
Abstract
Spin excitations play a fundamental role in understanding magnetic properties of materials, and have significant technological implications for magnonic devices. However, accurately modeling these in transition-metal and rare-earth compounds remains a formidable challenge. Here, we present a fully first-principles approach for calculating spin-wave spectra based on time-dependent (TD) density-functional perturbation theory (DFPT), using nonempirical Hubbard functionals. This approach is implemented in a general noncollinear formulation, enabling the study of magnons in both collinear and noncollinear magnetic systems. Unlike methods that rely on empirical Hubbard U parameters to describe the ground state, and Heisenberg Hamiltonians for describing magnetic excitations, the methodology developed here probes directly the dynamical spin susceptibility (efficiently evaluated with TDDFPT throught the Liouville-Lanczos approach), and treats the linear variation of the Hubbard augmentation (in itself calculated non-empirically) in full at a self-consistent level. Furthermore, the method satisfies the Goldstone condition without requiring empirical rescaling of the exchange-correlation kernel or explicit enforcement of sum rules, in contrast to existing state-of-the-art techniques. We benchmark the novel computational scheme on prototypical transition-metal monoxides NiO and MnO, showing remarkable agreement with experiments and highlighting the fundamental role of these newly implemented Hubbard corrections. The method holds great promise for describing collective spin excitations in complex materials containing localized electronic states.
Similar content being viewed by others
Introduction
In recent years, several intriguing research directions in spin excitations have attracted much attention; notably, these include magnons in two-dimensional materials1,2,3 and in altermagnets4,5,6,7, and the coupling of magnons with other quasiparticles like excitons8,9, phonons10,11,12,13, and plasmons14,15,16. These phenomena often occur in complex materials containing magnetic transition-metal and/or rare-earth ions, and characterized as Mott-Hubbard or charge-transfer insulators17. While there have been many experimental breakthroughs in studying collective spin excitations18,19,20,21,22, theoretical and computational investigations remain challenging. To model the ground state of this class of materials, density-functional theory (DFT)23,24 is typically used, where particular attention must be given to the selection of the exchange-correlation (xc) functional, as it strongly influences the accuracy of the results. While standard local spin-density approximation (LSDA) and spin-polarized generalized-gradient approximation (σ-GGA) provide satisfactory results for itinerant magnetic metals, they are inaccurate for insulating transition-metal and rare-earth compounds due to strong self-interaction errors (SIEs) for partially filled and localized d and f electrons25,26. To address these challenges, more advanced functionals have been developed, among which Hubbard-corrected DFT functionals (DFT + U)27,28,29 stand out for their capability to correct SIEs30,31 and low computational cost. The value of Hubbard U is critical, and empirical tuning of U based on experimental results is a popular strategy; still, not only it requires accurate reference data, which are not always available, but its tranferability to properties that are often not fitted is debatable. To overcome these limitations, several first-principles approaches to compute U have been developed, including constrained DFT (cDFT)32,33,34,35,36,37,38, Hartree-Fock-based methods39,40,41,42,43,44, and the constrained random phase approximation (cRPA)45,46,47,48. Machine learning techniques for determining Hubbard parameters have also emerged in recent years49,50,51,52,53, which provide a fast and attractive route. The linear-response formulation of cDFT54 has gained widespread popularity due to its simplicity and accuracy, and its recent reformulation using density-functional perturbation theory (DFPT)55,56,57 has further broadened its success. The physical rationale behind the linear-response determination of U relies on the heuristical imposition of piecewise linearity of the total energy of the system as a function of the occupation of the target Hubbard manifold54. Investigations of various magnetic materials using U from DFPT have proven to be accurate and effective58,59,60,61,62,63,64,65,66, making it an appealing approach for the description of spin waves.
The theoretical modeling of spin waves (magnons) can in general be achieved using a wide array of methodologies. One of the most popular techniques involves model spin Hamiltonians, particularly the Heisenberg model, which relies on the adiabatic assumption that the time scales of magnons and electrons differ enough to allow the local electronic structure to adapt to the presence of magnons. The Heisenberg Hamiltonian is parametrized with interatomic exchange interactions J and other magnetic interaction parameters; e.g., single-ion anisotropy and/or Dzyaloshinskii-Moriya (DM) interactions67. These parameters are often obtained empirically by fitting them to experimental magnon dispersions. From a theoretical perspective, they can also be calculated from first principles using established approaches such as total energy differences68, spin-spiral energy dispersions based on the generalized Bloch theorem69,70,71, and the infinitesimal-rotations method based on the magnetic force theorem72,73,74,75. For instance, in the prototypical transition-metal oxides NiO and MnO, the J parameters have been computed using some of the aforementioned methods on top of DFT calculations using different xc functionals and corrective methods to describe the ground state. These approaches include self-interaction correction methods76,77,78, hybrid functionals78, and DFT+U with either empirical or ab initio U values71,79,80. However, the J parameters exhibit strong sensitivity to the value of the Hubbard U, making comparisons across different studies and with experimental results challenging.
Once the Heisenberg Hamiltonian parametrization is set, magnon dispersions can be determined using linear spin-wave theory (LSWT)81,82,83. Mapping experimental magnon dispersions to lattice spin models has proven to be very effective, offering a valuable tool for investigating complex systems, particularly magnetic surfaces84 and skyrmions85. However, using Heisenberg Hamiltonians often requires prior knowledge of the specific magnetic interactions to include, which can be problematic if no experimental data are available. Moreover, Heisenberg Hamiltonians fail to account for the effects stemming from the low-frequency Stoner excitations of metals, which lead to Landau damping86 of magnons, and the application of the Heisenberg model to itinerant metallic magnets is in itself questionable. Additionally, in complex systems, the large number of J parameters and weak DM interactions can introduce intricacies in the determination of these quantities. Furthermore, modeling chiral magnons in altermagnets using Heisenberg Hamiltonians is particularly challenging, as it demands high-resolution accuracy for anisotropic splittings of exchange interactions between distant neighbors, such as the splitting of J parameters for the 7th nearest neighbors in MnF287 or the 10th nearest neighbors in MnTe88. Therefore, a first-principles approach for modeling magnons that is independent of underlying spin models is highly desirable.
An alternative approach for modeling spin waves is to calculate directly the electronic response to an external magnetic field perturbation, by evaluating explicitly the spin-spin susceptibility tensor. Two popular strategies to tackle this task are time-dependent density-functional theory (TDDFT)89 and many-body perturbation theory (MBPT)90. TDDFT equations are typically solved in the linear-response regime in the frequency domain, assuming a small external magnetic field perturbation, and can be addressed using Dyson91,92,93,94,95,96,97,98,99,100, Sternheimer101,102,103, or Liouville-Lanczos (LL)104 approaches. The strong perturbation regime can be accessed by solving TDDFT equations using real-time propagation, which enables the modeling of ultrafast phenomena105. MBPT techniques, consisting in the solution of the Bethe-Salpeter equation on top of the LSDA or GW ground state, have been applied for modeling magnons8,106,107,108. While MBPT is known to provide a more accurate description of absorption spectra of solids than TDDFT with adiabatic LSDA (ALSDA), it involves higher computational costs. However, for magnons a comprehensive comparison of the accuracy and computational costs of MBPT and TDDFT across a wide range of materials has not yet been conducted (previous studies have primarily focused on elementary ferromagnets such as Fe, Ni, and Co). Additionally, most MBPT and TDDFT implementations suffer from violations of the Goldstone condition (see, e.g., refs. 98,109). Improved versions of TDDFT embodying more advanced xc functionals are attractive because they could deliver accurate magnon predictions while preserving moderate computational costs. In this context, extending TDDFT to incorporate Hubbard U corrections has proven effective for absorption spectroscopy110,111,112. However, this extension113 to the case of magnons has only been explored in a few works, all using empirical U: one when solving the Sternheimer equation with a finite-difference scheme103, and another by solving the Dyson equation with an additional empirical rescaling of the xc kernel to enforce the Goldstone condition114,115, making these approaches not being entirely ab initio. Instead, a fully first-principles Hubbard approach is highly desirable, with U computed using one of the aforementioned methods and treated self-consistently and efficiently when solving the TDDFT equations, also satisfying the Goldstone theorem.
Here, we propose a first-principles methodology for the evaluation of spin-fluctuation spectra which is based on time-dependent density-functional perturbation theory (TDDFPT)89,116 with nonempirical Hubbard functionals57 in the LL scheme117 and in a general noncollinear formulation. The key features of the novel approach are: (i) the Hubbard U parameter is not arbitrarily adjusted, but is calculated using DFPT54,55,118 simultaneously optimizing also the crystal structure in a well-defined self-consistent protocol56,119, (ii) the approach probes explicitly the dynamical spin susceptibility, that is linked to the experimentally measurable double-differential cross section, thus providing a direct comparison with experiments104, (iii) the approach is implemented in a general noncollinear formulation, thus allowing to study systems with collinear and noncollinear ground states, and (iv) the Goldstone theorem is satisfied without the need in empirical rescaling of the xc kernel114,115 or explicit enforcement of sum rules92,93, in contrast to methods based on the solution of the Dyson equation. Besides avoiding any reference to the electronic empty states, as routinely done in DFPT118,120, the greatest advantage of the LL approach for step (ii) relies in the fact that a single linear-response calculation enables the evaluation of a column of the spin susceptibility \({\chi }_{\alpha {\alpha }^{{\prime} }}({\boldsymbol{q}},\omega )\) at a large number frequencies in an inexpensive way as a postprocessing step. This is a desirable feature—especially when the magnon spectrum of a material is not known a priori—because it allows a facile identification of the spin excitations along the frequency axis, without the need of scanning several values of ω, which instead would be required in the Sternheimer and Dyson approaches91,101. The LL method was already successfully applied to optical absorption spectroscopy117,121, electron-energy loss spectroscopy122, and inelastic neutron scattering spectroscopy104 (although limited to standard xc functionals), and it includes the self-consistent readjustment of the charge and magnetization densities. In this work, we explicitly account for the first-order variation of the Hubbard potential, similarly to the static phonon DFPT implementation59,123. However, in the present noncollinear dynamical case, we demonstrate that the linear response equation that is antiresonant with the frequency ω exhibits a reversal of the Hubbard magnetization—i.e., the magnetization projected onto the localized (3d or 4f) target manifold. Ultimately, the method proposed here works directly with the linearization of the Kohn-Sham (KS) Bloch states in reciprocal space, and thus it bypasses intermediate post-processing steps like Wannierizations75,91,124, improving user-friendliness and automation.
We apply this novel approach to the study of the magnon dispersions of NiO and MnO. By consistently addressing the electronic, structural, and magnetic degrees of freedom, we achieve a highly accurate determination of spin waves, comparable to results from other advanced methods like DMFT125 or GW3,126 but at more moderate computational cost. As a byproduct of the calculations, we fit the magnon dispersions in order to extract the magnetic exchange parameters, which are then used to explain features of the magnon dispersions, and link them to the different magnitude of the rhombohedral distortions detected in the two materials investigated. These structural distortions are shown to be correctly captured by the present approach, thanks to the use of an iterative evaluation of the Hubbard parameters and structural optimizations, yielding the self-consistent Hubbard U and crystal structure.
Results
Time-dependent density-functional perturbation theory with Hubbard corrections
TDDFPT is a dynamical generalization of static DFPT118,127, where the external perturbation is decomposed into monochromatic dynamical components characterized by wave-vectors q and frequency ω104. Due to the frequency dependence, two first-order (Sternheimer) equations must be solved: one resonant with the perturbation frequency ω at wave-vector q, and one antiresonant with −ω at wave-vector −q. Different methods have been developed to handle these equations. For example, in ref. 102, the antiresonant equation was explicitly solved at (−q, −ω), while ref. 104 applied the time-reversal operator \(\hat{{\mathcal{T}}}=\iota {\sigma }_{y}\hat{K}\) (with \(\hat{K}\) being the complex-conjugate operator) to the antiresonat equation, which restores the positive sign of ω and q in the response quantities while reversing the sign of the magnetic xc potential Bxc. The operator \(\hat{{\mathcal{T}}}\) has also been used in static DFPT to calculate phonons128 and the Hubbard U parameter57; given its formal elegance, in this work we follow this methodology.
The core of this paper is an extension of the TDDFPT formalism from ref. 104 to noncollinear Hubbard functionals57. We present the general formalism for both metals and insulators using norm-conserving pseudopotentials, and we use Hartree atomic units. It is worth noting that extending the current formalism to ultrasoft pseudopotentials129 and the projector augmented wave (PAW) method130 is straightforward but rather involved131,132, and could be addressed in future work.
Ground state
In a noncollinear DFT+U scheme, the Hubbard occupation is a 2 × 2 matrix in spin space112,133,134. The implementation of this formalism supports Löwdin-orthogonalized pseudo-atomic orbitals, which define the localized Hubbard subspace of interest57. For spinorial quantities, we adopt the notation \(| {\Psi }_{i}\left.\right\rangle ={\sum }_{\sigma }| {\psi }_{i}^{\sigma },\sigma \left.\right\rangle\) and \(| {\Phi }_{m}^{I}\left.\right\rangle ={\sum }_{\sigma }| {\phi }_{m}^{I\sigma },\sigma \left.\right\rangle\) for the KS spinor and the Hubbard atomic states, respectively (see ref. 57 for more detailed definitions). Here, i is the collective index for quasimomentum and KS band indices i = nk, I is the atomic site index, m is the magnetic quantum number, and σ is the spin index. The Hubbard occupation matrix is defined as \({N}_{m{m}^{{\prime} }}^{I}={\sum }_{i}{\tilde{\theta }}_{i}\langle {\Psi }_{i}| {\hat{P}}_{{m}^{{\prime} }m}^{I}| {\Psi }_{i}\rangle\), where \({\tilde{\theta }}_{i}\) are the electronic occupancies—which equal to 0 and 1 for empty and occupied states at zero temperature, respectively, and have intermediate values for metals around the Fermi level—and \({\hat{P}}_{{m}^{{\prime} }m}^{I}=| {\Phi }_{{m}^{{\prime} }}^{I}\left.\right\rangle \left\langle \right.{\Phi }_{m}^{I}|\) is the projector on the Hubbard subspace. In spin-resolved components, the Hubbard occupation matrix reads:
In terms of these quantities, the Hubbard energy is given by57:
where the trace (Tr) is taken over the spin degrees of freedom.
As was mentioned earlier, in order to exploit the time-reversal operator \(\hat{{\mathcal{T}}}\) in the antiresonant Sternheimer equation, we need to determine how \({N}_{m{m}^{{\prime} }}^{I}\) and \({\hat{P}}_{m{m}^{{\prime} }}^{I}\) transform under time reversal. To this aim, we use the completeness property135 of the Pauli matrices σ = (σx, σy, σz): \({{\boldsymbol{\sigma }}}_{\zeta {\zeta }^{{\prime} }}\cdot {{\boldsymbol{\sigma }}}_{\xi {\xi }^{{\prime} }}=2{\delta }_{\zeta {\xi }^{{\prime} }}{\delta }_{{\zeta }^{{\prime} }\xi }-{\delta }_{\zeta {\zeta }^{{\prime} }}{\delta }_{{\xi }^{{\prime} }\xi }\), which allows to rewrite the occupation matrix (1) as57:
where \({n}_{m{m}^{{\prime} }}^{I}={\rm{Tr}}\,\left[{N}_{m{m}^{{\prime} }}^{I}\right]\) and \({{\boldsymbol{m}}}_{m{m}^{{\prime} }}^{I}={\sum }_{\sigma {\sigma }^{{\prime} }}{N}_{m{m}^{{\prime} }}^{I\sigma {\sigma }^{{\prime} }}{{\boldsymbol{\sigma }}}^{{\sigma }^{{\prime} }\sigma }\) are the Hubbard occupation (or charge) and magnetization, respectively. In Eq. (3) and in the following, for the sake of convenience, we use a notation that explicitly highlights the dependence on the magnetization (indicated as a superscript in square brackets), which will be necessary later when we reverse the sign of the magnetization. From this representation, using the properties \(\hat{{\mathcal{T}}}\,{\hat{{\mathcal{T}}}}^{\dagger }=\hat{I}\) (where \(\hat{I}\) is the identity operator) and \(\hat{{\mathcal{T}}}\,{\boldsymbol{\sigma }}\,{\hat{{\mathcal{T}}}}^{\dagger }=-{\boldsymbol{\sigma }}\), it follows that:
which is the transpose to Eq. (3) but with the opposite sign in front of the magnetization matrix \({{\boldsymbol{m}}}_{m{m}^{{\prime} }}^{I}\). From this expression we can derive how the noncollinear Hubbard potential transforms under the time-reversal operation. Let us start from its definition57:
where the notation \({\hat{V}}_{U}^{[{\boldsymbol{m}}]}\) has to be understood as a 2 × 2 matrix operator in the spin space. Next, using Eq. (4), by inverting the dummy indices m and \({m}^{{\prime} }\), and since the application of \(\hat{{\mathcal{T}}}\) on Φ is immaterial (due to the spin-averaging procedure57), the desired transformation is:
which is identical to Eq. (5) but with \({N}_{m{m}^{{\prime} }}^{I[{\boldsymbol{m}}]}\) being replaced by \({N}_{m{m}^{{\prime} }}^{I[-{\boldsymbol{m}}]}\), which has been discussed above.
Dynamical linear response
Let us consider now an external weak perturbation of the system due to the magnetic dynamical potential with a finite q and ω modulation. This potential is given by the interaction energy of the system of electrons with an external magnetic field104: \({V}_{{\rm{ext}}}^{[{{\boldsymbol{B}}}_{\omega {\boldsymbol{q}}}]}({\boldsymbol{q}},\omega )=-{\mu }_{{\rm{B}}}\,{\boldsymbol{\sigma }}\cdot {{\boldsymbol{B}}}_{\omega {\boldsymbol{q}}}\). Hereafter, we indicate with \({B}_{\omega {\boldsymbol{q}}}^{\alpha }\) the Cartesian α-component of the vector amplitude of the external magnetic field. Next, we use the Bloch sum expression for the Hubbard atomic-like states55,136:
where \({\nu }_{m{\boldsymbol{k}}}^{s}({\boldsymbol{r}}+{{\boldsymbol{R}}}_{l})={\nu }_{m{\boldsymbol{k}}}^{s}({\boldsymbol{r}})\equiv {\sum }_{\sigma }{\nu }_{m{\boldsymbol{k}}}^{s\sigma }({\boldsymbol{r}})| \sigma \left.\right\rangle\) is the lattice-periodic spinorial part, and Nk is the number of points in the k-grid. Here, we used the notation I ≡ ls (RI ≡ Rl + τs), so that s identifies the atomic position within the lth cell. Going over to the (ω, q) space and differentiating with respect to the Cartesian α-component of Bωq, we get the linearized Hubbard potential:
for which a transformation law similar to Eq. (6) applies:
We now exploit Bloch’s theorem for the KS spinors: \(| {\Psi }_{i}\left.\right\rangle \equiv | {\Psi }_{n{\boldsymbol{k}}}\left.\right\rangle =\frac{{e}^{\iota {\boldsymbol{k}}\cdot {\boldsymbol{r}}}}{{\sqrt{N}}_{{\boldsymbol{k}}}}| {u}_{n{\boldsymbol{k}}}\left.\right\rangle\). In terms of the lattice-periodic spinorial part \({u}_{n{\boldsymbol{k}}}({\boldsymbol{r}}+{{\boldsymbol{R}}}_{l})={u}_{n{\boldsymbol{k}}}({\boldsymbol{r}})\equiv {\sum }_{\sigma }{u}_{n{\boldsymbol{k}}}^{\sigma }({\boldsymbol{r}})| \sigma \left.\right\rangle\), the first-order response Hubbard occupation matrix in Eq. (8) is written in terms of a first-order standard and time-reversed response KS wavefunctions:
where the scalar products between the lattice-periodic parts of the spinors are summed over the spin components: 〈u∣ν〉 ≡ ∑σ〈uσ∣νσ〉. Equation (10) is valid for finite q, while for q = 0 there is an extra term for metallic systems proportional to the derivative of the occupations \(\tilde{\theta }\)56,118,137. The implementation of TDDFPT+U does not currently support the case q = 0; hence, this term is omitted. Also, in Eq. (10) the prefactor \({\tilde{\theta }}_{n{\boldsymbol{k}}}\) does not occur due to the definition of the response KS wavefunctions for metallic systems137. Thanks to the relations:
it is readily seen that the time-reversed part of Eq. (10) has an inverted sign for the Hubbard magnetization m, similarly to what happens for the induced spin-resolved charge density128 (in analogy to Eqs. (26) and (27) of ref. 104). Finally, in Eq. (10) the resonant \({\Delta }_{\omega {\boldsymbol{q}}}^{\alpha }{u}_{n{\boldsymbol{k}}}\) and the time-reversed antiresonant \(\hat{{\mathcal{T}}}{\Delta }_{-\omega -{\boldsymbol{q}}}^{\alpha }{u}_{n-{\boldsymbol{k}}}\) wavefunctions can be obtained by solving the two coupled Sternheimer equations, which represent the core methodological development of this work:
where \({\hat{{\mathcal{P}}}}_{{\boldsymbol{k}}+{\boldsymbol{q}}}\) and \({\hat{\Pi }}_{{\boldsymbol{k}}+{\boldsymbol{q}}}=\hat{{\mathcal{T}}}{\hat{{\mathcal{P}}}}_{-{\boldsymbol{k}}-{\boldsymbol{q}}}{\hat{{\mathcal{T}}}}^{\dagger }\) are the standard and time-reversed projectors onto the empty electronic states manifold, which have a more complex expression for metallic systems104,118,137. Here, \({\hat{H}}_{{\boldsymbol{k}}+{\boldsymbol{q}}}^{[{{\boldsymbol{B}}}_{{\rm{xc}}}]}\) is the ground-state Hamiltonian defined in Eq. (21) in ref. 104, and \({\hat{H}}_{{\boldsymbol{k}}+{\boldsymbol{q}}}^{[-{{\boldsymbol{B}}}_{{\rm{xc}}}]}\) is its analog with the reversed sign of Bxc, while ϵnk and ϵn−k are the ground-state KS energies. This Hamiltonian does not include the ground-state lattice-periodic Hubbard potential \({\hat{V}}_{U,{\boldsymbol{k}}+{\boldsymbol{q}}}^{[{\boldsymbol{m}}]}\), which instead appears as a separate term in the equations above [see its definition in Eq. (A15) in ref. 55], and \({\hat{V}}_{U,{\boldsymbol{k}}+{\boldsymbol{q}}}^{[-{\boldsymbol{m}}]}\) is its analog with the reversed sign for the Hubbard magnetization m. Finally, the response Hartree and xc (Hxc), response Hubbard, and external potentials appear on the right-hand side of the equations above with the specified signs of Bxc, m, and Bωq. Equations (11) and (12) differ from analogs equations in ref. 104 by the presence of the ground-state and response Hubbard potentials tuned by the magnitude of the Hubbard U parameter. In addition, the ground-state KS energies, wavefunctions, and spin-resolved charge density are obtained from the DFT+U ground state. Therefore, by solving Eqs. (11) and (12) self-consistently it is possible to obtain magnon energies including the Hubbard corrections with full internal consistency.
In principle, the dynamical Sternheimer equations (11) and (12) can be solved at each frequency ω; this provides the response KS wavefunctions \({\Delta }_{\omega {\boldsymbol{q}}}^{\alpha }{u}_{n{\boldsymbol{k}}}\) and the corresponding time-reversed ones \(\hat{{\mathcal{T}}}{\Delta }_{-\omega -{\boldsymbol{q}}}^{\alpha }{u}_{n-{\boldsymbol{k}}}\). The evaluation of these quantities gives access to the dynamical response spin-charge density matrix operator, defined as:
The knowledge of this latter gives a complete description of the magnetic linear response of the system to a magnetic external perturbation, inasmuch as it delivers the spin susceptibility tensor through:
This dynamical Sternheimer approach was successfully employed in refs. 101,102 (without Hubbard corrections) to calculate magnons in elemental itinerant metallic magnets and in the context of lattice-dynamical properties138,139,140. However, the main disadvantage of this approach is its high computational cost coming from the need to solve these equations self-consistently for each value of the frequency ω. To avoid this drawback, we employ the LL approach104,116, that enables the determination of the target column of the spin susceptibility tensor along the ω axis at once with a single linear-response calculation. The technical details concerning the implementation of noncollinear Hubbard functionals within this methodology can be found in the “Methods” section.
Applications
In this section, we present the application of the TDDFPT+U implementation using the LL approach to the transition-metal monoxides NiO and MnO. We first discuss the structural and electronic properties of these materials using LSDA and LSDA+U, comparing them with experimental data. Next, we present the calculated magnon dispersions using TDDFPT (within ALSDA) and TDDFPT+U (within ALSDA+U), and compare these results with experimental estimates. Finally, we extract the Heisenberg exchange interaction parameters by fitting the calculated magnon spectrum and compare them with experimental values. Both NiO and MnO are widely investigated antiferromagnetic (AFM) type II insulators. They crystallize in a rocksalt-type structure in the paramagnetic phase. Below their respective Néel temperatures, 524 K for NiO141 and 120 K for MnO142, these materials exhibit a rhombohedral distortion along the [111] direction of the face-centered cubic (fcc) lattice.
We start our analysis by determining the electronic and crystal structure of the ground state of NiO and MnO. The U parameter employed for the Hubbard augmentation is calculated using linear-response theory54:
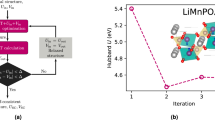
where \({n}^{I}\equiv {\sum }_{m}{\rm{Tr}}\,({N}_{mm}^{I})\) and \({n}_{0}^{I}\equiv {\sum }_{m}{\rm{Tr}}\,({N}_{0,mm}^{I})\) are the interacting and noninteracting response atomic occupations, respectively, which are decomposed into monochromatic components according to DFPT55,57: \(d{n}^{I}/d{\lambda }_{{I}^{{\prime} }}=\frac{1}{{N}_{{\boldsymbol{q}}}}\mathop{\sum }\nolimits_{{\boldsymbol{q}}}^{{N}_{{\boldsymbol{q}}}}{e}^{\iota {\boldsymbol{q}}\cdot ({{\boldsymbol{R}}}_{l}-{{\boldsymbol{R}}}_{{l}^{{\prime} }})}{\Delta }_{{\boldsymbol{q}}}^{{s}^{{\prime} }}{n}^{s}\) (detailed expression can be found in refs. 55,56,57). In Eq. (15), the Hubbard U parameter is defined by requiring that the second derivative of the DFT-LSDA total energy with respect to the occupation of the Hubbard manifold is zero. This condition eliminates the unphysical curvature of the total energy caused by SIEs, thereby ensuring the piecewise linearity of the LSDA+U total energy54. Thanks to the collinearity of the AFM ground state, and the neglect of the spin-orbit coupling due to the lightness of the elements, it is safely possible to restrict such a calculation to the collinear case, thus saving substantial computational effort. In order to simultaneously optimize the Hubbard parameters and the crystal structure, we employed the workflow proposed in refs. 56,119 and depicted in Fig. 1. This latter alternates variable-cell structural relaxations and determination of the U parameter until convergence is achieved. We obtained self-consistent U values of 6.26 and 4.29 eV for Ni-3d and Mn-3d states in NiO and MnO, respectively. Finally, these values are used to compute the magnon dispersions using TDDFPT+U.

Uin, Uout, and USC are the input, output, and self-consistent Hubbard parameters, respectively, while Δ is the convergence threshold.
Table 1 summarizes the equilibrium rhombohedral lattice parameter (a), rhombohedral angle (ϑ), magnetic moment (∣m∣), and band gap (Eg) as computed using LSDA, LSDA+U, and as measured in experiments (see also Fig. 4b). The experimental values for a and ϑ are derived from the cubic lattice143,144. We are not aware of any direct experimental reports of the rhombohedral lattice parameters for these materials, possibly because the rhombohedral distortions are small and hard to resolve experimentally. Therefore, the comparison of our theoretical rhombohedral lattice parameters with the experimental reference values should be considered somewhat peripheral. We find that the Hubbard correction systematically improves over LSDA. Still, non-negligible discrepancies are present even in the LSDA + U approach, which are to a large extent due to the limitation of the base xc LSDA functional that is known to cause excessive binding in crystal structures. In Sec. S1 of the supplemental information (SI), we show how the structural properties of the two systems change when the base xc functional is replaced with spin-polarized PBE145 or PBEsol146, combined with their respective self-consistent U values computed via DFPT. We find that PBE+U slightly overestimates the lattice parameter a, while PBEsol+U slightly underestimates it, with only minor variations in α. Overall, PBEsol+U provides more accurate predictions of the crystal structure than LSDA+U. Since our TDDFPT+U implementation currently does not support σ-GGA functionals, the remainder of this study focuses exclusively on LSDA+U. Concerning Eg, although its value generally improves when Hubbard corrections are included (see e.g., ref. 147), an underestimation of this quantity is to be expected, inasmuch as DFT + U is a theoretical framework that mainly corrects total energies. More precisely, when the band edges have the same orbital character of the Hubbard projectors, reasonable band gaps can be obtained with Hubbard-corrected functionals44,148,149. However, in the general case, for more accurate evaluations of spectral properties, methods like GW150,151,152, hybrid functionals153,154,155 or Koopmans functionals156,157,158 may be more appropriate.
Going over to the calculation of magnons, we recall that in the first Born approximation, there is a relation between the experimentally detectable double-differential cross section d2σ/(dΩdω) (measuring the scattering of neutrons) and the spin susceptibility tensor χ(q, ω) which is given by: \({d}^{2}\sigma /(d\Omega d\omega )=-\frac{{g}_{{\rm{n}}}^{2}}{4\pi }\frac{{k}_{{\rm{f}}}}{{k}_{{\rm{i}}}}\,{\mathcal{S}}({\boldsymbol{q}},\omega )\)104,159,160, where gn is the neutron g-factor, ki and kf are the initial and final wavevectors of the scattered neutrons, and
where \({T}_{\alpha {\alpha }^{{\prime} }}({\boldsymbol{q}})={\delta }_{\alpha {\alpha }^{{\prime} }}-{q}_{\alpha }{q}_{{\alpha }^{{\prime} }}/{q}^{2}\) is the projector to a plane transverse to the transferred momentum q, and q = ∣q∣. The poles of \({\mathcal{S}}({\boldsymbol{q}},\omega )\) occur at the frequencies ωm(q) of magnons and Stoner excitations. To illustrate how the determination of the magnon spectrum is carried out, we report in Fig. 2 an example of the calculated \({\mathcal{S}}({\boldsymbol{q}},\omega )\) for different values of the transferred momenta q for the magnon branch of NiO along the [111] direction. The sharp resonances in \({\mathcal{S}}({\boldsymbol{q}},\omega )\) indicate the frequency positions of the magnetic excitation of the system, which draw the spin-wave dispersions in the (ω, q) plane.

Calculated dynamical spin structure factor from Eq. (16) (in cyan color) as a function of the frequency ω (in meV) at several values of the transferred momentum q along the [111] direction (in units of 2π/a, where a is the rhombohedral lattice parameter obtained from the distorted cell within LSDA+U). Orange crosses mark the exact position of magnon peaks, and the green dashed line is a guide for the eye to highlight the magnon dispersion in the (ω, q) plane.
In Fig. 3, we show the calculated magnon spectra for NiO and MnO with and without the Hubbard correction, and the comparison with experimental measurements161,162. Plain TDDFPT using ALSDA significantly overestimates the magnon energies with respect to experiments for both NiO and MnO. This aligns with previous LSDA-based theoretical studies125,126. On the contrary, the effect of the Hubbard augmentation is substantial, and ALSDA+U significantly improves agreement with experiments, not only for the amplitude of the magnon dispersion, but also for their curvature. This outcome is remarkable since the TDDFPT+U calculations are fully first-principles and do not rely on any empirical parametrization for the lattice parameters, U value, or type and/or strength of magnetic interactions. For MnO, we remark that the inclusion of the rhombohedral distortion is crucial in order to obtain a nonzero magnon energy at the M point71,77. For NiO instead the rhombohedral distortion is much smaller, resulting in an almost vanishing magnon energy at q = M as compared to the magnon bandwidth. We observe that it is important to account for the rhombohedral distortion within the selection of the q–path across the Brillouin zone (BZ): the Γ−M high-symmetry direction undergoes a small rotation, while only the length along the Γ−X line is slightly modified. To do this, we specified the coordinates of the X and M points in the crystal framework (i.e., in the basis of the reciprocal lattice vectors of the BZ) and then transformed them to the Cartesian framework.

a NiO and b MnO computed using TDDFPT (gray dots), TDDFPT+U (cyan dots), and as measured in experiments (olive dots)161,162. Dashed lines along Γ−M and Γ−X are fit using Eq. (18) with the parameters \({J}_{1}^{+}\), \({J}_{1}^{-}\), and J2. The values of the magnetic exchange parameters resulting from the fit are summarized in Table 2.
The magnon energy at q → 0 requires special attention due to the Goldstone theorem, which dictates that the acoustic magnon energy must vanish in the absence of spin-orbit coupling163,164. However, in practice, numerical calculations often violate this condition due to approximations used to describe the ground and excited states, such as differing k-point grids or basis sets91,92,93,94,98,109. In contrast, our TDDFPT+U implementation based on the LL approach satisfies the Goldstone theorem with high accuracy and correctly reproduces the long-wavelength limit, as previously demonstrated for standard TDDFPT with ALSDA165. Since our current implementation does not support the exact q = 0 limit, we perform calculations using very small but finite q values near 0. As shown in Sec. S3 of the SI, we find that the magnon energy indeed vanishes as q → 0 in the absence of spin-orbit coupling. In contrast, the TDDFT+U implementation in refs. 114,115, which solves the Dyson equation, employs the so-called λALSDA+U, where λ is an empirical rescaling factor applied to the xc kernel to enforce the Goldstone condition. For antiferromagnetic insulators such as Cr2O3, a λ value of 1.4 was required to achieve a zero magnon energy at q = 0. However, when using empirical U values greater than 1 eV, unphysical behavior was observed in the acoustic magnon branch, including vanishing energy over extended portions of high-symmetry directions in the BZ114. This demonstrates the challenge of simultaneously satisfying the Goldstone condition and achieving magnon dispersions in close agreement with experimental results when using λALSDA+U. The approach in refs. 92,93, which enforces sum rules to satisfy the Goldstone condition, could potentially complement the TDDFT+U Dyson-based method of refs. 114,115 to address this issue. However, such corrections are unnecessary in our TDDFPT+U LL-based approach, which satisfies the Goldstone condition because the same computational parameters enter in both the DFT+U ground state and in the linear-response calculations. This highlights the robustness and accuracy of our novel method.
To gain further physical insights, we fit the magnon dispersions of Fig. 3 in order to have access to the Heisenberg exchange interaction parameters. For this analysis, we consider a Heisenberg model with nearest-neighbor (n.n.) and next-nearest-neighbor (n.n.n.) exchange interactions. The low-energy Hamiltonian reads162:
where the first sum is over nearest-neighbors with parallel spins (\({J}_{1}^{-}\)), the second sum is over nearest-neighbors with antiparallel spins (\({J}_{1}^{+}\)), and the third sum is over next-nearest-neighbors with antiparallel spins (J2) (see Fig. 4a). The notation \({J}_{1}^{-}\) and \({J}_{1}^{+}\), introduced by Lines and Jones166, highlights the difference in interaction strength for nearest neighbors due to rhombohedral distortions167. Without these distortions, \({J}_{1}^{-}={J}_{1}^{+}\). The Heisenberg Hamiltonian in Eq. (17) uses the same convention as in ref. 162, assuming that more remote exchange interactions are negligible. We also neglect DM and single-ion anisotropy magnetic interactions in this Hamiltonian. Here, i and j label magnetic atomic sites, and ∑i,j denotes the summation over pairs (i, j).

In the picture, the exchange interaction parameters \({J}_{1}^{+}\), \({J}_{1}^{-}\), and J2 introduced in Eq. (17), the lattice parameter a, and the angle ϑ quantifying the rhombohedral distortion are reported. Panel (a) shows the fcc cell, while (b) displays the rhombohedral cell employed in our first-principles calculations, and (c) is the Brillouin zone (BZ) corresponding to the rhombohedral cell. a1, a2, and a3 are the real-space primitive lattice vectors of the rhombohedral unit cell, while b1, b2, and b3 are the reciprocal-space primitive lattice vectors of the BZ. The high-symmetry points Γ, X, and M in the BZ are also highlighted.
From the Heisenberg Hamiltonian Eq. (17), using LSWT, it is possible to obtain an analytical expression for the magnon dispersion ωm(q) that explicitly depends on the exchange interaction parameters, which can then be used to fit the magnon spectrum (Fig. 3). The initial step involves transforming the Hamiltonian to represent each spin within its local reference frame, oriented along the z-direction. Next, a Holstein-Primakoff transformation is performed83, replacing spin operators with creation and annihilation bosonic operators similar to those in a harmonic oscillator. During this transformation, the Hamiltonian is linearized, retaining terms up to the second order in the Holstein-Primakoff bosonic quasiparticles. Finally, the equation of motion is solved by diagonalizing the dynamical matrix, and a Bogoliubov transformation ensures a diagonalizing basis that adheres to the usual commutation relations. The final stage delivers the analytical expression for the magnon frequencies, ωm(q). For a fcc lattice, this expression reads166:
where μ = 2S, i.e., it depends on the nominal magnetic moment S of the transition-metal ion (S = 1 for Ni2+ and S = 5/2 for Mn2+). The functions J11(q) and J12(q) are defined as161,162,168:
In Fig. 3, we show the fit of the magnon dispersions of NiO and MnO from TDDFPT and TDDFPT+U using Eq. (18) simultaneously along the Γ−M and Γ−X directions.Table 2 compares the theoretical Heisenberg exchange parameters obtained by fitting the TDDFPT and TDDFPT+U magnon dispersions in Fig. 3 with experimental values161,162. We extract the experimental Heisenberg exchange parameters by fitting the experimental magnon dispersions using Eq. (18), omitting single-ion anisotropy terms and using the S values given after Eq. (18). Our fitted values differ slightly from those in refs. 161,162 because the original works included single-ion anisotropy and used a slightly different S value for MnO. This refitting ensures a consistent comparison between theoretical and experimental exchange parameters under similar conditions and approximations169. As shown in Table 2, the \({J}_{1}^{+}\), \({J}_{1}^{-}\), and J2 parameters from TDDFPT within ALSDA are significantly overestimated compared to experimental values, while TDDFPT+U provides parameters much closer to experiments. Within TDDFPT, the theoretical exchange parameters deviate from the experimental ones by 300%, while within TDDFPT+U they deviate by 20%, drastically improving the accuracy of the predictions. It is worth stressing that the sign of these parameters is correct in both cases. Given the strong dependence of the parameters J on the structural properties, we observe once again that the residual disagreement obtained with the Hubbard corrections could be further lowered by using more advanced xc functionals146,170. It would be possible to bring the theoretical exchange parameters even closer to experimental values by slightly increasing the U parameter71, but this would introduce an adjustable parameter and, by doing so, the theory would not be unbiased anymore. This also highlights the strong sensitivity of the magnon dispersions and the corresponding J parameters to the value of the Hubbard U.
The difference between \({J}_{1}^{+}\) and \({J}_{1}^{-}\) serves as a useful measure of the impact of rhombohedral distortions on the magnon dispersions. We define \(\Delta {J}_{1}={J}_{1}^{+}-{J}_{1}^{-}\), and its relative strength compared to the average J1 parameter as ∣ΔJ1/J1∣. Table 2 shows that ΔJ1 and ∣ΔJ1/J1∣ are in much closer agreement with the experimental values when using TDDFPT+U as compared to TDDFPT. When ΔJ1 → 0, the magnon energy at the M point in the BZ vanishes, while increasing values of ΔJ1 lead to higher magnon energy at the M point, which can be verified using Eq. (18). This implies that ΔJ1 reflects the crystallographic inequivalence between two nearest-neighbor transition-metal ions, which is influenced by the rhombohedral distortion. Our first-principles calculations are in agreement with this picture, predicting a larger rhombohedral angle ϑ (indicating greater rhombohedral distortion) for MnO compared to NiO (see Table 1). Consistently, LSDA, which overestimates ϑ for both materials, displaying the largest values of both ΔJ1 and the magnon energy at the M point.
Discussion
We have presented a first-principles approach for calculating magnons based on time-dependent density-functional perturbation theory104 and the Liouville-Lanczos method augmenting the adiabatic exchange-correlation functional with nonempirical Hubbard corrections. This Hubbard-extended formulation of TDDFPT is fully ab initio, since the Hubbard U parameter is computed from first-principles using DFPT55,57, avoiding any empirical calibrations. Additionally, the dynamical spin susceptibility tensor is directly computed through linear-response theory, without assumptions about the type and strength of magnetic interactions, unlike spin models such as the Heisenberg Hamiltonian. The Hubbard U correction is included self-consistently when solving the ground-state DFT+U problem, and its linear response is included when solving the TDDFPT+U equations using the LL approach. We chose the LL approach over the Dyson or Sternheimer methods because it is computationally efficient and provides access to all frequencies at once, unlike the point-wise calculations required by the other methods. Similarly to the Sternheimer approach102, the LL approach avoids the computationally expensive summations over empty states104, commonly performed in static DFPT for phonons120. In all cases, the local spin density approximation (adiabatic in the time-dependent density-functional perturbation theory case) is used for the base exchange-correlation functional, then augmented with the Hubbard corrections. Crucially, the Goldstone theorem is satisfied within this computational framework.
To benchmark the TDDFPT+U formalism and ensure the correctness of its implementation, we applied it to the prototypical transition-metal monoxides NiO and MnO, including their rhombohedral lattice distortions. The computed magnon dispersions from TDDFPT+U show remarkable agreement with experimental data, unlike those from TDDFPT. Specifically, we accurately predict finite magnon energy at the M point in the BZ of MnO due to rhombohedral distortions, while this effect is almost negligible for NiO with respect to its magnon bandwidth, consistent with experimental observations. Using the Heisenberg Hamiltonian and the LSWT, we fit the magnon dispersions to extract the nearest-neighbor and next-nearest-neighbor Heisenberg interaction parameters. The parameters obtained from TDDFPT+U align well with experimental values, whereas those from TDDFPT are largely overestimated. A detailed comparison of Heisenberg exchange parameters from TDDFPT+U and those calculated via total energy differences or the infinitesimal-rotations method is of great interest and is presented in a separate study169.
Although the current implementation of TDDFPT+U using the LL approach yielded good results, it still has limitations. Indeed, this approach is based on the linear-response regime, meaning it cannot simulate ultrafast phenomena with strong external perturbations105. Moreover, when solving the dynamical TDDFPT+U equations, the response Hubbard potential is computed using the static Hubbard U parameter, neglecting its dynamical variations due to external perturbations. We believe that this is a reasonable approximation since the external perturbation is assumed to be weak. Investigating the effect of dynamical modulation of U on magnons could be interesting, as done in ref. 171 for studying high-harmonic generation. However, the U(ω) parameter cannot be treated within the LL approach because it does not allow for a frequency-dependent response potential. To explore U(ω), switching to the Dyson or Sternheimer approaches would be necessary. On a more technical level, the current implementation is limited to LSDA, with σ-GGA currently unsupported, and it works only with norm-conserving pseudopotentials. Extensions to ultrasoft pseudopotentials129 and the PAW method130 would be straightforward but they increase the computational complexity. In addition, the current implementation does not support symmetry, requiring the use of the full k point grid in the BZ. We plan to implement symmetries in future versions of the TDDFPT+U code, which will further reduce the computational cost. Finally, the current implementation runs only on central processing unit (CPU) architectures, and porting it to graphics processing unit (GPU) architectures would significantly boost the speed of magnon calculations172,173.
Finally, we discuss the outlook and future prospects. Our noncollinear TDDFPT+U implementation, based on ref. 104, supports spin-orbit coupling (SOC). This is particularly relevant for heavy elements such as e.g., rare earths, containing localized f electrons requiring Hubbard corrections. Moreover, incorporating SOC enables the study of its effect on magnons, e.g., through the magnon-phonon coupling, intrinsic damping, anisotropy and gaps in the magnon spectrum2,12. Thanks to the noncollinear extension of DFT+U and DFPT57, it is possible to evaluate the Hubbard U parameter within the noncollinear framework, fully including SOC and further employ it within TDDFPT+U. Moreover, the current TDDFPT+U implementation can be straighforwardly extended to incorporate inter-site Hubbard V corrections149, that have proven to be very accurate and effective for diverse materials and properties174,175,176,177,178,179,180,181. Another promising direction involves using TDDFPT+U in combination with MBPT182 to study the renormalization and damping of electronic band structures in antiferromagnetic insulators due to electron–magnon coupling108,183, with a particular focus on employing ab initio rather than empirical U values184. Additionally, the current TDDFPT+U implementation can be used for high-throughput calculations of magnons for hundreds or even thousands of materials using platforms like AiiDA185,186. This process can be further streamlined using equivariant neural networks to predict Hubbard parameters essentially at no cost, but with the accuracy close to that of DFPT52. We believe that the present TDDFPT+U extension opens the door to accurate modeling of magnons in complex transition-metal and rare-earth compounds, potentially leading to significant technological breakthroughs in spintronics and magnonics.
Methods
Liouville-Lanczos approach with Hubbard corrections
The LL approach aims to solve the quantum Liouville spinorial equation, which is equivalent to the coupled dynamical Sternheimer equations (11) and (12). This is done by linearizing the Liouville equation, and taking advantage of the batch representation116,117,122 to cast the equation in a matrix form that can be efficiently solved using the Lanczos algorithm187. The key advantage of using the LL approach over directly solving the dynamical Sternheimer equations is that the problem only needs to be solved once, regardless of frequency. The evaluation of the magnetic spectrum is then an inexpensive post-processing step121. The linearized quantum Liouville spinorial equation in the frequency domain reads104:
where \({\hat{\rho }}_{0}\) is the ground-state spin-charge density matrix operator, \(d{\hat{V}}_{{\rm{ext}}}/d{B}_{\omega {\boldsymbol{q}}}^{\alpha }\to d{\hat{V}}_{{\rm{ext}}}^{[\pm {{\boldsymbol{B}}}_{\omega {\boldsymbol{q}}}]}/d{B}_{\omega {\boldsymbol{q}}}^{\alpha }\) are the direct and reversed magnetic perturbations, and \({\hat{{\mathcal{L}}}}_{{\boldsymbol{q}}}\to {\hat{{\mathcal{L}}}}_{{\boldsymbol{q}}}^{\pm }\) is the Liouvillian superoperator, which action over a generic quantum-mechanical operator \(\hat{X}\) is defined as:
The first and third terms in the above equation are the noninteracting and interacting terms found in the standard TDDFPT formulation within ALSDA104. The second and fourth terms are new, referred to as the noninteracting and interacting Hubbard terms, which arise from the Hubbard U correction. These four terms also appear in the dynamical resonant and antiresonant Sternheimer equations (11) and (12) with the respective signs of Bxc and m.
To evaluate the spin susceptibility tensor using Eq. (14), we can use the solution of Eq. (20). For practical computation using the Lanczos algorithm, it is convenient to rewrite Eqs. (14) and (20) as a resolvent of the Liouvillian121:
where \(\left\langle \right.{{\mathsf{w}}}_{\alpha }| =\left\langle \right.\{{u}_{n{\boldsymbol{k}}}\}| {\mu }_{{\rm{B}}}{\sigma }_{\alpha }\) and \(\left\vert {{\mathsf{v}}}_{{\alpha }^{{\prime} }}\right\rangle =\left[\frac{d{\hat{V}}_{{\rm{ext}}}}{d{B}_{\omega {\boldsymbol{q}}}^{{\alpha }^{{\prime} }}},{\hat{\rho }}_{0}\right]| \{{u}_{n{\boldsymbol{k}}}\}\left.\right\rangle\), while {unk} denotes a set of the ground-state KS wavefunctions. Next, we introduce the dual basis \(\{\langle {{\mathsf{p}}}_{n}| ,| {{\mathsf{q}}}_{n}\rangle \}\) composed of “left” \(\left\langle \right.{{\mathsf{p}}}_{n}|\) and “right” \(| {{\mathsf{q}}}_{n}\left.\right\rangle\) Lanczos orthonormal vectors (\(\langle {{\mathsf{p}}}_{n}| {{\mathsf{q}}}_{m}\rangle ={\delta }_{n,m}\)) that tridiagonalizes the Liouvillian: \(\left\langle \right.{{\mathsf{p}}}_{n}| {\hat{{\mathcal{L}}}}_{{\boldsymbol{q}}}| {{\mathsf{q}}}_{m}\left.\right\rangle ={\alpha }_{n}\,{\delta }_{n,m}+{\beta }_{n}\,{\delta }_{n,m+1}+{\gamma }_{n}\,{\delta }_{n,m-1}\), with n and m being integer numbers (n ≥ 1, m ≥ 1). The Lanczos vectors are obtained by using the Lanczos recursive relations187:
where {αn, βn, γn} is a set of Lanczos coefficients that are defined as: \({\alpha }_{n}=\left\langle \right.{{\mathsf{p}}}_{n}| {\hat{{\mathcal{L}}}}_{{\boldsymbol{q}}}| {{\mathsf{q}}}_{n}\left.\right\rangle\), \({\beta }_{n+1}=\sqrt{\langle {{\mathsf{u}}}_{p}| {{\mathsf{u}}}_{q}\rangle }\), and \({\gamma }_{n+1}={\beta }_{n+1}{\rm{sign}}[\langle {{\mathsf{u}}}_{p}| {{\mathsf{u}}}_{q}\rangle ]\), where \(| {{\mathsf{u}}}_{q}\left.\right\rangle =| {{\mathsf{q}}}_{n+1}\left.\right\rangle {\beta }_{n+1}\) and \(| {{\mathsf{u}}}_{p}\left.\right\rangle =| {{\mathsf{p}}}_{n+1}\left.\right\rangle {\gamma }_{n+1}\)104. By setting \(| {{\mathsf{p}}}_{1}\left.\right\rangle =| {{\mathsf{q}}}_{1}\left.\right\rangle =| {{\mathsf{v}}}_{{\alpha }^{{\prime} }}\left.\right\rangle\), the resolvent of the Liouvillian in Eq. (22) can be expressed as a continued fraction117,131:
In practice, \({\chi }_{\alpha {\alpha }^{{\prime} }}({\boldsymbol{q}},\omega )\) must be converged with respect to the number of Lanczos iterations n when solving Eqs. (23) and (24) recursively.
We report in Sec. S2 in the SI the evolution of the average between the even and odd LL coefficients αn and βn along the Lanczos chain (the coefficient γn is essentially equal to βn and for conciseness we display only the latter). As it was reported in previous studies104,121,188, the αn coefficient is very small and oscillates around zero (when the batch rotation is performed, namely when time-reversal symmetry holds, αn = 0 by construction117), while βn is approximately equal to the half of the kinetic-energy cutoff in the wavefunction expansion (≈40 = 80/2 Ry). We mention that the number of LL iterations necessary to converge TDDFPT + U calculations is about 7000–8000, which is substantially smaller than the 16,000 iterations needed to converge TDDFPT. We attribute this difference to the fact that the + U correction widens the band gap (see Table 1). As a consequence, the energy of Stoner excitations is blue-shifted, and less electronic transitions contribute to the system’s response at low energies (where magnon excitations occur), stabilizing the convergence of the Lanczos chains.
Importantly, these equations are independent of the frequency ω, meaning that they need to be solved only once for each value of the transferred momentum q and each Cartesian α-component of the external magnetic field. The frequency only comes into play in the post-processing step, using the Lanczos coefficients {αn, βn, γn} to compute the spin susceptibility tensor according to Eq. (25). A small constant Lorentzian broadening η is added to the frequency ω → ω + iη to regularize the cases when the frequency of the perturbation resonates with electronic vertical transition processes in the system (see the left-hand sides of Eqs. (11) and (12)). Since only the transverse component of \({\chi }_{\alpha {\alpha }^{{\prime} }}({\boldsymbol{q}},\omega )\) is needed to compute magnons, just one or two Lanczos chains are sufficient in some systems, depending on the system’s symmetry and the direction of the ground-state magnetization.
Computational details
All calculations are performed using the Quantum ESPRESSO distribution172,189,190. The ground-state calculations are carried out with the PW code189 using LSDA191 as the base xc functional. Optimized norm-conserving scalar-relativistic pseudopotentials192 are taken from the PseudoDojo library193. We use a 80 Ry kinetic-energy cutoff for the plane-wave expansion of the KS wavefunctions and a 320 Ry cutoff for the charge density. The BZ is sampled with a Γ-centered 12 × 12 × 12 k-points grid. The spin-orbit coupling is neglected.
The Hubbard U parameters are computed using DFPT55,57 as implemented in the HP code194, with Löwdin-orthogonalized atomic orbitals for Hubbard projectors195. We employ uniform Γ-centered k- and q-point meshes of size 8 × 8 × 8 and 4 × 4 × 4, respectively, and use kinetic-energy cutoffs of 90 Ry for the KS wavefunctions and 360 Ry for the charge density, providing an accuracy for the Hubbard parameters of ~0.01 eV. The U parameters are computed iteratively in a self-consistent manner as described in refs. 56,119, which includes Hubbard forces and stresses in DFT+U structural optimizations196.
The magnon energies are computed using TDDFPT+U and the LL approach, as implemented in a modified version of the turboMagnon code165. We use ALSDA, both with and without Hubbard U. The TDDFPT and TDDFPT+U calculations are performed at their respective optimized rhombohedral lattice parameters reported in Table 1. The calculations employ the pseudo-Hermitian flavor of the Lanczos recursive algorithm2,12, which includes an extrapolation technique for the Lanczos coefficients121. A Lorentzian smearing with a broadening parameter of 0.5 meV is used to plot the magnetic excitation spectra. All calculations are performed without symmetries since these are not yet implemented.
Data availability
The data used to produce the results of this work is available in the Materials Cloud Archive at https://archive.materialscloud.org/record/2025.41. The turboMagnon code including Hubbard corrections is part of a customized version of Quantum ESPRESSO which will be made publicly available with the official future releases.
References
Avsar, A. et al. Colloquium: Spintronics in graphene and other two-dimensional materials. Rev. Mod. Phys. 92, 021003 (2020).
Gorni, T., Baseggio, O., Delugas, P., Timrov, I. & Baroni, S. First-principles study of the gap in the spin excitation spectrum of the CrI3 honeycomb ferromagnet. Phys. Rev. B 107, L220410 (2023).
Ke, L. & Katsnelson, M. I. Electron correlation effects on exchange interactions and spin excitations in 2D van der Waals materials. npj Comput. Mater. 7, 1–8 (2021).
Šmejkal, L. et al. Chiral magnons in altermagnetic RuO2. Phys. Rev. Lett. 131, 256703 (2023).
Cui, Q., Zeng, B., Cui, P., Yu, T. & Yang, H. Efficient spin Seebeck and spin Nernst effects of magnons in altermagnets. Phys. Rev. B 108, L180401 (2023).
Leenders, R. A., Afanasiev, D., Kimel, A. & Mikhaylovskiy, R. Canted spin order as a platform for ultrafast conversion of magnons. Nature 630, 335 (2024).
Costa, A. T., Henriques, J. & Fernández-Rossier, J. Giant spatial anisotropy of magnon lifetime in altermagnets. Preprint at https://arxiv.org/abs/2405.12896 (2024).
Olsen, T. Unified treatment of magnons and excitons in monolayer CrI3 from many-body perturbation theory. Phys. Rev. Lett. 127, 166402 (2021).
Bae, Y. J. et al. Exciton-coupled coherent magnons in a 2D semiconductor. Nature 609, 282–286 (2022).
Bistoni, O., Mauri, F. & Calandra, M. Intrinsic vibrational angular momentum from nonadiabatic effects in noncollinear magnetic molecules. Phys. Rev. Lett. 126, 225703 (2021).
Bonini, J. et al. Frequency splitting of chiral phonons from broken time-reversal symmetry in CrI3. Phys. Rev. Lett. 130, 086701 (2023).
Delugas, P., Baseggio, O., Timrov, I., Baroni, S. & Gorni, T. Magnon-phonon interactions enhance the gap at the Dirac point in the spin-wave spectra of CrI3 two-dimensional magnets. Phys. Rev. B 107, 214452 (2023).
Ren, S., Bonini, J., Stengel, M., Dreyer, C. E. & Vanderbilt, D. Adiabatic Dynamics of Coupled Spins and Phonons in Magnetic Insulators. Phys. Rev. X 14, 011041 (2024).
Ghosh, S., Menichetti, G., Katsnelson, M. & Polini, M. Plasmon-magnon interactions in two-dimensional honeycomb magnets. Phys. Rev. B 107, 195302 (2023).
Dyrdal, A., Qaiumzadeh, A., Brataas, A. & Barnaś, J. Magnon-plasmon hybridization mediated by spin-orbit interaction in magnetic materials. Phys. Rev. B 108, 045414 (2023).
Falch, V., Danon, J., Qaiumzadeh, A. & Brataas, A. Impact of spin torques and spin-pumping phenomena on magnon-plasmon polaritons in antiferromagnetic insulator-semiconductor heterostructures. Phys. Rev. B 109, 214436 (2024).
Imada, M., Fujimori, A. & Tokura, Y. Metal-insulator transitions. Rev. Mod. Phys. 70, 1039 (1998).
Tóth, S. et al. Electromagnon dispersion probed by inelastic X-ray scattering in LiCrO2. Nat. Commun. 7, 13547 (2016).
Pershoguba, S. et al. Dirac magnons in honeycomb ferromagnets. Phys. Rev. X 8, 011010 (2018).
Chen, L. et al. Topological spin excitations in honeycomb ferromagnet CrI3. Phys. Rev. X 8, 041028 (2018).
Scheie, A. et al. Spin waves and magnetic exchange Hamiltonian in CrSBr. Adv. Sci. 9, 2202467 (2022).
Huang, C. et al. Extreme terahertz magnon multiplication induced by resonant magnetic pulse pairs. Nat. Commun. 15, 3214 (2024).
Hohenberg, P. & Kohn, W. Inhomogeneous electron gas. Phys. Rev. 136, B864–B871 (1964).
Kohn, W. & Sham, L. J. Self-consistent equations including exchange and correlation effects. Phys. Rev. 140, A1133–A1138 (1965).
Perdew, J. & Zunger, A. Self-interaction correction to density-functional approximations for many-electron systems. Phys. Rev. B 23, 5048 (1981).
Mori-Sánchez, P., Cohen, A. & Yang, W. Many-electron self-interaction error in approximate density functionals. J. Chem. Phys. 125, 201102 (2006).
Anisimov, V., Zaanen, J. & Andersen, O. Band theory and Mott insulators: Hubbard U instead of Stoner I. Phys. Rev. B 44, 943 (1991).
Liechtenstein, A., Anisimov, V. & Zaanen, J. Density-functional theory and strong interactions: orbital ordering in Mott-Hubbard insulators. Phys. Rev. B 52, R5467 (1995).
Dudarev, S., Botton, G., Savrasov, S., Humphreys, C. & Sutton, A. Electron-energy-loss spectra and the structural stability of nickel oxide: an LSDA+U study. Phys. Rev. B 57, 1505 (1998).
Kulik, H., Cococcioni, M., Scherlis, D. & Marzari, N. Density functional theory in transition-metal chemistry: a self-consistent Hubbard U approach. Phys. Rev. Lett. 97, 103001 (2006).
Kulik, H. & Marzari, N. A self-consistent Hubbard U density-functional theory approach to the addition-elimination reactions of hydrocarbons on bare FeO+. J. Chem. Phys. 129, 134314 (2008).
Dederichs, P., Blügel, S., Zeller, R. & Akai, H. Ground states of constrained systems: application to cerium impurities. Phys. Rev. Lett. 53, 2512 (1984).
McMahan, A., Martin, R. & Satpathy, S. Calculated effective Hamiltonian for La2CuO4 and solution in the impurity Anderson approximation. Phys. Rev. B 38, 6650 (1988).
Gunnarsson, O., Andersen, O., Jepsen, O. & Zaanen, J. Density-functional calculation of the parameters in the Anderson model: Application to Mn in CdTe. Phys. Rev. B 39, 1708 (1989).
Hybertsen, M., Schlüter, M. & Christensen, N. Calculation of Coulomb-interaction parameters for La2CuO4 using a constrained-density-functional approach. Phys. Rev. B 39, 9028 (1989).
Gunnarsson, O. Calculation of parameters in model Hamiltonians. Phys. Rev. B 41, 514 (1990).
Pickett, W., Erwin, S. & Ethridge, E. Reformulation of the LDA+U method for a local-orbital basis. Phys. Rev. B 58, 1201 (1998).
Shishkin, M. & Sato, H. Self-consistent parametrization of DFT+U framework using linear response approach: application to evaluation of redox potentials of battery cathodes. Phys. Rev. B 93, 085135 (2016).
Mosey, N. & Carter, E. Ab initio evaluation of Coulomb and exchange parameters for DFT+U calculations. Phys. Rev. B 76, 155123 (2007).
Mosey, N., Liao, P. & Carter, E. Rotationally invariant ab initio evaluation of Coulomb and exchange parameters for DFT+U calculations. J. Chem. Phys. 129, 014103 (2008).
Andriotis, A., Sheetz, R. & Menon, M. LSDA+U method: a calculation of the U values at the Hartree-Fock level of approximation. Phys. Rev. B 81, 245103 (2010).
Agapito, L., Curtarolo, S. & Buongiorno Nardelli, M. Reformulation of DFT+U as a pseudohybrid Hubbard density functional for accelerated materials discovery. Phys. Rev. X 5, 011006 (2015).
Tancogne-Dejean, N. & Rubio, A. Parameter-free hybridlike functional based on an extended Hubbard model: DFT+U+V. Phys. Rev. B 102, 155117 (2020).
Lee, S.-H. & Son, Y.-W. First-principles approach with a pseudohybrid density functional for extended Hubbard interactions. Phys. Rev. Res. 2, 043410 (2020).
Springer, M. & Aryasetiawan, F. Frequency-dependent screened interaction in Ni within the random-phase approximation. Phys. Rev. B 57, 4364 (1998).
Kotani, T. Ab initio random-phase-approximation calculation of the frequency-dependent effective interaction between 3d electrons: Ni, Fe, and MnO. J. Phys.: Condens. Matter 12, 2413 (2000).
Aryasetiawan, F. et al. Frequency-dependent local interactions and low-energy effective models from electronic structure calculations. Phys. Rev. B 70, 195104 (2004).
Aryasetiawan, F., Karlsson, K., Jepsen, O. & Scönberger, U. Calculations of Hubbard U from first-principles. Phys. Rev. B 74, 125106 (2006).
Yu, M., Yang, S., Wu, C. & Marom, N. Machine learning the Hubbard U parameter in DFT+U using Bayesian optimization. npj Comput. Mater. 6, 180 (2020).
Yu, W. et al. Active learning the high-dimensional transferable Hubbard U and V parameters in the DFT+U+V scheme. J. Chem. Theory Comput. 19, 6425 (2023).
Cai, G. et al. Predicting structure-dependent Hubbard U parameters via machine learning. Mater. Futures 3, 025601 (2024).
Uhrin, M., Zadoks, A., Binci, L., Marzari, N. & Timrov, I. Machine learning Hubbard parameters with equivariant neural networks. npj Comput. Mater. 11, 19 (2025).
Das, R. BMach: a Bayesian machine for optimizing Hubbard U parameters in DFT+U with machine learning. Preprint at https://arxiv.org/abs/2407.20848 (2024).
Cococcioni, M. & de Gironcoli, S. Linear response approach to the calculation of the effective interaction parameters in the LDA + U method. Phys. Rev. B 71, 035105 (2005).
Timrov, I., Marzari, N. & Cococcioni, M. Hubbard parameters from density-functional perturbation theory. Phys. Rev. B 98, 085127 (2018).
Timrov, I., Marzari, N. & Cococcioni, M. Self-consistent Hubbard parameters from density-functional perturbation theory in the ultrasoft and projector-augmented wave formulations. Phys. Rev. B 103, 045141 (2021).
Binci, L. & Marzari, N. Noncollinear DFT + U and Hubbard parameters with fully relativistic ultrasoft pseudopotentials. Phys. Rev. B 108, 115157 (2023).
Ricca, C., Timrov, I., Cococcioni, M., Marzari, N. & Aschauer, U. Self-consistent site-dependent DFT+U study of stoichiometric and defective SrMnO_3. Phys. Rev. B 99, 094102 (2019).
Floris, A. et al. Hubbard-corrected density functional perturbation theory with ultrasoft pseudopotentials. Phys. Rev. B 101, 064305 (2020).
Zhou, J.-J. et al. Ab initio electron-phonon interactions in correlated electron systems. Phys. Rev. Lett. 127, 126404 (2021).
Grassano, D., Binci, L. & Marzari, N. Type-I antiferromagnetic Weyl semimetal InMnTi2. Phys. Rev. Res. 6, 013140 (2024).
Bonfà, P. et al. Magnetostriction-driven muon localisation in an antiferromagnetic oxide. Phys. Rev. Lett. 132, 046701 (2024).
Macke, E., Timrov, I., Marzari, N. & Ciacchi, L. C. Orbital-resolved dft+u for molecules and solids. J. Chem. Theory Comput. 20, 4824–4843 (2024).
Ponet, L., Di Lucente, E. & Marzari, N. The energy landscape of magnetic materials. npj Comput. Mater 10, 51 (2024).
Gelin, S. et al. Ternary oxides of s- and p-block metals for photocatalytic solar-to-hydrogen conversion. PRX Energy 3, 013007 (2024).
Chang, B. et al. First-principles electron-phonon interactions and polarons in the parent cuprate La2CuO4. Phys. Rev. Res. 7, L012073 (2025).
Szilva, A. et al. Quantitative theory of magnetic interactions in solids. Rev. Mod. Phys. 95, 035004 (2023).
Šabani, D., Bacaksiz, C. & Milošević, M. V. Ab initio methodology for magnetic exchange parameters: Generic four-state energy mapping onto a Heisenberg spin Hamiltonian. Phys. Rev. B 102, 014457 (2020).
Halilov, S. V., Eschrig, H., Perlov, A. Y. & Oppeneer, P. M. Adiabatic spin dynamics from spin-density-functional theory: Application to Fe, Co, and Ni. Phys. Rev. B 58, 293–302 (1998).
Gebauer, R. & Baroni, S. Magnons in real materials from density-functional theory. Phys. Rev. B 61, R6459–R6462 (2000).
Jacobsson, A., Sanyal, B., Ležaić, M. & Blügel, S. Exchange parameters and adiabatic magnon energies from spin-spiral calculations. Phys. Rev. B 88, 134427 (2013).
Liechtenstein, A. I., Katsnelson, M. I., Antropov, V. P. & Gubanov, V. A. Local spin density functional approach to the theory of exchange interactions in ferromagnetic metals and alloys. J. Magn. Magn. Mater. 67, 65–74 (1987).
Solovyev, I., Hamada, N. & Terakura, K. Crucial role of the lattice distortion in the magnetism of LaMnO3. Phys. Rev. Lett. 76, 4825–4828 (1996).
Pajda, M., Kudrnovský, J., Turek, I., Drchal, V. & Bruno, P. Ab initio calculations of exchange interactions, spin-wave stiffness constants, and Curie temperatures of Fe, Co, and Ni. Phys. Rev. B 64, 174402 (2001).
Korotin, D. M., Mazurenko, V. V., Anisimov, V. I. & Streltsov, S. V. Calculation of exchange constants of the Heisenberg model in plane-wave-based methods using the Green’s function approach. Phys. Rev. B 91, 224405 (2015).
Ködderitzsch, D. et al. Exchange interactions in NiO and at the NiO(100) surface. Phys. Rev. B 66, 064434 (2002).
Fischer, G. et al. Exchange coupling in transition metal monoxides: electronic structure calculations. Phys. Rev. B 80, 014408 (2009).
Archer, T. et al. Exchange interactions and magnetic phases of transition metal oxides: Benchmarking advanced ab initio methods. Phys. Rev. B 84, 115114 (2011).
Solovyev, I. & Terakura, K. Effective single-particle potentials for MnO in light of interatomic magnetic interactions: Existing theories and perspectives. Phys. Rev. B 58, 15496 (1998).
Gopal, P. et al. Improved electronic structure and magnetic exchange interactions in transition metal oxides. J. Phys.: Condens. Matter 29, 444003 (2017).
Bloch, F. Zur Theorie des Ferromagnetismus. Z. Phys. 61, 206 (1930).
Slater, J. C. Cohesion in monovalent metals. Phys. Rev. 35, 509 (1930).
Holstein, T. & Primakoff, H. Field dependence of the intrinsic domain magnetization of a ferromagnet. Phys. Rev. 58, 1098 (1940).
Zakeri, K., Zhang, Y., Chuang, T.-H. & Kirschner, J. Magnon lifetimes on the Fe(110) surface: the role of spin-orbit coupling. Phys. Rev. Lett. 108, 197205 (2012).
Heinze, S. et al. Spontaneous atomic-scale magnetic skyrmion lattice in two dimensions. Nat. Phys. 7, 713–718 (2011).
Landau, L. On the vibrations of the electronic plasma. J. Phys. USSR 10, 25 (1946).
Morano, V. C. et al. Absence of altermagnetic magnon band splitting in MnF2. Preprint at https://arxiv.org/abs/2412.03545 (2024).
Liu, Z., Ozeki, M., Asai, S., Itoh, S. & Masuda, T. Chiral split magnon in altermagnetic MnTe. Phys. Rev. Lett. 133, 156702 (2024).
Gross, E. K. U. & Kohn, W. Local density-functional theory of frequency-dependent linear response. Phys. Rev. Lett. 55, 2850–2852 (1985).
Hedin, L. New method for calculating the one-particle Green’s function with application to the electron-gas problem. Phys. Rev. 139, A796 (1965).
Rousseau, B., Eiguren, A. & Bergara, A. Efficient computation of magnon dispersions within time-dependent density functional theory using maximally localized Wannier functions. Phys. Rev. B 85, 054305 (2012).
Lounis, S., Costa, A. T., Muniz, R. B. & Mills, D. L. Dynamical magnetic excitations of nanostructures from first principles. Phys. Rev. Lett. 105, 187205 (2010).
Lounis, S., Costa, A., Muniz, R. & Mills, D. Theory of local dynamical magnetic susceptibilities from the Korringa-Kohn-Rostoker Green function method. Phys. Rev. B 83, 035109 (2011).
Buczek, P., Ernst, A. & Sandratskii, L. Different dimensionality trends in the Landau damping of magnons in iron, cobalt, and nickel: time-dependent density functional study. Phys. Rev. B 84, 174418 (2011).
dos Santos Dias, M., Schweflinghaus, B., Blügel, S. & Lounis, S. Relativistic dynamical spin excitations of magnetic adatoms. Phys. Rev. B 91, 075405 (2015).
Wysocki, A. et al. Spin-density fluctuations and the fluctuation-dissipation theorem in 3d ferromagnetic metals. Phys. Rev. B 96, 184418 (2017).
Singh, N., Elliott, P., Nautiyal, T., Dewhurst, J. & Sharma, S. Adiabatic generalized gradient approximation kernel in time-dependent density functional theory. Phys. Rev. B 99, 035151 (2019).
Skovhus, T. & Olsen, T. Dynamic transverse magnetic susceptibility in the projector augmented-wave method: application to Fe, Ni, and Co. Phys. Rev. B 103, 245110 (2021).
Sandratskii, L. M. & Buczek, P. Lifetimes and chirality of spin waves in antiferromagnetic and ferromagnetic ferh from the perspective of time-dependent density functional theory. Phys. Rev. B 85, 020406 (2012).
Odashima, M. M., Marmodoro, A., Buczek, P., Ernst, A. & Sandratskii, L. Chirality-dependent magnon lifetime in a compensated half-metallic ferrimagnet. Phys. Rev. B 87, 174420 (2013).
Savrasov, S. Y. Linear response calculations of spin fluctuations. Phys. Rev. Lett. 81, 2570–2573 (1998).
Cao, K., Lambert, H., Radaelli, P. G. & Giustino, F. Ab initio calculation of spin fluctuation spectra using time-dependent density functional perturbation theory, plane waves, and pseudopotentials. Phys. Rev. B 97, 024420 (2018).
Liu, X., Lin, Y. & Feng, J. Implementation of the density functional perturbation theory for generalized susceptibility in the projector augmented wave framework. Phys. Rev. B 108, 094405 (2023).
Gorni, T., Timrov, I. & Baroni, S. Spin dynamics from time-dependent density functional perturbation theory. Eur. Phys. J. B 91, 249 (2018).
Tancogne-Dejean, N., Eich, F. G. & Rubio, A. Time-dependent magnons from first principles. J. Chem. Theory Comput. 16, 1007–1017 (2020).
Karlsson, K. & Aryasetiawan, F. Spin-wave excitation spectra of nickel and iron. Phys. Rev. B 62, 3006–3009 (2000).
Şaşıoglu, E., Schindlmayr, A., Friedrich, C., Freimuth, F. & Blügel, S. Wannier-function approach to spin excitations in solids. Phys. Rev. B 81, 054434 (2010).
Müller, M. C. T. D., Blügel, S. & Friedrich, C. Electron-magnon scattering in elementary ferromagnets from first principles: Lifetime broadening and band anomalies. Phys. Rev. B 100, 045130 (2019).
Müller, M. C. T. D., Friedrich, C. & Blügel, S. Acoustic magnons in the long-wavelength limit: investigating the Goldstone violation in many-body perturbation theory. Phys. Rev. B 94, 064433 (2016).
Lee, C.-C., Hsueh, H. C. & Ku, W. Dynamical linear response of TDDFT with LDA+U functional: strongly hybridized Frenkel excitons in NiO. Phys. Rev. B 82, 081106 (2010).
Orhan, O. K. & O’Regan, D. D. TDDFT + U: a critical assessment of the Hubbard U correction to exchange-correlation kernels and potentials. Phys. Rev. B 99, 165120 (2019).
Tancogne-Dejean, N., Oliveira, M. J. T. & Rubio, A. Self-consistent DFT + U method for real-space time-dependent density functional theory calculations. Phys. Rev. B 96, 245133 (2017).
To clarify potential misunderstandings, we note that the TDDFT method described in refs. 92,93 does not include the Hubbard U correction from the DFT+U framework. In these references, the letter “U” refers to the exchange-correlation kernel of the Dyson equation, not the “Hubbard U” parameter. The TDDFT method in refs. 92,93 is based on LSDA, and should not be confused with TDDFT+U.
Skovhus, T. & Olsen, T. Magnons in antiferromagnetic bcc Cr and Cr2O3 from time-dependent density functional theory. Phys. Rev. B 106, 085131 (2022).
Skovhus, T., Olsen, T. & Rønnow, H. Influence of static correlation on the magnon dynamics of an itinerant ferromagnet with competing exchange interactions: First-principles study of MnBi. Phys. Rev. Mater. 6, 054402 (2022).
Baroni, S. & Gebauer, R. The Liouville-Lanczos Approach to Time-Dependent Density-Functional Perturbation Theory, 375–390 (Springer, 2012).
Walker, B., Saitta, A. M., Gebauer, R. & Baroni, S. Efficient approach to time-dependent density-functional perturbation theory for optical spectroscopy. Phys. Rev. Lett. 96, 113001 (2006).
Baroni, S., de Gironcoli, S., Dal Corso, A. & Giannozzi, P. Phonons and related crystal properties from density-functional perturbation theory. Rev. Mod. Phys. 73, 515–562 (2001).
Cococcioni, M. & Marzari, N. Energetics and cathode voltages of LiMPO4 olivines (M = Fe, Mn) from extended Hubbard functionals. Phys. Rev. Mater. 3, 033801 (2019).
Baroni, S., Giannozzi, P. & Testa, A. Green’s-function approach to linear response in solids. Phys. Rev. Lett. 58, 1861–1864 (1987).
Rocca, D., Gebauer, R., Saad, Y. & Baroni, S. Turbo charging time-dependent density-functional theory with Lanczos chains. J. Chem. Phys. 128, 154105 (2008).
Timrov, I., Vast, N., Gebauer, R. & Baroni, S. Electron energy loss and inelastic x-ray scattering cross sections from time-dependent density-functional perturbation theory. Phys. Rev. B 88, 064301 (2013).
Floris, A., De Gironcoli, S., Gross, E. K. U. & Cococcioni, M. Vibrational properties of MnO and NiO from DFT + U-based density functional perturbation theory. Phys. Rev. B 84, 161102 (2011).
Zhu, X., Edström, A. & Ederer, C. Magnetic exchange interactions in SrMnO3. Phys. Rev. B 101, 064401 (2020).
Wan, X., Yin, Q. & Savrasov, S. Y. Calculation of magnetic exchange interactions in Mott-Hubbard systems. Phys. Rev. Lett. 97, 266403 (2006).
Kotani, T. & Schilfgaarde, M. V. Spin wave dispersion based on the quasiparticle self-consistent GW method: NiO, MnO and α-MnAs. J. Phys.: Condens. Matter 20, 295214 (2008).
Gonze, X. & Lee, C. Dynamical matrices, Born effective charges, dielectric permittivity tensors, and interatomic force constants from density-functional perturbation theory. Phys. Rev. B 55, 10355–10368 (1997).
Urru, A. & Dal Corso, A. Density functional perturbation theory for lattice dynamics with fully relativistic ultrasoft pseudopotentials: The magnetic case. Phys. Rev. B 100, 045115 (2019).
Vanderbilt, D. Soft self-consistent pseudopotentials in a generalized eigenvalue formalism. Phys. Rev. B 41, 7892–7895 (1990).
Blöchl, P. E. Projector augmented-wave method. Phys. Rev. B 50, 17953–17979 (1994).
Walker, B. & Gebauer, R. Ultrasoft pseudopotentials in time-dependent density-functional theory. J. Chem. Phys. 127, 164106 (2007).
Motornyi, O. et al. Electron energy loss spectroscopy of bulk gold with ultrasoft pseudopotentials and the Liouville-Lanczos method. Phys. Rev. B 102, 035156 (2020).
Dudarev, S. L. et al. Parametrization of LSDA + U for noncollinear magnetic configurations: Multipolar magnetism in UO2. Phys. Rev. Mater 3, 083802 (2019).
Savrasov, S. Y. & Kotliar, G. Ground State Theory of δ-Pu. Phys. Rev. Lett. 84, 3670 (2000).
Sakurai, J. J. & Napolitano, J. Modern Quantum Mechanics (Cambridge University Press, 2020),
Cococcioni, M. Accurate and efficient calculations on strongly correlated minerals with the LDA+U method: review and perspectives. Rev. Mineral. Geochem. 71, 147–167 (2010).
de Gironcoli, S. Lattice dynamics of metals from density-functional perturbation theory. Phys. Rev. B 51, 6773–6776 (1995).
Calandra, M., Profeta, G. & Mauri, F. Adiabatic and nonadiabatic phonon dispersion in a Wannier function approach. Phys. Rev. B 82, 165111 (2010).
Giustino, F., Cohen, M. L. & Louie, S. G. GW method with the self-consistent Sternheimer equation. Phys. Rev. B 81, 115105 (2010).
Binci, L., Barone, P. & Mauri, F. First-principles theory of infrared vibrational spectroscopy of metals and semimetals: Application to graphite. Phys. Rev. B 103, 134304 (2021).
Srinivasan, G. & Seehra, M. Magnetic susceptibilities, their temperature variation, and exchange constants of NiO. Phys. Rev. B 29, 6295 (1984).
Blech, I. & Averbach, B. Spin correlations in MnO. Physics 1, 31 (1964).
Schmahl, N., Barthel, J. & Eikerling, G., Rontgenographische Untersuchungen an den Systemen MgO-CuO und NiO-CuO, Z. Anorg. Allg. Chem. 332, 230 (1964).
Sasaki, S., Fujino, K., Taki-Uchi, Y. & Sadanag, R. On the Estimation of Atomic Charges by the X-ray Method for Some Oxides and Silicates. Acta Cryst. A 36, 904 (1980).
Perdew, J. P., Burke, K. & Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 77, 3865 (1996).
Perdew, J. P. et al. Restoring the density-gradient expansion for exchange in solids and surfaces. Phys. Rev. Lett. 100, 136406 (2008).
Kirchner-Hall, N., Zhao, W., Xiong, Y., Timrov, I. & Dabo, I. Extensive benchmarking of DFT+U calculations for predicting band gaps. Appl. Sci. 11, 2395 (2021).
Lambert, D. S. & O’Regan, D. D. Use of DFT+U+J with linear response parameters to predict non-magnetic oxide band gaps with hybrid-functional accuracy. Phys. Rev. Res. 5, 013160 (2023).
Leiria Campo Jr, V. & Cococcioni, M. Extended DFT + U + V method with on-site and inter-site electronic interactions. J. Phys.: Condens. Matter 22, 055602 (2010).
van Setten, M. J. et al. Gw100: Benchmarking g0w0 for molecular systems. J. Chem. Theory Comput. 11, 5665–5687 (2015).
Grumet, M., Liu, P., Kaltak, M., Klimeš, Jcv & Kresse, G. Beyond the quasiparticle approximation: fully self-consistent GW calculations. Phys. Rev. B 98, 155143 (2018).
Bonacci, M. et al. Towards high-throughput many-body perturbation theory: efficient algorithms and automated workflows. npj Comput. Mater. 9, 74 (2023).
Wing, D. et al. Band gaps of crystalline solids from Wannier-localization-based optimal tuning of a screened range-separated hybrid functional. Proc. Natl Acad. Sci. USA 118, e2104556118 (2021).
Skone, J. H., Govoni, M. & Galli, G. Nonempirical range-separated hybrid functionals for solids and molecules. Phys. Rev. B 93, 235106 (2016).
Chen, W., Miceli, G., Rignanese, G.-M. & Pasquarello, A. Nonempirical dielectric-dependent hybrid functional with range separation for semiconductors and insulators. Phys. Rev. Mater. 2, 073803 (2018).
Dabo, I. et al. Koopmans’ condition for density-functional theory. Phys. Rev. B 82, 115121 (2010).
Nguyen, N. L., Colonna, N., Ferretti, A. & Marzari, N. Koopmans-compliant spectral functionals for extended systems. Phys. Rev. X 8, 021051 (2018).
Linscott, E. B. et al. Koopmans: an open-source package for accurately and efficiently predicting spectral properties with Koopmans functionals. J. Chem. Theory Comput. 19, 7097 (2023).
Van Hove, L. Correlations in space and time and born approximation scattering in systems of interacting particles. Phys. Rev. 95, 249–262 (1954).
Blume, M. Polarization effects in the magnetic elastic scattering of slow neutrons. Phys. Rev. 130, 1670–1676 (1963).
Hutchings, M. T. & Samuelsen, E. J. Measurement of spin-wave dispersion in NiO by inelastic neutron scattering and its relation to magnetic properties. Phys. Rev. B 6, 3447–3461 (1972).
Pepy, G. Spin waves in MnO; from 4∘K to temperatures close to TN. J. Phys. Chem. Solids 35, 433–444 (1974).
Goldstone, J., Salam, A. & Weinberg, S. Broken symmetries. Phys. Rev. 127, 965 (1962).
Watanabe, H. & Murayama, H. Unified description of Nambu-Goldstone bosons without Lorentz invariance. Phys. Rev. Lett. 108, 251602 (2012).
Gorni, T., Baseggio, O., Delugas, P., Baroni, S. & Timrov, I. turboMagnon—a code for the simulation of spin-wave spectra using the Liouville-Lanczos approach to time-dependent density-functional perturbation theory. Comput. Phys. Commun. 280, 108500 (2022).
Lines, M. E. & Jones, E. D. Antiferromagnetism in the face-centered cubic lattice. II. Magnetic properties of MnO. Phys. Rev. 139, A1313–A1327 (1965).
Kohgi, M., Ishikawa, Y. & Endoh, Y. Inelastic neutron scattering study of spin waves in MnO. Solid State Commun. 11, 391 (1972).
Note that in the equations for J11(q) and J12(q), there are double sums over the Cartesian indices (α and \(\alpha {\prime}\)), which results in each term appearing twice. For example, \({\sum }_{\alpha \ne \alpha {\prime} }\cos \pi ({q}_{\alpha }+{q}_{\alpha ^{\prime} })=2\cos \pi ({q}_{x}+{q}_{y})+2\cos \pi ({q}_{x}+{q}_{z})+2\cos \pi ({q}_{y}+{q}_{z})\).
dos Santos, F. et al. Comparative study of magnetic-exchange parameters and magnon dispersions in NiO and MnO using first-principles methods, in preparation (2025).
Sun, J., Ruzsinszky, A. & Perdew, J. Strongly constrained and appropriately normed semilocal density functional. Phys. Rev. Lett. 115, 036402 (2015).
Tancogne-Dejean, N., Sentef, M. A. & Rubio, A. Ultrafast modification of Hubbard U in a strongly correlated material: ab initio high-harmonic generation in NiO. Phys. Rev. Lett. 121, 097402 (2018).
Giannozzi, P. et al. Quantum ESPRESSO toward the exascale. J. Chem. Phys. 152, 154105 (2020).
Carnimeo, I. et al. Quantum ESPRESSO: one further step toward the exascale. J. Chem. Theory Comput. 19, 6292 (2023).
Timrov, I. et al. Electronic structure of Ni-substituted LaFeO3 from near edge x-ray absorption fine structure experiments and first-principles simulations. Phys. Rev. Res. 2, 033265 (2020).
Mahajan, R., Timrov, I., Marzari, N. & Kashyap, A. Importance of intersite Hubbard interactions in β-MnO2: a first-principles DFT+U+V study. Phys. Rev. Mater. 5, 104402 (2021).
Mahajan, R., Kashyap, A. & Timrov, I. Pivotal role of intersite Hubbard interactions in Fe-doped α-MnO2. J. Phys. Chem. C. 126, 14353 (2022).
Timrov, I., Aquilante, F., Cococcioni, M. & Marzari, N. Accurate electronic properties and intercalation voltages of olivine-type Li-ion cathode materials from extended Hubbard functionals. PRX Energy 1, 033003 (2022).
Timrov, I., Kotiuga, M. & Marzari, N. Unraveling the effects of inter-site Hubbard interactions in spinel Li-ion cathode materials. Phys. Chem. Chem. Phys. 25, 9061 (2023).
Binci, L., Kotiuga, M., Timrov, I. & Marzari, N. Hybridization driving distortions and multiferroicity in rare-earth nickelates. Phys. Rev. Res. 5, 033146 (2023).
Gebreyesus, G., Bastonero, L., Kotiuga, M., Marzari, N. & Timrov, I. Understanding the role of Hubbard corrections in the rhombohedral phase of BaTiO3. Phys. Rev. B 108, 235171 (2023).
Haddadi, F., Linscott, E., Timrov, I., Marzari, N. & Gibertini, M. On-site and inter-site Hubbard corrections in magnetic monolayers: the case of FePS3 and CrI3. Phys. Rev. Mater. 8, 014007 (2024).
Paischer, S., Vignale, G., Katsnelson, M. I., Ernst, A. & Buczek, P. A. Nonlocal correlation effects due to virtual spin-flip processes in itinerant electron ferromagnets. Phys. Rev. B 107, 134410 (2023).
Müller, M. C. T. D., Blügel, S. & Friedrich, C. Electron-plasmon and electron-magnon scattering in ferromagnets from first principles by combining GW and GT self-energies. npj Comput. Mater. 7, 178 (2021).
Paischer, S. et al. Correlations, disorder, and multimagnon processes in terahertz spin dynamics of magnetic nanostructures: a first-principles investigation. Phys. Rev. B 109, L220405 (2024).
Huber, S. P. et al. AiiDA 1.0, a scalable computational infrastructure for automated reproducible workflows and data provenance. Sci. Data 7, 300 (2020).
Bastonero, L. et al. Marzari, First-principles Hubbard parameters with automated and reproducible workflows, arXiv:2503.01590 (2025).
Saad, Y. Iterative Methods for Sparse Linear Systems (SIAM, 2003).
Malcıoğlu, O. B., Gebauer, R., Rocca, D. & Baroni, S. turboTDDFT—a code for the simulation of molecular spectra using the Liouville-Lanczos approach to time-dependent density-functional perturbation theory. Comput. Phys. Commun. 182, 1744–1754 (2011).
Giannozzi, P. et al. QUANTUM ESPRESSO: a modular and open-source software project for quantum simulations of materials. J. Phys.: Condens. Matter 21, 395502 (2009).
Giannozzi, P. et al. Advanced capabilities for materials modelling with Quantum ESPRESSO. J. Phys.: Condens. Matter 29, 465901 (2017).
Perdew, J. P. & Wang, Y. Accurate and simple analytic representation of the electron-gas correlation energy. Phys. Rev. B 45, 13244–13249 (1992).
Hamann, D. R. Optimized norm-conserving Vanderbilt pseudopotentials. Phys. Rev. B 88, 085117 (2013).
Van Setten, M. et al. The PseudoDojo: Training and grading a 85 element optimized norm-conserving pseudopotential table. Comput. Phys. Commun. 226, 39–54 (2018).
Timrov, I., Marzari, N. & Cococcioni, M. HP—a code for the calculation of Hubbard parameters using density-functional perturbation theory. Comput. Phys. Commun. 279, 108455 (2022).
Löwdin, P.-O. On the non-orthogonality problem connected with the use of atomic wave functions in the theory of molecules and crystals. J. Chem. Phys. 18, 365 (1950).
Timrov, I., Aquilante, F., Binci, L., Cococcioni, M. & Marzari, N. Pulay forces in density-functional theory with extended Hubbard functionals: from nonorthogonalized to orthogonalized manifolds. Phys. Rev. B 102, 235159 (2020).
Fender, B., Jacobson, A. & Wedgwood, F. Covalency Parameters in MnO, α‐MnS, and NiO. J. Chem. Phys. 48, 990 (1968).
Cheetham, A. & Hope, D. Magnetic ordering and exchange effects in the antiferromagnetic solid solutions MnxNi1-xO. Phys. Rev. B 27, 6964 (1983).
Kurmaev, E. et al. Oxygen x-ray emission and absorption spectra as a probe of the electronic structure of strongly correlated oxides. Phys. Rev. B 77, 165127 (2008).
Sawatzky, G. & Allen, J. Magnitude and Origin of the Band Gap in NiO. Phys. Rev. Lett. 53, 2339 (1984).
van Elp, J., Potze, R., Eskes, H., Berger, R. & Sawatzky, G. Electronic structure of MnO. Phys. Rev. B 44, 1530 (1991).
Acknowledgements
We thank Tommaso Gorni, Flaviano dos Santos, and Francesco Mauri for fruitful discussions. We acknowledge support by the NCCR MARVEL, a National Centre of Competence in Research, funded by the Swiss National Science Foundation (Grant number 205602). L.B. acknowledges the Fellowship from the EPFL QSE Center “Many-body neural simulations of quantum materials” (Grant number 10060). This work was supported by a grant from the Swiss National Supercomputing Centre (CSCS) under project ID s1073 and mr33 (Piz Daint).
Author information
Authors and Affiliations
Contributions
L.B. derived the equations and implemented the novel approach in Quantum ESPRESSO using preliminary I.T.'s routines. L.B. and I.T. tested the implementation and applied it to NiO and MnO. L.B., N.M., and I.T. analyzed the results and equally contributed to writing the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Binci, L., Marzari, N. & Timrov, I. Magnons from time-dependent density-functional perturbation theory and nonempirical Hubbard functionals. npj Comput Mater 11, 100 (2025). https://doi.org/10.1038/s41524-025-01570-0
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41524-025-01570-0
This article is cited by
-
First-principles Hubbard parameters with automated and reproducible workflows
npj Computational Materials (2025)
