
Abstract
Accurately predicting patient outcomes is essential for optimizing treatment and improving outcomes in pediatric acute myeloid leukemia (AML). In recent years, microRNAs have emerged as a promising prognostic marker, with a growing body of evidence supporting their potential predictive value. We systematically reviewed all previous studies that have analyzed the expression of microRNAs as predictors of survival in pediatric AML and found 16 microRNAs and 4 microRNA signatures previously proposed as predictors of survival. We then used a public access cohort of 1414 pediatric AML patients from the TARGET project to develop a new predictive model using penalized lasso Cox regression based on microRNA expression. Here we propose a new score based on a 37-microRNA signature that is associated with AML and is able to predict survival more accurately than previous microRNA-based methods.
Similar content being viewed by others


Introduction
Childhood acute myeloid leukemia (AML) is a rare and heterogeneous disease that represents 25% of childhood leukemias. The outcome of children with AML has improved over the last years, reaching the complete remission of 90% in pediatric patients, but overall survival (OS) and event-free survival (EFS) rates remain low, around 70% and 50%, respectively, due to the high frequency of relapse and the treatment-related risk of death1,2,3.
Acute myeloid leukemia appears mostly in adults. Therefore, most of the research has been based on adult patients; however, pediatric and adult AML are different in the molecular and mutational landscape, as well as in the tolerance to treatment4,5,6. Pediatric AML patients present a larger proportion of cryptic gene fusions and recurrent focal deletions, while adults have more somatic mutations impacting DNA methylation4. Furthermore, the cytogenetic abnormalities used to identify AML subtypes following the WHO classification are more commonly found in adults than pediatric patients; consequently, a larger portion of pediatric patients is classified as “AML not otherwise specified”, which limits the applicability of the WHO classification in AML children7,8,9. These differences between children and adults emphasize the importance of the study of children-specific treatments and risk stratification.
An adequate risk assessment is key in the selection of a proper treatment for AML, and risk stratification criteria have been improved in the last few years. In children, current risk prediction assessment is mainly based on the identification of genomic alterations or mutations1, but there remains a considerable scope for further improvement as individual genomic features cannot explain disease outcomes, therefore it is necessary to use whole genomic data in order to refine and improve risk stratification.
DNA methylation, microRNA expression, and genomic alteration data can be used to stratify pediatric patients with AML according to their expected survival4.
Micro RNAs (miRNAs) are a class of small non-coding RNAs that play an important role in regulating gene expression and participate in cellular processes, such as differentiation, proliferation, migration, and apoptosis. MiRNAs can also participate in AML pathogenesis and can be used as biomarkers, risk predictors, or even therapeutic targets10,11,12, although their mechanisms of action still remain complex and unclear due to their wide range of target genes and signaling pathways13.
In the last years, many studies have focused on the characterization and identification of miRNAs in AML14,15,16,17,18,19, and some of them have demonstrated that MiRNAs are useful in the diagnosis of AML and AML subtypes19,20,21,22. Regular gene expression data have been used to develop prognostic risk-prediction signatures for AML in adults and children23,24,25,26; however, there is a lack of a consensus miRNA signature for risk assessment and marked differences among studies27. To critically evaluate available studies in this field and identify common and different findings across them, we have performed a systematic review of the literature that analyzes the potential role of miRNAs as predictive biomarkers for survival in pediatric AML. In addition, using available data, we have identified a new miRNA signature for predicting survival and we have conducted a comparative analysis to evaluate our model together with other signatures and individual microRNAs.
Results
Systematic review
We identified 203 non-duplicate records by a systematic search of existing literature. After reviewing the titles, abstracts, and full texts of each record, we excluded 181 articles that did not meet our inclusion criteria, finally selecting 22 articles for this review (Fig. 1).

Systematic review of microRNA-based predictors of survival, predictive model construction, and systematic validation and evaluation of predictors.
From these 22 articles, we found 4 miRNA signatures and 16 single miRNAs with survival prediction capability, assessed by 426,28,29,30 and 1814,15,16,17,31,32,33,34,35,36,37,38,39,40,41,42,43,44 studies, respectively (Table 1). From the 18 articles that analyzed the expression of single miRNAs, we found 11 that performed a multivariate Cox regression analysis to prove if the selected miRNA was an independent prognosis predictor. All these tests were significant, confirming their capability to predict overall survival. Only one article31 did not test the predictor (miR-381) for event-free survival. We assembled all the studies that associated miRNA expression with survival in pediatric AML in Table 1.
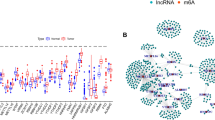
Although these studies demonstrated the association between the expression level of each miRNA or miRNA signature and the patient outcome, there is little miRNA overlap among signatures. MiRNAs hsa-let-7g-5p and hsa-miR-181c-5p appeared in both the 24 miRNA signature (miR24) from Esperanza-Cebollada et al. 26 and the 36 miRNA signature (AMLmiR36) from Lim et al. 30, being the second one also found in the 3 miRNA signature (miR3) from R. Zhu et al. 29 (Fig. 2). Hsa-miR-146a-5p was present in both the AMLmiR36 and the 4 miRNA signature (miR4) from R. Zhu et al. 28.

small gray capsules indicate individually analyzed miRNAs, whereas larger ellipses contain miRNA. signatures. Mature miRNA IDs are displayed when a mature-based study and an immature-based study overlap.
Construction of a new predictive signature
Afterward, we developed a new survival predictive model based on miRNA expression using the penalized lasso CoxPH in the Discovery set of TARGET-AML patients. The resulting model was composed of 37 miRNAs (miR37 signature) that were used to calculate a predictive score for each patient. We assessed the similarities between our model and the previously described miRNAs with survival prediction capability, and we found that our signature shared two miRNAs with the miR24 signature26 and nine miRNAs with the AMLmiR36 signature30, four of which had also been individually associated with survival (Fig. 2).
Systematic evaluation of predictors
We compiled all previously described microRNAs and signatures, along with our new miR37 signature, and analyzed their association with prognosis in the TARGET-AML cohort. We analyzed the association of the 16 selected miRNAs and 5 miRNA signature scores with overall survival and event-free survival separately.
By univariate Cox proportional hazards regression analysis, we found that all five scores were significantly associated with OS (Fig. 3A) and EFS (Supplementary Fig. 3), along with hsa-mir-199a, hsa-mir-335, hsa-mir-196b, hsa-mir-100, hsa-miR-139-5p and hsa-mir-195 (p-value < 0.05). However, only the miR37 score, AMLmiR36 score, miR24 score, hsa-mir-199a, and hsa-mir-335 showed a marked difference in Hazard ratio (HR), indicating that high signature scores were associated with an increased risk of death, while high expression of hsa-mir-199a and hsa-mir-335 was the most representative predictors of good prognosis.

a Univariate and multivariate Cox proportional hazards model parameters for each predictor. b Kaplan–Meier survival plots of the miRNA predictive signatures, representing survival curves of high (blue) and low (yellow) signature scores.
Multivariate CoxPH showed that the five signature scores and hsa-mir-199a, hsa-mir-100, hsa-mir-34b, hsa-mir-195 and hsa-mir-381 were independent predictors of OS and the miR37 score, AMLmiR36 score, miR24 score, miR4 score, hsa-mir-335, hsa-mir-100, hsa-miR-139-5p, hsa-mir-34b and hsa-mir-195 were independent predictors of EFS (p-value < 0.05).
We then categorized the expression of each miRNA and signature score in high and low expression and we tested the ability of each predictor to separate the two outcome cohorts (Fig. 3B, Supplementary Figs. 2 and 3B). We found that high miR37, miR24, AMLmiR36, and miR4 scores and high expression of hsa-mir-196b, hsa-mir-155, and hsa-mir-34b were significantly associated with lower OS (log-rank test p-value < 0.05) and, with the exception of hsa-mir-34b, with lower EFS. High expression of hsa-mir-335, hsa-mir-100, hsa-miR-139-5p, and hsa-mir-199a was significantly associated with higher OS and EFS (log-rank test p-value < 0.05). The increased expression of hsa-mir-34b was significantly linked to lower OS but not lower EFS.
We estimated the survival prediction accuracy of the 16 selected miRNAs, 5 scores, and a random signature score by calculating the Concordance index and time-dependent AUC in the validation cohort. Our model score showed a median C-index of 0.63 for OS, outperforming previous signatures in predictive capability (Fig. 4A). The AUC calculated for each time point revealed that although miR4 has a good predictive performance for the early stage, our model outperforms all other signatures in predictive accuracy for OS after approximately the first year (Fig. 4B). Similar results were found for EFS, with a median C-index of 0.64 and the same trend in AUC over time (Supplementary Fig. 4).

a C-index of each predictor to measure the predictive accuracy. b Time-dependent area under the receiver operating characteristic curve (AUC) for each of the miRNA predictive signatures.
After separating patients by their assigned cytogenetic risk, the C-index indicated that our miR37 score still outperformed other predictors in patients of the standard risk group when predicting OS but not in EFS, where the miR4 score was the best predictor (Supplementary Fig. 5A). In low-risk patients hsa-mir-195 showed the best predictive performance. Accuracy on high-risk patients was inconclusive due to the low number of patients in this risk group. When comparing the accuracy of signatures by time-dependent AUC (Supplementary Fig. 5B), we found that miR24 predicted better in low-risk patients, in standard-risk patients, miR4 predicted better at early disease stage (approximately 1 year from diagnosis) and miR37 at late disease stage (approximately more than 1 year from diagnosis), and miR4 signature had a good predictive performance in high-risk patients.
Enrichment analysis
In order to characterize our miR37 signature, we assessed its functional characteristics through miRNA set enrichment analysis. We discovered that Hematopoiesis and Immune Response were the more overrepresented functions in the miRNA signature, with 11 and 13 miRNAs associated with these functions, respectively (Fig. 5A, B). We also analyzed the diseases overrepresented in our miRNAs and we found that acute myeloid leukemia was the most significantly associated disease, with 15 AML-related miRNAs present in our signature (Fig. 5C, D). We also analyzed the level of association of each miRNA and miRNA set to test which miRNA presented the strongest association with each term (Supplementary Fig. 6). We found that hsa-mir-378c had the strongest association with the Hematopoietic function (Association Score = 7.8) and hsa-mir-27a was strongly associated with Immune Response (Association Score = 7.5).

Top 10 overrepresented functions (a) and diseases (c) in the signature. Enrichment network for the top 10 overrepresented functions (b) and diseases (d). MiRNA names are not displayed for clarity.
Signature quality control
Finally, to assess the robustness of our miR37 signature, we performed a quality control analysis by comparing it with a randomly selected miRNA signature of the same length. To conduct this analysis, we added an external miRNA expression microarray dataset (GSE97135). We observed relatively high concordance between the quality control metrics calculated for our miR37 signature in both the TARGET-AML and the GSE97135 datasets (Supplementary Fig. 7A) and large differences in the metrics compared to the negative controls in both cases (Supplementary Fig. 7B) suggesting that our signature is highly applicable across different sequencing platforms. We saw that the miRNAs in the miR37 signature exhibited higher expression levels and variability compared to the random signature in both the TARGET-AML validation and the GSE97135 datasets (Supplementary Fig. 8), supporting the idea that the miR37 signature might have greater clinical utility. We also observed that, in the external GSE97135 dataset, the correlations of scoring metrics were higher for the miR37 signature than for the random signature (Supplementary Fig. 9), suggesting that the miR37 signature can be summarized consistently, unlike the random signature. When analyzing the intra-signature correlation, we observed that the miRNAs in our miR37 signature were slightly more correlated with each other than the random miRNAs in the GSE97135 dataset (Supplementary Fig. 10), supporting the potential of our signature to be summarized into a score or used in approaches like GSEA. It was observed that the random signature contained more miRNAs with low expression than miR37 (Supplementary Fig. 11), as expected due to the inherent right-skewed distribution of the miRNA expression data.
Finally, we calculated all quality control metrics provided by the sigQC pipeline on the GSE97135 dataset. The miR37 signature outperformed the random signature on almost all metrics (Supplementary Fig. 12A) and most of the test results were different from the negative controls, unlike the random signature (Supplementary Fig. 12B). This ultimately confirmed the good quality of our signature.
Discussion
In our systematic review, we searched for studies that assessed the association between microRNA expression and survival. Although many of these studies also analyzed the expression of miRNAs as a diagnosis marker, we focused on their capability as a prognostic factor. The selected studies routinely performed Kaplan–Meier and Cox regression analysis to assess the association of miRNA expression with survival and test the independence of the predictors. However, the ability of these miRNAs to predict survival has rarely been adequately measured. Only the performance of miR3 and miR4 signatures were calculated by classical ROC curves and AUC28,29 and Esperanza-Cebollada et al. 26 calculated the predictive accuracy of their signature by estimating the C-index on the CoxPH-fitted patients. Nevertheless, reliable predictive performance indicators are needed in order to ensure replicable results.
Here we developed a new survival prediction model based on miRNA expression and systematically evaluated our model together with all previous existing models using standard analyses as well as new robust methods.
When analyzing coincident miRNAs between signatures we saw that our signature had several miRNAs matched to AMLmiR36, up to 24% of the signature. This could be attributed to the fact that we developed our signature using an approximation of the methods of Lim et al. 30 and a similar patient distribution. Particularly, among these matched microRNAs we can find four miRNAs that have been independently validated as survival predictors. Of particular note is hsa-miR-155-5p, whose high expression is associated with poor prognosis in our analysis and previous studies14,41,42, and has several associated functions related to hematopoiesis (Supplementary Fig. 6). MiR-155 was studied as an oncogene in B cell lymphoma45 and was estimated to block myeloid differentiation in normal human hematopoietic stem cells46. However, other studies have shown that it can promote apoptosis and cell differentiation in AML47,48. According to our results, we could hypothesize that hsa-miR-155-5p functions as an oncogene in pediatric AML patients, blocking myeloid differentiation.
When analyzing our new miR37 signature, we found that, as expected, several miRNAs were related to hematopoiesis and immune response. Among the hematopoiesis-related miRNAs, hsa-mir-378c has been shown to be down-regulated in AML when apoptosis is induced by cycle-dependent kinase inhibitor SNS-03249. Immune response miRNAs included hsa-mir-27a, which is a known tumor suppressor in acute leukemia50.
Our systematic evaluation revealed some disparities with previous studies. In particular, hsa-mir-100 and hsa-mir-335 had previously been associated with poor prognosis; however, our results indicated an association with good prognosis, which has been found in other cancers as well51,52.
In this study, we identify significant associations between the expression of certain miRNAs and survival, that failed to achieve significance in previous studies. This is the case for hsa-mir-199a, miR4 signature, and AMLmiR36 signature, which we have shown to be significantly associated with OS, as well as hsa-miR-139-5p and hsa-mir-196b, which exhibit significant associations with EFS.
We found that certain individual miRNAs, specifically hsa-mir-199a, hsa-miR-139-5p, hsa-mir-100, hsa-mir-335, and hsa-mir-196b, were consistently associated with survival, as confirmed by significance in both hazard ratio and survival curve analysis. In particular, we observed that hsa-mir-199a was the best predictor of survival in high-risk patients, and its high expression was associated with better survival, possibly due to its ability to enhance chemotherapy efficacy by repressing DRAM1 and WNT2. These genes are involved in protective autophagy, which can lead to chemotherapy resistance in AML and chronic myeloid leukemia53,54. Furthermore, it has been reported that hsa-mir-199a enhances the sensitivity of chronic myeloid leukemia cells to chemotherapy by down-regulating mTOR signaling55. Nevertheless, the reliability of the results observed in high-risk patients is constrained by the limited number of samples within this risk group, since the insufficient sample size resulted in increased error rates.
In low-risk patients, the most accurate predictor of survival was hsa-mir-195, even though its association with survival was not significant in the analysis of survival curves. The expression of hsa-mir-195 has previously been linked to a good prognosis in pediatric AML34. Various targets and mechanisms have been proposed to explain this effect. Among these targets, we highlight BMI1, which has been studied as an oncogene in myeloid leukemias56, RET, a proto-oncogene overexpressed in t(8;16) AML57, and MYB, whose silencing has been shown to alleviate chemotherapy resistance in pediatric AML58.
Another interesting miRNA is hsa-mir-335, which was the best individual predictor of survival among all patients, outperforming all predictive signature scores except for miR4 and our miR37 signature. Furthermore, we observed that its expression was consistently linked to increased survival in all of our analyses. Previous research has shown that hsa-mir-335 can function as either an oncogene or tumor suppressor by regulating different pathways59. Recently, several studies have proposed different mechanisms for its therapeutic effect in AML. Zhang et al. 60 demonstrated that has-miR-335-3p directly inhibited the EIF3E gene, activating apoptosis and cell cycle arrest and ultimately reducing AML cell proliferation. Liu et al. 61 showed that has-miR-335-5p down-regulated NFS1, improving ferroptosis-based antitumor treatment in AML.
Regarding the comparison of predictive capacities between miRNAs and miRNA signatures, we observed notable differences between the use of individual miRNAs and miRNA signatures for survival prediction, with the latter generally being better predictors. However, as previously mentioned, we have identified that some individual miRNAs can predict outcomes better than signature scores when patients are stratified by cytogenetic risk groups. This indicates the necessity to develop new, specific miRNA signatures for survival prediction in these risk groups, particularly in patients classified as standard risk, where known predictors perform poorly and risk assessment is often inconclusive.
Conclusions
We have systematically collected and evaluated all microRNA-based predictors of survival in pediatric AML patients and we have proposed a new predictor model that is more accurate than previous ones, testing its predictive ability with robust methods to ensure its reproducibility in new patients. We have shown that scores calculated from miRNA signatures tend to be more effective than individual miRNAs in the prognosis of pediatric AML. However, neither our model, nor any other existing model, can yet be considered sufficiently accurate to be used as an independent predictor of survival in the clinic, and further research is needed in the field of microRNA signatures to find a viable miRNA model that accurately predicts disease outcome and that will ultimately guide treatment decisions. Moreover, how our signature and the other predictors perform when used in conjunction with other widely established predictor variables still needs to be evaluated.
This study contributes to the investigation of the role of miRNAs in proper risk estimation in pediatric AML patients and provides a new predictive model that would allow refinement of the current risk stratification.
Methods
Systematic review strategy
The systematic review was conducted in accordance with the Preferred Reporting Items for Systematic Reviews and Meta-Analysis (PRISMA) statement62. A detailed search was carried out from three different citation databases: Pubmed, Scopus and Web of Science. Our search query included the terms “pediatric”, “microRNA” and “Acute Myeloid Leukemia”, as well as every synonym added either manually or automatically by each search algorithm. Detailed queries are described in supplementary methods. As exclusion criteria, we used the following: we excluded reviews, articles studying predictors for specific subtypes of AML, and studies with patients whose miRNA expression was measured after treatment or transplantation. According to our inclusion criteria, we only selected articles that tested pediatric AML patients, and we looked for studies with miRNAs or miRNA signatures tested as independent predictors or whose expression was correlated with survival.
Patient selection
We used miRNA expression data from a set of 1441 pediatric AML patients from the Therapeutically Applicable Research to Generate Effective Treatments (TARGET) project4,63, publicly available in the Genomic Data Commons repository64. We excluded 27 patients who were older than 21 years old at diagnosis, as an upper age limit identified by the American Academy of Pediatrics, keeping 1414 patients for our analysis.
Development of miRNA predictive model
In order to establish a miRNA signature capable of predicting survival, we organized the patients into Discovery and Validation cohorts. As the TARGET data were originally obtained from patients with different protocols, we selected the patients from the protocols AAML0531 (n = 393), AAML03P1 (n = 66), and CCG2961 (n = 34) for the Discovery set (n = 493) and the patients from the protocol AAML1031 (n = 921) for the Validation set, as these patients had not previously been used to train any of the predictive models. The protocol and data distribution used to build the model were similar to Lim et al. 30 with additional steps. We downloaded the expression of 2280 mature miRNAs and filtered the low expressed counts, keeping 267 miRNAs that were subjected to the Lasso Cox regression method. This method allowed the selection of the 37 miRNAs with the greatest impact on survival and the fitting of a Cox model in the Discovery cohort using these 37 miRNAs, with a Lasso penalty applied to the obtained regression coefficients. Lasso Cox regression was internally subjected to a 5-fold cross-validation. This process was carried out using the biospear R package (version 1.0.2)65 with default parameters. The model was fitted to predict OS. The miRNAs selected in the final model comprised our new predictive miRNA signature (miR37).
Quality control
We conducted quality control of our new predictive signature using the sigQC R package (version 0.1.21)66. SigQC provides a systematic approach for the quality control of previously obtained gene signatures across multiple expression datasets by evaluating their expression, variability and structure. For comparison purposes, we generated an equal-length signature of 37 randomly selected miRNAs. We compared our miR37 signature with the random signature in both our TARGET-AML Validation cohort and a new pediatric AML cohort obtained from the Gene Expression Omnibus study GSE9713567. The GSE97135 dataset included miRNA expression data from 39 pediatric AML patients. The data were sequenced using a non-coding RNA microarray (Affymetrix GeneChip miRNA 4.0). Although survival data was not provided for the patients in GSE97135, it was not necessary for performing quality control using sigQC. We performed our analysis following the sigQC guidelines from Dhawan et al. 66.
Model evaluation
To evaluate the performance of our signature, along with other miRNA signatures and individual miRNAs found by systematic search, we first transformed the unfiltered miRNA expression counts to log2(counts per million). The trimmed mean of M-values normalization method was implemented when computing the counts per million values by edgeR R package (version 3.38.4). Log2 values were returned adding a small count of 2 to avoid zero values. Next, for comparative purposes, we calculated a score for each signature for each of the patients.
In the assessment of AMLmiR36 signature30, miR3 signature29, and miR4 signature28 scores using expression data, we followed the formulas provided by authors, where each score is calculated by the summatory of the given coefficient multiplied by each miRNA log(counts per million). For the miR24 signature, the coefficients are not provided, so we calculated them by fitting a Cox proportional hazards (CoxPH) model on OS with the 24 miRNAs in all 1441 patients. For our new miR37 signature, we used the coefficients of the Lasso Cox regression model fitted on OS in all the Discovery cohorts. Since the miR3 signature, miR4 signature, and some individual miRNAs were originally mapped to precursor miRNAs, but we have mature miRNA data in our patients, we estimated the expression of each immature miRNA by summing the counts of both the 5p and 3p mature miRNAs. In all studies where the miRNA maturation stage was not specified explicitly or by identifiers, the immature form was assumed. All miRNAs and coefficients used to calculate a score for each signature are available in Supplementary Table 1.
In order to analyze the association of each candidate with survival, we compared the patients with high or low miRNA expression. We used the median expression value to separate the patients with high and low miRNA expression or score. The estimation of survival curves was done with Kaplan–Meier and log-rank test to evaluate the differences in survival between high and low-expressed miRNAs.
We also fitted univariate and multivariate CoxPH models for each continuous miRNA expression value or miRNA signature score to assess if the miRNA was an independent predictor of survival. For the univariate analysis, only miRNA expression or signature score was analyzed. In the multivariate analysis, in addition to expression, we added as covariates the stem cell transplant (SCT) in the first complete remission, minimal residual disease (MRD) at the end of the first course of primary therapy, NPM mutation, CEBPA mutation, t(8:21) and inv(16), as they were the most significantly correlated with survival with a p-value below 0.005 (Supplementary Fig. 1A, B). For this CoxPH regression, we used all 1414 patients.
To assess the capability of each miRNA or signature score to predict survival, we used the validation cohort. To calculate the C-index, the dataset was randomly split using a 10-fold cross-validation, which increases the confidence in our results as it helps to avoid overfitting by providing an unbiased estimate of the model’s performance. We fitted a Cox proportional hazards regression model for each continuous miRNA expression or miRNA signature score using the validation-train cohort. We then extracted survival probability predictions by adjusting the Cox regression model to the validation-test cohort and calculated the C-index68 to measure predictive accuracy. The C-index (Harrell’s C index or Concordance index) is a commonly used measure to evaluate the performance of a model in predicting outcomes. We also calculated the time-dependent area under the receiver operating characteristic curve (AUC) to evaluate the predictive power of the miRNA signatures in all validation patients over time. A higher value of the two indicators represented a higher accuracy. Predictive accuracy assessment analyses were also performed on subsets of patients of different assigned cytogenetic risks: high risk (n = 174), standard risk (n = 678), and low risk (n = 561). As provided by the TARGET-AML project, risk group assignment was carried out at diagnosis and based on the cytogenetic and molecular abnormalities of each patient30. 5-Fold cross-validation was applied when calculating the C-index of high-risk patients. The signature of 37 randomly selected miRNAs was added to the predictive accuracy analyses as negative control.
miRNA enrichment analysis
We performed an enrichment analysis of our new predictive miRNA signature using TAM 2.069, a database of miRNA-disease and miRNA-function associations based on curated information. We performed an overrepresentation analysis of the 37 miRNAs and used the p-value method to select the best results.
Data availability
We used miRNA expression data from patients that is freely available through the Therapeutically Applicable Research to Generate Effective Treatments (https://ocg.cancer.gov/programs/target) initiative4,63, phs000465, at the Genomic Data Commons repository64 (https://portal.gdc.cancer.gov/projects). For signature quality control we used an external gene expression dataset, publicly available at the Gene Expression Omnibus repository (GSE97135)67.
Code availability
The underlying code for this study is available on GitHub and can be accessed via this link https://github.com/IvanEllson/miRNA-predictive-signature-survival.git.
References
Quessada, J. et al. Cytogenetics of pediatric acute myeloid leukemia: a review of the current knowledge. Genes (Basel) 12, https://doi.org/10.3390/genes12060924 (2021).
Slats, A. M. et al. Causes of death—other than progressive leukemia—in childhood acute lymphoblastic (ALL) and myeloid leukemia (AML): the Dutch Childhoold Oncology Group experience. Leukemia 19, 537–544 (2005).
Zwaan, C. M. et al. Collaborative efforts driving progress in pediatric acute myeloid leukemia. J. Clin. Oncol. 33, 2949–2962 (2015).
Bolouri, H. et al. The molecular landscape of pediatric acute myeloid leukemia reveals recurrent structural alterations and age-specific mutational interactions. Nat. Med. 24, 103–112 (2018).
Elgarten, C. W. & Aplenc, R. Pediatric acute myeloid leukemia: updates on biology, risk stratification, and therapy. Curr. Opin. Pediatr. 32, 57–66 (2020).
Conneely, S. E. & Stevens, A. M. Acute myeloid leukemia in children: emerging paradigms in genetics and new approaches to therapy. Curr. Oncol. Rep. 23, https://doi.org/10.1007/s11912-020-01009-3 (2021).
Nunes, A. et al. Cytogenetic abnormalities, WHO classification, and evolution of children and adolescents with acute myeloid leukemia. Hematol. Transfus. Cell Ther. 41, 236–243 (2019).
Sandahl, J. D. et al. The applicability of the WHO classification in paediatric AML. A NOPHO-AML study. Br. J. Haematol. 169, 859–867 (2015).
Lonetti, A., Pession, A. & Masetti, R. Targeted therapies for pediatric AML: gaps and perspective. Front. Pediatr. 7, 1–11 (2019).
Morris, K. V. & Mattick, J. S. The rise of regulatory RNA. Nat. Rev. Genet. 15, 423–437 (2014).
Anelli, L., Zagaria, A., Specchia, G., Musto, P. & Albano, F. Dysregulation of miRNA in leukemia: exploiting miRNA expression profiles as biomarkers. Int. J. Mol. Sci. 22, https://doi.org/10.3390/ijms22137156 (2021).
de Carvalho, I. N. S. R., de Freitas, R. M. & Vargas F. R. Translating microRNAs into biomarkers: what is new for pediatric cancer? Med. Oncol. 33, https://doi.org/10.1007/s12032-016-0766-4 (2016).
Liu, Y. et al. Role of microRNAs, circRNAs and long noncoding RNAs in acute myeloid leukemia. J. Hematol. Oncol. 12, 1–20 (2019).
Narayan, N. et al. Functionally distinct roles for different miR-155 expression levels through contrasting effects on gene expression, in acute myeloid leukaemia. Leukemia 31, 808–820 (2017).
Qi, X. & Zhang, Y. MicroRNA-199a deficiency relates to higher bone marrow blasts, poor risk stratification and worse prognostication in pediatric acute myeloid leukemia patients. Pediatr. Hematol. Oncol. 39, 500–507 (2022).
Liu, H., Wu, H. & Qin, X. MicroRNA-206 serves as a tumor suppressor in pediatric acute myeloid leukemia by targeting Cyclin D1. Pathol. Res. Pract. 215, https://doi.org/10.1016/J.PRP.2019.152554 (2019).
Emmrich, S. et al. miR-139-5p controls translation in myeloid leukemia through EIF4G2. Oncogene 35, 1822–1831 (2016).
Daschkey, S. et al. MicroRNAs distinguish cytogenetic subgroups in pediatric AML and contribute to complex regulatory networks in AML-relevant pathways. PLoS ONE 8, 1–16 (2013).
Yan, W. et al. MicroRNA biomarker identification for pediatric acute myeloid leukemia based on a novel bioinformatics model. Oncotarget 6, 26424–26436 (2015).
Obulkasim, A. et al. Classification of pediatric acute myeloid leukemia based on miRNA expression profiles. Oncotarget 8, 33078–33085 (2017).
Vanhooren, J. et al. Deciphering the non-coding RNA landscape of pediatric acute myeloid leukemia. Cancers (Basel) 14, https://doi.org/10.3390/cancers14092098 (2022).
Kumar, S. & Bakhshi, S. Diagnostic & prognostic role of microRNAs in paediatric acute myeloid leukaemia. Indian J. Med. Res. 144, 807 (2016).
Ng, S. W. K. et al. A 17-gene stemness score for rapid determination of risk in acute leukaemia. Nature 540, 433–437 (2016).
Elsayed, A. H. et al. A six-gene leukemic stem cell score identifies high risk pediatric acute myeloid leukemia. Leukemia 34, 735–745 (2020).
Shi, H., Gao, L., Zhang, W. & Jiang, M. Identification and validation of a siglec-based and aging-related 9-gene signature for predicting prognosis in acute myeloid leukemia patients. BMC Bioinform. 23, https://doi.org/10.1186/S12859-022-04841-5 (2022).
Esperanza-Cebollada, E. et al. A miRNA signature related to stemness identifies high-risk patients in paediatric acute myeloid leukaemia. Br. J. Haematol. 202, https://doi.org/10.1111/BJH.18746 (2023).
Zebisch, A. & Sill, H. How do non-coding RNAs impact treatment regimens currently being used in AML? Expert Rev. AntiCancer Ther. 22, 331–333 (2022).
Zhu, R. et al. A 4-microRNA signature for survival prognosis in pediatric and adolescent acute myeloid leukemia. J. Cell. Biochem. 120, 3958–3968 (2019).
Zhu, R. et al. A 3-miRNA signature predicts prognosis of pediatric and adolescent cytogenetically normal acute myeloid leukemia. Oncotarget 8, 38902–38913 (2017).
Lim, E. L. et al. MicroRNA expression-based model indicates event-free survival in pediatric acute myeloid leukemia. J. Clin. Oncol. 35, 3964–3977 (2017).
Zhang, P., Sun, D., Sun, X. & Li, H. Clinical significance of dysregulation of miR-381 in pediatric acute myeloid leukemia. Eur. J. Med. Res. 25, https://doi.org/10.1186/S40001-020-00442-1 (2020).
Bhayadia, R. et al. Endogenous tumor suppressor microRNA-193b: therapeutic and prognostic value in acute myeloid leukemia. J. Clin. Oncol. 36, 1007–1016 (2018).
Kuai, W., Bai, J., Guo, A. & Hong, Z. Upregulation of microRNA-100 predicts poor prognosis in patients with pediatric acute myeloid leukemia. Onco Targets Ther. 5, 213–219 (2012).
Hong, Z., Zhang, R. & Qi, H. Diagnostic and prognostic relevance of serum miR-195 in pediatric acute myeloid leukemia. Cancer Biomark. 21, 269–275 (2018).
Wang, Z., Hong, Z., Gao, F. & Feng, W. Upregulation of microRNA-375 is associated with poor prognosis in pediatric acute myeloid leukemia. Mol. Cell Biochem. 383, 59–65 (2013).
Lin, X., Wang, Z., Zhang, R. & Feng, W. High serum microRNA-335 level predicts aggressive tumor progression and unfavorable prognosis in pediatric acute myeloid leukemia. Clin. Transl. Oncol. 17, 358–364 (2015).
Lin, X., Wang, Z., Wang, Y. & Feng, W. Serum MicroRNA-370 as a potential diagnostic and prognostic biomarker for pediatric acute myeloid leukemia. Int. J. Clin. Exp. Pathol. 8, 14658 (2015).
Zhu, C. et al. Prognostic value of miR-29a expression in pediatric acute myeloid leukemia. Clin. Biochem. 46, 49–53 (2013).
Yang, J., Yuan, Y., Yang, X., Hong, Z. & Yang, L. Decreased expression of microRNA-122 is associated with an unfavorable prognosis in childhood acute myeloid leukemia and function analysis indicates a therapeutic potential. Pathol. Res. Pract. 213, 1166–1172 (2017).
Tian, C. et al. Low miR-192 expression predicts poor prognosis in pediatric acute myeloid leukemia. Cancer Biomark. 22, 209–215 (2018).
Ramamurthy, R. et al. miR-155 expression and correlation with clinical outcome in pediatric AML: a report from Children’s Oncology Group. Pediatr. Blood Cancer 63, 2096–2103 (2016).
Xu, L. H. et al. Overexpressed miR-155 is associated with initial presentation and poor outcome in Chinese pediatric acute myeloid leukemia. Eur. Rev. Med. Pharm. Sci. 19, 4841–4850, (2015).
Xu, L. et al. High level of miR-196b at newly diagnosed pediatric acute myeloid leukemia predicts a poor outcome. EXCLI J. 16, 197–209 (2017).
Qi, H. X. et al. MicroRNA 34b inhibits cell proliferation in pediatric acute myeloid leukemia via regulating LDHA. Eur. Rev. Med. Pharm. Sci. 23, 5351–5359 (2019).
Eis, P. S. et al. Accumulation of miR-155 and BIC RNA in human B cell lymphomas. Proc. Natl Acad. Sci. USA 102, 3627–3632 (2005).
Georgantas, R. W. et al. CD34+ hematopoietic stem-progenitor cell microRNA expression and function: a circuit diagram of differentiation control. Proc. Natl Acad. Sci. USA 104, 2750–2755 (2007).
Forrest, A. R. R. et al. Induction of microRNAs, mir-155, mir-222, mir-424 and mir-503, promotes monocytic differentiation through combinatorial regulation. Leukemia 24, 460–466 (2010).
Palma, C. A. et al. MicroRNA-155 as an inducer of apoptosis and cell differentiation in acute myeloid leukaemia. Mol. Cancer 13, 1–15 (2014).
Yan-Xia Han et al. Apoptosis of acute myeloid leukemia HL-60 cells induced by CDK inhibitor SNS-032 and its molecular mechanisms. J. Zhejiang Univ. (Med. Sci.) 174–178 https://doi.org/10.3785/J.ISSN.1008-9292.2015.03.009 (2015).
Scheibner, K. A. et al. MiR-27a functions as a tumor suppressor in acute leukemia by regulating 14-3-3θ. PLoS ONE 7, https://doi.org/10.1371/JOURNAL.PONE.0050895 (2012)
Azizmohammadi, S. et al. The role and expression of miR-100 and miR-203 profile as prognostic markers in epithelial ovarian cancer. Am. J. Transl. Res. 8, 2403–2410 (2016).
Cao, J. et al. miR-335 represents an independent prognostic marker in epithelial ovarian cancer. Am. J. Clin. Pathol. 141, 437–442 (2014).
Li, Y., Zhang, G., Wu, B., Yang, W., Liu, Z. miR-199a-5p represses protective autophagy and overcomes chemoresistance by directly targeting DRAM1 in acute myeloid leukemia. J. Oncol. https://doi.org/10.1155/2019/5613417 (2019).
Chen, P. H. et al. microRNA-199a/b-5p enhance imatinib efficacy via repressing WNT2 signaling-mediated protective autophagy in imatinib-resistant chronic myeloid leukemia cells. Chem. Biol. Interact. 291, 144–151 (2018).
Singh, P. MicroRNA based combinatorial therapy against TKIs resistant CML by inactivating the PI3K/Akt/mTOR pathway: a review. Med. Oncol. 40, https://doi.org/10.1007/S12032-023-02161-Z (2023).
Saudy, N. S. et al. BMI1 gene expression in myeloid leukemias and its impact on prognosis. Blood Cells Mol. Dis. 53, 194–198 (2014).
Díaz-Beyá, M. et al. Acute myeloid leukemia with translocation (8;16)(p11;p13) and MYST3-CREBBP rearrangement harbors a distinctive microRNA signature targeting RET proto-oncogene. Leukemia 27, 595–603 (2013).
Li, Q. & Wang, J. Long noncoding RNA ZFAS1 enhances adriamycin resistance in pediatric acute myeloid leukemia through the miR-195/Myb axis. RSC Adv. 9, 28126–28134 (2019).
Ye, L. et al. Functions and targets of miR-335 in cancer. Onco Targets Ther. 14, 3335–3349 (2021).
Zhang, L., Wang, X., Wu, J., Xiao, R. & Liu, J. MiR-335-3p inhibits cell proliferation and induces cell cycle arrest and apoptosis in acute myeloid leukemia by targeting EIF3E. Biosci. Biotechnol. Biochem. 85, 1953–1961 (2021).
Liu, J., Gao, W., Sheng, Y., Sun, J. & Wen, D. Resveratrol drives ferroptosis of acute myeloid leukemia cells through Hsa-miR-335-5p/NFS1/ GPX4 pathway in a ROS-dependent manner. Cell. Mol. Biol. (Noisy-le.-Gd.) 69, 131–137 (2023).
Page, M. J. et al. The PRISMA 2020 statement: an updated guideline for reporting systematic reviews. BMJ 372, https://doi.org/10.1136/BMJ.N71 (2021).
Farrar, J. E. et al. Genomic profiling of pediatric acute myeloid leukemia reveals a changing mutational landscape from disease diagnosis to relapse. Cancer Res. 76, 2197–2205 (2016).
Grossman, R. L. et al. Toward a shared vision for cancer genomic data. N. Engl. J. Med. 375, 1109–1112 (2016).
Ternès, N., Rotolo, F. & Michiels, S. biospear: an R package for biomarker selection in penalized Cox regression. Bioinformatics 34, 112–113 (2018).
Dhawan, A. et al. Guidelines for using sigQC for systematic evaluation of gene signatures. Nat. Protoc. 14, 1377–1400 (2019).
Zampini, M. et al. A three-miRNA-based expression signature at diagnosis can predict occurrence of relapse in children with t(8;21) RUNX1-RUNX1T1 acute myeloid leukaemia. Br. J. Haematol. 183, 298–301 (2018).
Harrell, F. E., Lee, K. L. & Mark, D. B. Multivariable prognostic models: issues in developing models, evaluating assumptions and adequacy, and measuring and reducing errors. Stat. Med. 15, 361–387 (1996).
Li, J. et al. TAM 2.0: tool for microRNA set analysis. Nucleic Acids Res. 46, https://doi.org/10.1093/nar/gky509 (2018).
Acknowledgements
This work is part of Ivan Ellson's doctoral thesis. Ivan Ellson is enrolled in the Ph.D. program in Biomedicine at the University of Granada, Spain. Ivan Ellson is supported by the Spanish Ministry of Science and Innovation (PEJ2018-004549-A). The Ramos-Mejía’s lab is supported by the Asociación “El Mundo de Namu”, Asociación Grupo Girasoles and Asociación de Madres y Padres de Niños Oncológicos de Granada (AUPA).
Author information
Authors and Affiliations
Contributions
Ivan Ellson conceptualized and designed the study, designed the data collection, collected data, carried out the analyses and critically reviewed and critically reviewed and revised the manuscript. Jordi Martorell-Marugán supervised data collection and data analysis and critically reviewed and revised the manuscript. Verónica Ramos-Mejia and Pedro Carmona-Saez conceptualized and designed the study, drafted the initial manuscript, coordinated, and supervised data collection and data analysis, and critically reviewed and revised the manuscript. All authors approved the final manuscript as submitted and agreed to be accountable for all aspects of the work.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Ellson, I., Martorell-Marugán, J., Carmona-Sáez, P. et al. MiRNA expression as outcome predictor in pediatric AML: systematic evaluation of a new model. npj Genom. Med. 9, 40 (2024). https://doi.org/10.1038/s41525-024-00424-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41525-024-00424-w
