US), Washington (DC), 2003).
National Research Council (US) Committee on Hormonally Active Agents in the Environment. Hormonally Active Agents in the Environment. (National Academies Press (US), Washington (DC), 1999).
Faroon, O. Addendum for Polychlorinated Biphenyls. https://www.atsdr.cdc.gov/toxprofiles/pcbs_addendum.pdf (2011).
Branchi, I., Alleva, E. & Costa, L. G. Effects of perinatal exposure to a polybrominated diphenyl ether (pbde 99) on mouse neurobehavioural development. NeuroToxicology 23, 375–384 (2002).
Hites, R. A. Polybrominated diphenyl ethers in the environment and in people: a meta-analysis of concentrations. Environ. Sci. Technol. 38, 945–956 (2004).
Fäys, F. et al. Biomonitoring of fast-elimination endocrine disruptors - results from a 6 month follow up on human volunteers with repeated urine and hair collection. Sci. Total Environ. 778, 146330 (2021).
Stanfield, Z. et al. Characterizing chemical exposure trends from NHANES urinary biomonitoring data. Environ. Health Perspect. 132, 17009 (2024).
Young, A. S. et al. Hormone receptor activities of complex mixtures of known and suspect chemicals in personal silicone wristband samplers worn in office buildings. Chemosphere 315, 137705 (2023).
Samon, S. M. et al. Measuring semi-volatile organic compound exposures during pregnancy using silicone wristbands. Chemosphere 339, 139778 (2023).
Pike, M. C., Krailo, M. D., Henderson, B. E., Casagrande, J. T. & Hoel, D. G. ‘Hormonal’ risk factors, ‘breast tissue age’ and the age-incidence of breast cancer. Nature 303, 767–770 (1983).
Dutta, S. et al. Reproductive toxicity of combined effects of endocrine disruptors on human reproduction. Front. Cell Dev. Biol. 11, 1162015 (2023).
Sehl, M. E., Henry, J. E., Storniolo, A. M., Ganz, P. A. & Horvath, S. DNA methylation age is elevated in breast tissue of healthy women. Breast Cancer Res. Treat. 164, 209–219 (2017).
Kostecka, A. et al. High prevalence of somatic PIK3CA and TP53 pathogenic variants in the normal mammary gland tissue of sporadic breast cancer patients revealed by duplex sequencing. Npj Breast Cancer 8, 1–10 (2022).
Cereser, B. et al. The mutational landscape of the adult healthy parous and nulliparous human breast. Nat. Commun. 14, 5136 (2023).
Sehl, M. E. et al. Systematic dissection of epigenetic age acceleration in normal breast tissue reveals its link to estrogen signaling and cancer risk. bioRxiv https://doi.org/10.1101/2024.10.31.619735 (2024).
Oltra, S. S. et al. Acceleration in the DNA methylation age in breast cancer tumours from very young women. Sci. Rep. 9, 14991 (2019).
Söderqvist, G. et al. Proliferation of breast epithelial cells in healthy women during the menstrual cycle. Am. J. Obstet. Gynecol. 176, 123–128 (1997).
Navarrete, M. A. H. et al. Assessment of the proliferative, apoptotic and cellular renovation indices of the human mammary epithelium during the follicular and luteal phases of the menstrual cycle. Breast Cancer Res. BCR 7, R306–R313 (2005).
Bodicoat, D. H. et al. Timing of pubertal stages and breast cancer risk: the breakthrough generations study. Breast Cancer Res. BCR 16, R18 (2014).
Kelsey, J. L., Gammon, M. D. & John, E. M. Reproductive factors and breast cancer. Epidemiol. Rev. 15, 36–47 (1993).
Ahlgren M, Melbye M, Wohlfahrt J, Sorensen TI. Growth patterns and the risk of breast cancer in women. N. Engl. J. Med. 351, 1619-26 (2004).
Fernandez, S. V., Russo, I. H. & Russo, J. Estradiol and its metabolites 4-hydroxyestradiol and 2-hydroxyestradiol induce mutations in human breast epithelial cells. Int. J. Cancer 118, 1862–1868 (2006).
Li, K.-M. et al. Metabolism and DNA binding studies of 4-hydroxyestradiol and estradiol-3,4-quinone in vitro and in female ACI rat mammary gland in vivo. Carcinogenesis 25, 289–297 (2004).
Miao, S. et al. 4-Hydroxy estrogen metabolite, causing genomic instability by attenuating the function of spindle-assembly checkpoint, can serve as a biomarker for breast cancer. Am. J. Transl. Res. 11, 4992–5007 (2019).
Lu, F., Zahid, M., Saeed, M., Cavalieri, E. L. & Rogan, E. G. Estrogen metabolism and formation of estrogen-DNA adducts in estradiol-treated MCF-10F cells. The effects of 2,3,7,8-tetrachlorodibenzo-p-dioxin induction and catechol-O-methyltransferase inhibition. J. Steroid Biochem. Mol. Biol. 105, 150–158 (2007).
Gaikwad, N. W. et al. The molecular etiology of breast cancer: Evidence from biomarkers of risk. Int. J. Cancer J. Int. Cancer 122, 1949–1957 (2008).
Gaikwad, N. W. et al. Urine biomarkers of risk in the molecular etiology of breast cancer. Breast Cancer Basic Clin. Res 3, 1–8 (2009).
Cavalieri, E. L. & Rogan, E. G. Depurinating estrogen–DNA adducts in the etiology and prevention of breast and other human cancers. Future Oncol. Lond. Engl. 6, 75–91 (2010).
Kidane, D. et al. Interplay between DNA repair and inflammation, and the link to cancer. Crit. Rev. Biochem. Mol. Biol. 49, 116–139 (2014).
Cavalieri, E. L. & Rogan, E. G. Depurinating estrogen-DNA adducts, generators of cancer initiation: their minimization leads to cancer prevention. Clin. Transl. Med. 5, 12 (2016).
Kay, J., Thadhani, E., Samson, L. & Engelward, B. Inflammation-induced DNA damage, mutations and cancer. DNA Repair 83, 102673 (2019).
Ozasa, K. Epidemiological research on radiation-induced cancer in atomic bomb survivors. J. Radiat. Res. (Tokyo) 57, i112–i117 (2016).
Bhatia, S. et al. Breast cancer and other second neoplasms after childhood Hodgkin’s disease. N. Engl. J. Med. 334, 745–751 (1996).
Land, C. E. Studies of cancer and radiation dose among atomic bomb survivors. Ex. Breast Cancer JAMA 274, 402–407 (1995).
Chatterjee, N. & Walker, G. C. Mechanisms of DNA damage, repair, and mutagenesis. Environ. Mol. Mutagen. 58, 235–263 (2017).
Poetsch, A. R. The genomics of oxidative DNA damage, repair, and resulting mutagenesis. Comput. Struct. Biotechnol. J. 18, 207–219 (2020).
Zhou, X., Speer, R. M., Volk, L., Hudson, L. G. & Liu, K. J. Arsenic co-carcinogenesis: inhibition of DNA repair and interaction with zinc finger proteins. Semin. Cancer Biol. 76, 86–98 (2021).
Santa-Gonzalez, G. A., Gomez-Molina, A., Arcos-Burgos, M., Meyer, J. N. & Camargo, M. Distinctive adaptive response to repeated exposure to hydrogen peroxide associated with upregulation of DNA repair genes and cell cycle arrest. Redox Biol. 9, 124–133 (2016).
Choudhury, S. et al. Molecular profiling of human mammary gland links breast cancer risk to a p27(+) cell population with progenitor characteristics. Cell Stem Cell 13, 117–130 (2013).
Blakely, C. M. et al. Hormone-induced protection against mammary tumorigenesis is conserved in multiple rat strains and identifies a core gene expression signature induced by pregnancy. Cancer Res. 66, 6421–6431 (2006).
Sivaraman, L., Conneely, O. M., Medina, D. & O’Malley, B. W. p53 is a potential mediator of pregnancy and hormone-induced resistance to mammary carcinogenesis. Proc. Natl. Acad. Sci. USA. 98, 12379–12384 (2001).
Yeo, W. et al. High frequency of promoter hypermethylation of RASSF1A in tumorous and non-tumourous tissue of breast cancer. Pathology 37, 125–130 (2005).
Fackler, M. J. et al. DNA methylation of RASSF1A, HIN-1, RAR-β, Cyclin D2 and twist in in situ and invasive lobular breast carcinoma. Int. J. Cancer 107, 970–975 (2003).
Honorio, S. et al. Detection of RASSF1A aberrant promoter hypermethylation in sputum from chronic smokers and ductal carcinoma in situ from breast cancer patients. Oncogene 22, 147–150 (2003).
García, J. M. et al. Promoter methylation of the PTEN gene is a common molecular change in breast cancer. Genes. Chromosomes Cancer 41, 117–124 (2004).
Zhang, H.-Y., Liang, F., Jia, Z.-L., Song, S.-T. & Jiang, Z.-F. PTEN mutation, methylation and expression in breast cancer patients. Oncol. Lett. 6, 161–168 (2013).
He, K., Zhang, L. & Long, X. Quantitative assessment of the association between APC promoter methylation and breast cancer. Oncotarget 7, 37920–37930 (2016).
Holm, K. et al. Molecular subtypes of breast cancer are associated with characteristic DNA methylation patterns. Breast Cancer Res. 12, R36 (2010).
Liu, J. et al. CDH1 promoter methylation correlates with decreased gene expression and poor prognosis in patients with breast cancer. Oncol. Lett. 11, 2635–2643 (2016).
Zou, D. et al. Epigenetic silencing in non-neoplastic epithelia identifies E-cadherin (CDH1) as a target for chemoprevention of lobular neoplasia. J. Pathol. 218, 265–272 (2009).
Miyake, T. et al. Association of GSTP1 methylation with aggressive phenotype in ER-positive breast cancer. Anticancer Res 33, 5617–5623 (2013).
Bediaga, N. G. et al. DNA methylation epigenotypes in breast cancer molecular subtypes. Breast Cancer Res. BCR 12, R77 (2010).
Song, N., Xi, X., Yang, K., Pei, C. & Zhao, L. Effects of endocrine disrupting chemicals, blood metabolome, and epigenetics on breast cancer risk: a multi-dimensional mendelian randomization study. Ecotoxicol. Environ. Saf. 291, 117791 (2025).
Bode, H. F., He, L., Hjelmborg, J. V. B., Kaprio, J. & Ollikainen, M. Pre-diagnosis blood DNA methylation profiling of twin pairs discordant for breast cancer points to the importance of environmental risk factors. Clin. Epigenetics 16, 160 (2024).
Cohn, B. A., Wolff, M. S., Cirillo, P. M. & Sholtz, R. I. DDT and breast cancer in young women: new data on the significance of age at exposure. Environ. Health Perspect. 115, 1406–1414 (2007).
Martina Plioskova, M., Vondracek, J. Vojtesek, B., Kozubík, A., Machala, M. Deregulation of cell proliferation by polycyclic aromatic hydrocarbons in human breast carcinoma mcf-7 cells reflects both genotoxic and nongenotoxic events. Toxicol. Sci. 83, 246–256 (2005).
Williams, G. P. & Darbre, P. D. Low-dose environmental endocrine disruptors, increase aromatase activity, estradiol biosynthesis and cell proliferation in human breast cells. Mol. Cell. Endocrinol. 486, 55–64 (2019).
Soto, A. M. & Sonnenschein, C. Environmental causes of cancer: endocrine disruptors as carcinogens. Nat. Rev. Endocrinol. 6, 363–370 (2010).
Diamanti-Kandarakis, E. et al. Endocrine-disrupting chemicals: an endocrine society scientific statement. Endocr. Rev. 30, 293–342 (2009).
Cohn, B. A., Cirillo, P. M. & Terry, M. B. DDT and Breast cancer: prospective study of induction time and susceptibility windows. J. Natl Cancer Inst. 111, 803–810 (2019).
Nettore, I. C., Franchini, F., Palatucci, G., Macchia, P. E. & Ungaro, P. Epigenetic mechanisms of endocrine-disrupting chemicals in obesity. Biomedicines 9, 1716 (2021).
Bernal, A. J. & Jirtle, R. L. Epigenomic disruption: the effects of early developmental exposures. Birt. Defects Res. A. Clin. Mol. Teratol. 88, 938–944 (2010).
Natarajan, R. et al. Environmental exposures during puberty: window of breast cancer risk and epigenetic damage. Int. J. Environ. Res. Public. Health 17, 493 (2020).
Feil, R. & Fraga, M. F. Epigenetics and the environment: emerging patterns and implications. Nat. Rev. Genet. 13, 97–109 (2012).
Li, Z., Lyu, C., Ren, Y. & Wang, H. Role of TET Dioxygenases and DNA hydroxymethylation in bisphenols-stimulated proliferation of breast cancer cells. Environ. Health Perspect. 128, 27008 (2020).
Deng, G., Lu, Y., Zlotnikov, G., Thor, A. D. & Smith, H. S. Loss of heterozygosity in normal tissue adjacent to breast carcinomas. Science 274, 2057–2059 (1996).
Ciwinska, M. et al. Mechanisms that clear mutations drive field cancerization in mammary tissue. Nature 633, 198–206 (2024).
Heaphy, C. M., Griffith, J. K. & Bisoffi, M. Mammary field cancerization: molecular evidence and clinical importance. Breast Cancer Res. Treat. 118, 229–239 (2009).
Gao, Y., Widschwendter, M. & Teschendorff, A. E. DNA Methylation patterns in normal tissue correlate more strongly with breast cancer status than copy-number variants. EBioMedicine 31, 243–252 (2018).
Bedrosian, I. et al. Abstract 3433: identification of a subtype specific molecular field across the mammary gland of breast cancer patients. Cancer Res 80, 3433 (2020).
Troester, M. A. et al. DNA defects, epigenetics, and gene expression in cancer-adjacent breast: a study from The Cancer Genome Atlas. Npj Breast Cancer 2, 1–7 (2016).
Blanck, H. M. et al. Age at menarche and tanner stage in girls exposed in utero and postnatally to polybrominated biphenyl. Epidemiol. Camb. Mass 11, 641–647 (2000).
Ayyanan, A. et al. Perinatal exposure to bisphenol a increases adult mammary gland progesterone response and cell number. Mol. Endocrinol. Baltim. Md. 25, 1915–1923 (2011).
Vandenberg, L. N. et al. Exposure to environmentally relevant doses of the xenoestrogen bisphenol-A alters development of the fetal mouse mammary gland. Endocrinology 148, 116–127 (2007).
Moral, R. et al. Effect of prenatal exposure to the endocrine disruptor bisphenol A on mammary gland morphology and gene expression signature. J. Endocrinol. 196, 101–112 (2008).
Wadia, P. R. et al. Low-dose BPA exposure alters the mesenchymal and epithelial transcriptomes of the mouse fetal mammary gland. PloS ONE 8, e63902 (2013).
Kim, J. B., Stein, R. & O’Hare, M. J. Tumour-stromal interactions in breast cancer: the role of stroma in tumourigenesis. Tumour Biol. J. Int. Soc. Oncodev. Biol. Med. 26, 173–185 (2005).
Oskarsson, T. Extracellular matrix components in breast cancer progression and metastasis. Breast Edinb. Scotl. 22, S66–S72 (2013).
Durando, M. et al. Prenatal bisphenol a exposure induces preneoplastic lesions in the mammary gland in wistar rats. Environ. Health Perspect. 115, 80–86 (2007).
Wang, Z. et al. Identification of key genes linking bisphenols exposure and breast cancer. Toxicology 514, 154123 (2025).
Wolf, N. K., Kissiov, D. U. & Raulet, D. H. Roles of natural killer cells in immunity to cancer, and applications to immunotherapy. Nat. Rev. Immunol. 23, 90–105 (2023).
Raskov, H., Orhan, A., Christensen, J. P. & Gögenur, I. Cytotoxic CD8 + T cells in cancer and cancer immunotherapy. Br. J. Cancer 124, 359–367 (2021).
Weigelin, B. et al. Cytotoxic T cells are able to efficiently eliminate cancer cells by additive cytotoxicity. Nat. Commun. 12, 5217 (2021).
Waldman, A. D., Fritz, J. M. & Lenardo, M. J. A guide to cancer immunotherapy: from T cell basic science to clinical practice. Nat. Rev. Immunol. 20, 651–668 (2020).
Besteman, E. G., Zimmerman, K. L. & Holladay, S. D. Diethylstilbestrol (DES)-induced fetal thymic atrophy in C57BL/6 mice: inhibited thymocyte differentiation and increased apoptotic cell death. Int. J. Toxicol. 24, 231–239 (2005).
Rees Clayton, E. M., Todd, M., Dowd, J. B. & Aiello, A. E. The impact of bisphenol A and triclosan on immune parameters in the U.S. population, NHANES 2003–2006. Environ. Health Perspect. 119, 390–396 (2011).
Kuo, C.-H., Yang, S.-N., Kuo, P.-L. & Hung, C.-H. Immunomodulatory effects of environmental endocrine disrupting chemicals. Kaohsiung J. Med. Sci. 28, S37–S42 (2012).
Gostner, J. M. et al. Bisphenol A suppresses Th1-type immune response in human peripheral blood mononuclear cells in vitro. Immunol. Lett. 168, 285–292 (2015).
Nowak, K., Jabłońska, E. & Ratajczak-Wrona, W. Immunomodulatory effects of synthetic endocrine disrupting chemicals on the development and functions of human immune cells. Environ. Int. 125, 350–364 (2019).
Renz, H. et al. An exposome perspective: early-life events and immune development in a changing world. J. Allergy Clin. Immunol. 140, 24–40 (2017).
Carpenter, D. O., Ma, J. & Lessner, L. Asthma and infectious respiratory disease in relation to residence near hazardous waste sites. Ann. N. Y. Acad. Sci. 1140, 201–208 (2008).
Weisglas-Kuperus, N. et al. Immunologic effects of background prenatal and postnatal exposure to dioxins and polychlorinated biphenyls in dutch infants. Pediatr. Res. 38, 404–410 (1995).
Zhao, H. et al. Inflammation and tumor progression: signaling pathways and targeted intervention. Signal Transduct. Target. Ther. 6, 263 (2021).
Kassab, R. B., Lokman, M. S. & Essawy, E. A. Neurochemical alterations following the exposure to di-n-butyl phthalate in rats. Metab. Brain Dis. 34, 235–244 (2019).
Bjørklund, G. et al. Linking environmental chemicals to neuroinflammation and autism spectrum disorder: mechanisms and implications for prevention. Mol. Neurobiol. 61, 6328–6340 (2024).
Rebuli, M. E., Gibson, P., Rhodes, C. L., Cushing, B. S. & Patisaul, H. B. Sex differences in microglial colonization and vulnerabilities to endocrine disruption in the social brain. Gen. Comp. Endocrinol. 238, 39–46 (2016).
Jaiswal, S., Chao, M. P., Majeti, R. & Weissman, I. L. Macrophages as mediators of tumor immunosurveillance. Trends Immunol. 31, 212–219 (2010).
Nishioka, J. et al. Di-(2-ethylhexyl) phthalate induces production of inflammatory molecules in human macrophages. Inflamm. Res. J. Eur. Histamine Res. Soc. Al 61, 69–78 (2012).
Wang, X. et al. Macrophage polarization as a novel endpoint for assessing combined risk of phthalate esters. Environ. Int. 190, 108835 (2024).
Mehta, A. K., Kadel, S., Townsend, M. G., Oliwa, M. & Guerriero, J. L. Macrophage biology and mechanisms of immune suppression in breast cancer. Front. Immunol. 12, 643771 (2021).
Lee, J.-W., Han, H.-K., Park, S. & Moon, E.-Y. Nonylphenol increases tumor formation and growth by suppressing gender-independent lymphocyte proliferation and macrophage activation. Environ. Toxicol. 32, 1679–1687 (2017).
Lee-Sarwar, K. et al. Prenatal and early-life triclosan and paraben exposure and allergic outcomes. J. Allergy Clin. Immunol. 142, 269–278.e15 (2018).
Ait Bamai, Y. et al. Exposure to house dust phthalates in relation to asthma and allergies in both children and adults. Sci. Total Environ. 485–486, 153–163 (2014).
Quirós-Alcalá, L., Hansel, N. N., McCormack, M. C. & Matsui, E. C. Paraben exposures and asthma-related outcomes among children from the US general population. J. Allergy Clin. Immunol. 143, 948–956.e4 (2019).
Buckley, J. P. et al. Associations of prenatal environmental phenol and phthalate biomarkers with respiratory and allergic diseases among children aged 6 and 7 years. Environ. Int. 115, 79–88 (2018).
Lee, M. H. et al. Enhanced interleukin-4 production in CD4 + T cells and elevated immunoglobulin E levels in antigen-primed mice by bisphenol A and nonylphenol, endocrine disruptors: involvement of nuclear factor-AT and Ca2 + . Immunology 109, 76–86 (2003).
Iwata, M., Eshima, Y., Kagechika, H. & Miyaura, H. The endocrine disruptors nonylphenol and octylphenol exert direct effects on T cells to suppress Th1 development and enhance Th2 development. Immunol. Lett. 94, 135–139 (2004).
Little, A. C. et al. IL-4/IL-13 stimulated macrophages enhance creast cancer invasion via rho-GTPase regulation of synergistic VEGF/CCL-18 signaling. Front. Oncol. 9, 456 (2019).
Shiao, S. L. et al. TH2-polarized CD4( + ) T cells and macrophages limit efficacy of radiotherapy. Cancer Immunol. Res. 3, 518–525 (2015).
Hong, C.-C., Shimomura-Shimizu, M., Muroi, M. & Tanamoto, K. Effect of endocrine disrupting chemicals on lipopolysaccharide-induced tumor necrosis factor-alpha and nitric oxide production by mouse macrophages. Biol. Pharm. Bull. 27, 1136–1139 (2004).
Papalou, O., Kandaraki, E. A., Papadakis, G. & Diamanti-Kandarakis, E. Endocrine disrupting chemicals: an occult mediator of metabolic disease. Front. Endocrinol. 10, 112 (2019).
Tang-Péronard, J. L., Andersen, H. R., Jensen, T. K. & Heitmann, B. L. Endocrine-disrupting chemicals and obesity development in humans: a review. Obes. Rev. J. Int. Assoc. Study Obes. 12, 622–636 (2011).
Gladen, B. C., Ragan, N. B. & Rogan, W. J. Pubertal growth and development and prenatal and lactational exposure to polychlorinated biphenyls and dichlorodiphenyl dichloroethene. J. Pediatr. 136, 490–496 (2000).
Masuno, H. et al. Effect of 4-nonylphenol on cell proliferation and adipocyte formation in cultures of fully differentiated 3T3-L1 cells. Toxicol. Sci. J. Soc. Toxicol. 75, 314–320 (2003).
Sakurai, K. et al. Bisphenol A affects glucose transport in mouse 3T3-F442A adipocytes. Br. J. Pharmacol. 141, 209–214 (2004).
Alonso-Magdalena, P. et al. Low doses of bisphenol A and diethylstilbestrol impair ca2+ signals in pancreatic α-cells through a nonclassical membrane estrogen receptor within intact islets of langerhans. Environ. Health Perspect. 113, 969–977 (2005).
Le Magueresse-Battistoni, B. A. diposeT. issue and endocrine-disrupting chemicals: does sex matter?. Int. J. Environ. Res. Public. Health 17, 9403 (2020).
Key, T. J. et al. Body mass index, serum sex hormones, and breast cancer risk in postmenopausal women. J. Natl Cancer Inst. 95, 1218–1226 (2003).
Eliassen, A. H. et al. Endogenous steroid hormone concentrations and risk of breast cancer among premenopausal women. J. Natl. Cancer Inst. 98, 1406–1415 (2006).
Walker, B. E., Haven, M. I. Intensity of multigenerational carcinogenesis from diethylstilbestrol in mice. Carcinogenesis 18, 791–793 (1997).
Titus, L. et al. Reproductive and hormone-related outcomes in women whose mothers were exposed in utero to diethylstilbestrol (DES): a report from the us national cancer institute des third generation study. Reprod. Toxicol. Elmsford N. 84, 32–38 (2019).
Silk, K. J. & Totzkay, D. The Breast Cancer and Environment Research Program.https://epi.grants.cancer.gov/bcerp/(2017).
Kooijman, M. N. et al. The generation R study: design and cohort update 2017. Eur. J. Epidemiol. 31, 1243–1264 (2016).
Rauner, G. et al. Breast tissue regeneration is driven by cell-matrix interactions coordinating multi-lineage stem cell differentiation through DDR1. Nat. Commun. 12, 7116 (2021).
Rauner, G. et al. Single-cell organogenesis captures complex breast tissue formation in three dimensions. Development 152, dev204813 (2025).
Acknowledgements
This work was funded by Find The Cause Breast Cancer Foundation.
Author information
Authors and Affiliations
Contributions
M.P., N.T., M.S. and C.K. wrote the main manuscript text and C.K. prepared figures 1–2. All authors reviewed the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Parrish, M., Traugh, N., Seraj, M. et al. Field cancerization, accelerated aging, and immunosuppression: the rapid rise of hormone-sensitive and early-onset breast cancer. npj Breast Cancer 11, 128 (2025). https://doi.org/10.1038/s41523-025-00840-w
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41523-025-00840-w
