
Abstract
Deep space represents a challenging environment for human exploration and can be accompanied by harmful health-related risks. We aimed to assess the effect of simplified galactic cosmic ray simulated (simGCRsim) and gamma (γ) ionizing radiation (IR) on transcriptome changes in right ventricular (RV) tissue after a single low dose (0.5 Gy, 500 MeV/nucleon) full body exposure in C57BL/6J male and female mice. In females, no differentially expressed genes (DEGs) and only 2 upregulated genes in males exposed to γ-IR were revealed. In contrast, exposure to simGCRsim-IR resulted in 4 DEGs in females and 371 DEGs in males, suggesting longer-lasting and sex-biased DEGs after simGCRsim-IR. Overrepresentation analysis of DEGs in simGCRsim-IR males revealed significant enrichment in pathways related to muscle contraction, hypertrophic cardiomyopathy, oxytocin release, the regulation of cytoskeleton, and genes associated with Alzheimer’s, Huntington’s, and Parkinson’s diseases. Our results suggested the RV transcriptome exhibits distinct responses after exposure based on both the IR and sex.
Similar content being viewed by others

Introduction
Deep space represents a challenging environment for human exploration with unique factors like microgravity and high (H) atomic (Z) number and energy (E) particle radiation (HZE) among other hazards. One of the major concerns for astronauts on long-term missions is deep space is radiation exposure, particularly from high-energy galactic cosmic rays (GCR) and particle bursts, which could lead to serious health-related issues1. GCR primarily comprises of very high-energy protons and alpha particles, with a smaller amount of high-charge, high-energy (HZE) nuclei traveling at nearly the speed of light. GCR nuclei, originating from beyond our solar system, are high linear energy transfer (LET) particles with sufficient energy to penetrate current spacecraft shielding. Although there are no direct studies on the use of GCR in humans, several lines of evidence suggest that GCR can be harmful to human cells. GCR can induce DNA damage and subsequent development of mutations, potentially leading to a broad spectrum of diseases. Extended exposure to microgravity adds further health concerns, including viral reactivation, space motion sickness, muscle and bone degradation, and orthostatic intolerance2. Earlier studies showed that short-term space missions affect cardiovascular physiology by decreasing blood circulation volume, diastolic pressure, left ventricular (LV) mass, and heart contractility, while extended exposure might cause cardiac atrophy, all were attributed to microgravity in space as well as gravitational forces while re-entering the Earth’s gravity3.
Our recent study on LV transcriptome analysis of mice exposed to gamma (γ) and simGCRsim-IR indicated that mice sex and post-IR time points significantly affect gene expression variability in LV heart tissue, with simGCRsim-IR causing more persistent transcriptome changes4. In LV tissue, we found that - (i) there were drastically different sex-dependent gene expression responses to the IR-type, i.e., females had decreased expression of inflammatory and TGF-beta-associated gene signatures, whereas males showed upregulation of fibrosis-related gene, both agreeing with the sex-associated cardiac function alteration in the same group of mice; (ii) we also reported that radiation effects in the heart can persist up to 550–660 days post-IR, with overwhelmingly sex-biased responses at the level of individual gene expression4.
To date, most of the studies on the effect of terrestrial and space-type IR on cardiovascular alterations have been reported for the LV, whereas alterations in right ventricular (RV) structure, function, and underlying molecular mechanisms have not been well studied. Our current study aimed to assess the effect of simGCRsim and γ-IR on transcriptome changes in RV tissue after a single full-body dose exposure in male and female C57BL/6J mice, respectively.
Results
Transcriptome landscape
RNA-seq of RV tissues of male and female mice after exposure to a single full-body dose of simGCRsim- and γ-IR was performed with sham/non-IR mice serving as a control group. To evaluate the global transcriptome variability among samples based on their gene expression profiles, we performed Principal Component Analysis (PCA) on the normalized expression data (Fig. 1). The largest source of transcriptomic variability was attributed to sex differences, where we observed the strongest separation along the first principal component (PC1). Female groups formed more compact clusters, indicating lower intra-group variability, whereas male groups exhibited a broader spread, suggesting greater variability in gene expression within this group. We investigated the contribution of different experimental factors with an ANOVA test. PC1 was significantly associated with sex differences, confirming that sex is the primary driver of transcriptomic variability, while PC12 was significantly associated with radiation exposure. RNA integrity values were associated with PC8, indicating that they contribute to a portion of the expression variability. To ensure the accuracy and reliability of our analysis, we removed all differentially expressed genes that were significantly associated with RIN values (Supplementary Fig. 1A, B) from the downstream analysis. This step was taken to minimize potential biases introduced by RNA fragmentation and to avoid false-positive findings.

Scatter plot of the first two principal components from PCA of normalized gene expression data. Each dot represents a sample colored by sex and radiation exposure type. The primary source of variability in gene expression is associated with sex differences. Male experimental groups (yellow, orange, and purple dots) are located in the upper right side of the plot and display a wider spread, indicating greater expression variability. In contrast, female groups (green, blue, and red dots) are more tightly clustered and located on the left side of the plot.
Differentially expressed genes and functional mining
DEGs were stratified and analyzed using the DESeq2 R package. To uncover DEGs between IR-exposed groups versus controls, we performed comparisons based on sex and IR-type for the 4 experimental groups. Our analysis indicated that differential expression of genes is dependent on sex and IR-type (Fig. 2A–D). It must be noted that there were no DEGs in female mice and only 2 upregulated genes in the male group (Fig. 2A, C). In contrast, exposure to simGCRsim-IR resulted in 4 DEGs in females (Fig. 2B) and 371 DEGs in males (Fig. 2D). Notably, simGCRsim-IR was associated with a higher number of DEGs compared to γ-IR, a pattern also observed in our previous transcriptomic analysis of the LV chamber4.

Volcano plots showing differentially expressed genes between the two irradiation (IR) groups (γ- and simGCRsim) for both females (A, C) and males (B, D). Each row in the plot grid corresponds to an IR type, and each column represents a specific gender. Red dots indicate genes with an adjusted p-value (FDR) < 0.05 and an average log₂ fold change > 0.5. Blue dots represent genes with an adjusted p-value (FDR) < 0.05 but with a log₂ fold change ≤ 0.5.
As no biological pathways using overrepresentation analyses (ORA) were identified, we used mouse-focused tools such as MGI Mouse Genome Informatics and gProfiler to find any significant associations with biological processes and signaling pathways5. In the simGCRsim-IR female group, 4 DEGs were identified, namely, Ier3, Kansl1, Gpbp1, and Hspb6. Ier3 is an early response protein taking part in apoptosis and cell cycle6, while Kansl1 encodes a nuclear protein mediating histone H4 acetylation and is implicated in a cellular processes such as enhancer regulation, cell proliferation, and mitosis7. In turn, Gpbp1 is a transcription factor targeting GC-rich regions, directing myofibril formation8 and is differentially expressed in human atherogenesis9. Hspb6 plays an important role in smooth muscle relaxation10 and mitochondrial biogenesis11,12.
Further, the two upregulated genes in the γ-IR male group were Mapt and Iigp1, that are associated with microtubule stabilization13 and interferon-induced immune responses14, respectively. Overrepresentation analysis of DEGs in male simGCRsim-IR animals revealed significant enrichment in pathways related to vascular smooth (padj = 0.031) and cardiac muscle contraction (padj = 0.014), hypertrophic cardiomyopathy (borderline association with padj = 0.061), oxytocin release (padj = 0.013), and the regulation of actin cytoskeleton (padj = 0.03). Additionally, pathways associated with neurodegenerative diseases, including Alzheimer’s (padj = 0.0005), Huntington’s (padj = 0.002), and Parkinson’s diseases (padj = 0.005), were also enriched. Among genes contributing to the above-mentioned pathways, those associated with mitochondrial respiration and functioning (Ndufa6, Ndufs7, Cox411, Ndufs5, Ndufa4l2, Ndufs2, Atp5g3, etc), mRNA synthesis (Polr2j), bone formation (Ppp3ca), and intracellular organelles and channels (Cltc, Itpr1, etc) were identified. Furthermore, pathways reflecting non-alcoholic fatty liver disease (padj = 5.49 × 10−6), diabetic cardiomyopathy (padj = 2.499 × 10−4) as well as abnormalities in mitochondrial functioning (padj = 1.27 × 10−2), myocardium morphology (padj = 1.207 × 10−2), respiration (padj = 5.01 × 10−3) were reported using gProfiler. In spite of no fibrosis-related pathway being identified in male RVs, significant associations of TGF-beta1 with morphogenesis (padj = 0.000005), tube, connective tissue and system development (padj = 0.00018), cell adhesion (padj = 0.00039), angiogenesis (padj = 0.011), and blood vessel morphogenesis (padj = 0.013) were detected. In female mice exposed to γ-IR, DEGs related to apoptosis (Cdk5rap3) and cell cycle (Ier3), angiogenesis (Tsp2, Baiap2), fibrosis and hypertension (Thbs2)15,16,17, transportation (Slc6a6, Ift122), ion transport (Scn7a), oxidative stress (Mapk14)18, ribosomal function (Rpl17ps3), and DNA damage (Ddit3) were detected. However, after correction, no significant DEGs were found.
Among the top upregulated genes in the male simGCRsim-IR group were Myh7, Myot, and Ifitm1, each showing more than a two-fold increased value compared to the control mice. The top downregulated genes, ranked by fold change values, were Myl4, Nppb, Stard10, Cytl1, and Gpx3 (Table 1). Interestingly, the Myh7 gene that encodes β-Myosin heavy chain (βMyHC), is expressed in slow, type 1 muscle fibers and in the heart ventricles19. In simGCRsim-IR male mice group, the Fbn1, belonging to the fibrillin family of proteins and involved in tissue homeostasis and fiber formation was downregulated20,21.
The heatmap visualization of significant DEGs across all samples revealed a clear expression pattern in the male simGCRsim-IR group, with distinct transcriptomic alterations compared to the sham and other groups both in expression direction and magnitude (Fig. 3). In contrast, male γ-IR samples exhibited fewer expression changes relative to their respective sham controls. In female animals, there was no clear pattern of expression changes for these genes. Overall, these results highlight sex-specific differences in transcriptome signatures in response to radiation exposure in the heart RV tissue, with males showing stronger and more defined expression changes, particularly after simGCRsim-IR exposure.

Heatmap of normalized expression values of DEGs identified across all comparison groups (256 genes). The color bar at the top of the heatmap represents sample groups, with colors corresponding to the legend on the right side of the plot. Sample clustering was turned off to preserve the original sample order and facilitate direct comparison across experimental conditions.
To identify common and unique changes between differentially expressed genes (DEGs) in the LV and RV chambers, we compared the DEGs from this study with those from our previous LV study4 for both genders (Fig. 4A, B). This comparison revealed a total of 16 overlapping genes, 12 of which were shared between the simGCRsim-IR groups of LV and RV (Supplementary Table 1). Among the overlapping genes were Tspan4, Rhobtb1, Herpud1, Glul, Cebpb, Bcl6, Rhoj, and Mknk2 (Fig. 4B) underlying signal transduction (padj = 0.0127) and exercise-induced circadian regulation (padj = 0.025). These overlapping genes suggested shared response to IR between the two chambers, particularly in male animals and reflect processes associated with circadian rhythm (Tspan4), signal transduction (Rhobtb1), endoplasmic reticulum stress response (Herpud1), glutamine synthesis (Glul), transcription (Cebpb), apoptosis (Bcl6), and cell adhesion (Rhoj). Interestingly, apoptotic and cell adhesion signaling pathways were both downregulated in LV and RV simGCRsim-IR male groups, indicating potential shared altered mechanisms caused by IR. All 12 overlapping genes were consistently deregulated in both direction and magnitude across both chambers. To avoid false positives, we removed 4 genes from the overlapping list that were also significantly differentially expressed due to variations in sample RIN values. In turn, there were also up- and downregulated genes, being present only in the RV transcriptome of simGCRsim-IR male mice. The top upregulated genes found in RV only were Myh7, Myot, Gm29015, Ifitm1, Gm50092, Ddit4, Sf3a2, Ldhb-ps, Cox5a, and Gm57414. Those genes were associated with cardiac muscle contraction pathway (padj = 0.044) and metabolic processes (padj = 0.03), while downregulated genes reflected enzymatic activity-underlying processes (padj = 0.05). Overall, it can be suggested that RV response to simGCRsim-IR is mainly considered to uniquely affect muscle contraction and metabolism.

Overlap between the groups is presented as upset plots for females (A) and males (B). Single dots in the matrix indicate group-specific DEGs, with the corresponding gene counts displayed in the top bar plots. Connected dots across matrix rows indicate overlapping genes shared between comparison groups. The total number of DEGs for each experimental group are shown in the horizontal bar plots on the left side of the matrix. A Overlapping and group-specific DEGs in female comparison groups. Only 2 overlapping genes were observed: one between the γ- and simGCRsim-IR groups in the LV, and the second between simGCRsim-IR groups of the LV and RV. B Overlapping and group-specific DEGs in male comparison groups. Overlap was observed only between the simGCRsim-IR groups of LV and RV tissues, with 12 genes in common.
Finally, Masson’s trichrome analysis revealed a two-fold substantial increase in perivascular fibrosis in the simGCRsim-IR male groups (Supplementary Fig. 2) compared to controls, whereas fibrosis remained unchanged in the γ-IR group. However, it must be noted that overrepresentation analysis performed have not identified significance (p = 0.82) in fibrosis-associated TGFb signalling pathway in simGCRsim-IR male mice (Supplementary Table 2), yet associations of altered genes with different biological processes have been detected.
Discussion
In male mice exposed to γ-IR, only 2 DEGs, Mapt and IIgp1, were significantly upregulated, whereas no DEGs were detected in female mice exposed to the same IR. Previous studies have shown that Mapt knockout mice exhibit a hypertrophic phenotype and a reduced right atrial rate22. Another report indicated that γ-IR had no significant effect on increased Mapt expression in male mice while a significant effect on increasing phosphorylation of tau, coded by the Mapt gene, has been detected13.
The absence of DEGs in female mice exposed to γ-IR, might be associated with protective hormonal effects against IR at younger age. Previous studies suggested that the treatment with estrogen and estrogen-like molecules might have a neuroprotective effect on radiation-induced brain degeneration23. Another report also demonstrated the protective role of sex hormones in terms of decreasing the mortality rate of irradiated animals24. Recently, Shedid et al. (2025) identified melatonin as a protective agent minimizing radiation impact in rats exposed to γ-IR25.
In female mice exposed to simGCRsim-IR, DEGs related to muscle contraction and hypertrophic cardiomyopathy were detected. Interestingly, the Ier3 gene was differentially expressed in both male and female animals in the simGCRsim-IR group. Ier3 is a radiation-induced immediate response gene known to inhibit apoptosis and play a crucial role in cell cycle6. Its downregulation has been observed during the progression of heart failure in mouse models26. Notably, in this study, Ier3 exhibited opposite expression patterns between sexes, being downregulated in female animals (FC = 0.69) while upregulated in males (FC = 1.33). This sex-specific Ier3 expression in opposite directions suggests potential differences in the biological response to simGCRsim-IR between sexes. Further, based on our previous results, we performed a comparative analysis to identify the same DEGs and also overlapping genes regardless of IR and sex between RV and LV heart chambers. The Tspan4, Rhobtb1, Herpud1, Glul, Cebpb, Bcl6, Rhoj, and Mknk2 genes were found to be differentially expressed in both RV and LV. It must be noted that Tspan4, a circadian rhythm regulator, was upregulated both in RV and LV males exposed to simGCRsim-IR, and has a rhythmic expression pattern in multiple organs27. A recent report has indicated RhoBTB1 as a protector against hypertension and arterial stiffness28. Herpud1, a component of the endoplasmic reticulum-associated degradation (ERAD) complex, has been proposed to negatively regulate pathological cardiac hypertrophy29. Another overlapping gene, the glutamate-ammonia ligase (Glul), is associated with the increased risk of cardiovascular diseases in type 2 diabetes30. In contrast, the Cebpb gene, coding CCAAT enhancer binding protein beta, is considered to have a protective role against pathological cardiac remodeling31. At the same time, the Bcl6 and Rhoj in both heart chambers were downregulated. The Bcl6 is responsible for the regulation (inhibition) of apoptotic processes32 and suppresses cardiac fibroblast activation33, while Rhoj is associated with focal adhesions in endothelial cells and regulates their angiogenesis34,35. Finally, the Mknk2, coding MAPK interacting serine/threonine kinase 2, has been discovered as a target for heart regenerative therapy; moreover, its overexpression has been linked to enhanced proliferation of cardiomyocytes both in vitro and in vivo36. Taken together, the functions of overlapping genes might suggest that IR affects genes related to cardiovascular function and heart remodeling similarly, irrespective of the heart chambers explored.
The heatmap visualization of significant DEGs across all samples showed a distinct expression pattern in the male simGCRsim-IR group, while male γ-IR samples exhibited fewer expression changes relative to their respective sham controls. Meanwhile, in female mice, no clear pattern of expression changes was observed. Overall, these findings emphasize sex-specific differences in transcriptome signatures following radiation exposure, with males displaying longer-lasting and well-defined expression changes, particularly in response to simGCRsim-IR.
In conclusion, this study confirmed the effects of a single-dose γ-IR and simGCRsim-IR on transcriptome changes in both male and female mice within the RV. Notably, the simGCRsim-IR seems to affect more DEGs than γ-IR. Meanwhile, sex had a significant contribution to IR-induced transcriptomic changes.
One of the key limitations of our study related to differential gene expression analysis is the presence of low RIN values in several samples. Low RIN values indicate RNA fragmentation/degradation, which can lead to inaccurate quantification of gene expression. This can potentially result in biased or false-positive differentially expressed genes (DEGs). To minimize these biases and improve the accuracy of our analysis, we implemented a custom read-counting method that only considers reads from a defined region starting from the 3′ end of the genes. Additionally, to further reduce the risk of false positives and enhance statistical power, we combined animals from two collection time points and analyzed them as independent of collection time. This approach allowed us to incorporate higher RIN value samples into the experimental groups, thereby improving the robustness of the analysis. However, this methodological adjustment also imposed a limitation on our study, as it precluded the investigation of age-dependent IR effects. Due to the necessity of merging time points, we were unable to distinguish potential time-dependent transcriptional responses to radiation exposure.
Methods
Animals and ionizing radiation
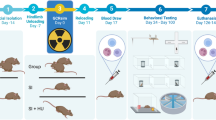
The detailed experimental design is shown in Fig. 5A, and the radiation ions, doses, energy, and animal procedures have been previously described37. All animal procedures were performed in accordance with the guidelines of the Guide for the Care and Use of Laboratory Animals for the National Institutes of Health and approved by the Animal Care and Use Committees at Brookhaven National Laboratory (BNL) (Upton, NY, USA) (BNL IACUC Protocol #502) and the Icahn School of Medicine at Mount Sinai (New York, NY, USA) (ISMMS IACUC Protocol #2019-0017). Sixty 3-month-old C57Bl/6J mice (30 males and 30 females) were used. We exposed 3-month-old male and female age-matched mice to 100 cGy, 0.662 MeV of 137Cs-γ-IR (20 animals, male/female: 10/10) and 50 cGy 500 MeV/nucleon simGCRsim-IR (20 animals, male/female: 10/10), whereas control mice (20 animals, male/female: 10/10) were not irradiated. Animals were monitored at least once a day for any physical or behavioral changes that might indicate distress, discomfort, pain, or injury. Only mice showing general signs of poor health and distress (e.g., rapid heart/respiratory rate, oral/nasal discharge, wound dehiscence, marked swelling, ulcerating neoplasms >2 cm or ulcerating, inability to eat/drink, or weight loss >15%) were immediately euthanized using 100% CO2 at 20% air replacement per minute rate, followed by cervical dislocation. Animals found spontaneously dead in their cages were grossly examined, and their carcasses were frozen. In this study, mice were euthanized via exsanguination at 440/440, and 660/550 (male/female) days after irradiation. Fresh snap-frozen RV tissues from male and female mice were collected at 440 and 660/550 days from control, 100 cGy γ and simGCRsim 50 cGy (5/group, respectively), and stored at −80 °C until further use. All animal procedures were performed in accordance with the ARRIVE guidelines. Among 20 non-IR mice, 4 samples were not sequenced due to low RIN numbers (RIN < 4), and the rest 56 samples were sequenced and analyzed. We did not sequence RV tissue of 1 mouse from the non-irradiated sham group at 440 days female, 1 mouse from gamma-IR females at 440 days, and 2 male mice exposed to simGCRsim-IR at 440 days. Only one group (simGCRsim IR males 440 days) contained 3 animals. The IR doses of 100 cGy-γ and 50 cGy-simGCRsim were selected based on the recommendations of The Space Radiation Element of NASA’s Human Research Program.

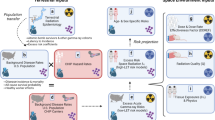
A Experimental design. Male and female mice were exposed to γ- and simGCRsim-irradiation. RV tissues were collected for sequencing at two timepoints (440 days and 660/550 days, male/female, respectively). B Sequencing and analysis workflow. Total RNA was extracted and sequenced using Illumina technology. The resulting reads were aligned to the mouse reference genome, and raw read counts were used for differential gene expression analysis. The diagram and the Workflow in this figure were created by using BioRender for our earlier published paper on transcriptomic changes in the left ventricular tissue4 of the same set of mice used in this study for the transcriptome changes in right ventricular tissue. Therefore, Fig. 5 is being reused in this paper under the Creative Commons Attribution (CC-BY) license.
Long term survival
Autopsies of mice that were found spontaneously dead or euthanized between 365- and 440-days post-IR showed a large distribution of organ-specific pathologic diagnoses, but none had a high prevalence to provide a unifying etiology for the higher mortality observed within the female mouse cohort. Therefore, we limited the collection of female samples to 550 days post-IR to ensure an adequate sample size for analysis of females, but males collection was extended to 660 days.
RNA extraction and sequencing
Total RNA was extracted from RV tissues using the RNeasy Mini Kit (Qiagen, USA) according to the manufacturer’s protocol and stored at −80 °C until further use. All animal procedures were performed in accordance with the ARRIVE guidelines. A poly-A selected mRNA library was prepared using the NEBNextUltra™II RNA Library Prep Kit. The sequencing was performed on the Illumina NovaSeq-6000 platform to collect an average of 48 million 2x150bp paired-end reads per sample.
Raw read processing, QC, and alignment
FastQC (available online at https://www.bioinformatics.babraham.ac.uk/projects/fastqc/ - version 0.11.9) was used for the quality assessment of raw sequencing reads (Q30% was greater than 94%). Next, we aligned the reads to the mouse reference genome (mm10) with the STAR aligner (version 2.7.8a). On average, 90.48% of the reads were mapped to the reference genome. We observed low RIN values in a number of samples from the control groups of both males and females, as well as at 660 days in the male simGCRsim-IR group (Supplementary Fig. S1). Due to these group-specific low RIN values, standard adjustment methods in differential expression analysis would not sufficiently address the issue and could lead to biased or inaccurate results. To minimize the impact of RNA degradation and reduce the likelihood of false positives, we applied a custom read-counting method as suggested by Sigurgeirsson et al.38. The principle of this method is that RNA fragmentation typically begins at the 5′ end. By counting the reads of each gene from a specific region starting from the 3′ end, it is possible to reduce the impact of fragmentation and obtain more accurate gene expression measurements. Based on our RIN values and RNA length distributions, we performed read counting from a 500 bp region starting from the 3′ end of each gene. To implement this approach, we generated a custom General Feature Format (GTF) annotation file in which gene lengths were truncated from the 5′ end, retaining only 500 bp from the 3′ end. Trimming was performed within exonic regions to ensure accurate read mapping. Subsequently, htseq-count (version 1.99.2) was used with the modified GTF file to count reads39.
Differential gene expression analysis and functional annotation
To assess the effects of γ- and simGCRsim-IR, we performed differential gene expression analysis across comparison groups. Raw gene count data were filtered to exclude genes with counts lower than 20 in any sample. Differential gene expression analysis was conducted using the DESeq2 R package (version 1.42.0)40. Due to the presence of multiple samples with low RNA integrity numbers (RIN) within the experimental groups, samples from different time points were combined in order to increase the number of high-RIN samples included in each group. This approach was taken to mitigate the impact of RNA degradation on downstream analyses and to ensure more reliable differential expression results by minimizing the risk of false-positive findings. Following normalization, scaling, and global variance estimation, we evaluated differential gene expression in 4 groups, comparing both γ- and simGCRsim-IR with their respective control groups in males and females. Genes with an FDR-adjusted p-value (padj) < 0.05 were considered as differentially expressed. Additionally, we conducted an overrepresentation analysis (ORA) using the enrichR R package (version 3.2) to identify pathways and biological processes significantly enriched with differentially expressed genes41. The overall study workflow is illustrated in Fig. 5B.
Masson Trichrome staining
Formalin-fixed paraffin-embedded longitudinal sections of RV (to include mostly the trabecular area and some inlet of RV) from 5 mice per group were stained with Masson’s trichrome (HT15, Sigma, St. Louis, MO, USA) according to the manufacturer’s instructions. Images were acquired using a DMi8 microscope (Leica Microsystems Inc., Deerfield, IL, USA). Fiji software (version 2.0.0-rc-68/1.52w for Mac) was used for image analysis and quantification.
PCA analysis of gene expression data
To understand the contribution of various parameters to the variability in gene expression in our samples, we performed principal component analysis (PCA) on normalized expression data. The eigenvectors derived from the PCA analysis were then utilized to conduct ANOVA statistical tests between groups for each clinical variable, thereby elucidating which variable factor can explain the most variability in gene expression. ANOVA was performed for each eigenvector across the groups for every variable, and subsequently, one eigenvector was selected per variable based on the lowest significance.
Data availability
The cited in the main text supporting Supplementary Figures and Data can be downloaded at https://doi.org/10.5281/zenodo.15624414 and the datasets can be downloaded using the following accession numbers GSE272168 for the left ventricle RNAseq dataset and GSE300189 for the right ventricle RNAseq dataset. Any additional datasets used and/or analyzed in this study are available from the corresponding authors upon request at the reasonable timeframe. Preprocessed raw gene expression count matrices used to perform the analysis of this study can be accessed from the GitHub repository at this link: https://github.com/hakobyansiras/RV_diff_exp_analysis.
Code availability
The underlying code to reproduce the results of this study is available on GitHub and can be accessed via the following link: https://github.com/hakobyansiras/RV_diff_exp_analysis.
References
Tomsia, M. et al. Long-term space missions’ effects on the human organism: what we do know and what requires further research. Front. Physiol. 15, 1284644 (2024).
Garrett-Bakelman, F. E. et al. The NASA Twins Study: A multidimensional analysis of a year-long human spaceflight. Science 364, https://doi.org/10.1126/science.aau8650 (2019).
Levine, B. D. et al. Maximal exercise performance after adaptation to microgravity. J. Appl. Physiol. (1985) 81, 686–694 (1996).
Zakharyan, R. et al. Long-lasting sex-specific alteration in left ventricular cardiac transcriptome following gamma and simGCRsim radiation. Sci. Rep. 15, 5963 (2025).
Raudvere, U. et al. G:Profiler: a web server for functional enrichment analysis and conversions of gene lists (2019 update). Nucleic Acids Res. 47, W191–W198 (2019).
Arlt, A. & Schafer, H. Role of the immediate early response 3 (IER3) gene in cellular stress response, inflammation and tumorigenesis. Eur. J. Cell Biol. 90, 545–552 (2011).
Meunier, S. et al. An epigenetic regulator emerges as microtubule minus-end binding and stabilizing factor in mitosis. Nat. Commun. 6, 7889 (2015).
Revert-Ros, F. et al. Goodpasture antigen-binding protein (GPBP) directs myofibril formation: identification of intracellular downstream effector 130-kDa GPBP-interacting protein (GIP130). J. Biol. Chem. 286, 35030–35043 (2011).
Bijnens, A. P. et al. Vasculin, a novel vascular protein differentially expressed in human atherogenesis. Blood 102, 2803–2810 (2003).
de Oliveira, A. A. & Nunes, K. P. An additional physiological role for HSP70: Assistance of vascular reactivity. Life Sci. 256, 117986 (2020).
Labbadia, J. et al. Mitochondrial stress restores the heat shock response and prevents proteostasis collapse during aging. Cell Rep. 21, 1481–1494 (2017).
Voos, W. & Rottgers, K. Molecular chaperones as essential mediators of mitochondrial biogenesis. Biochim Biophys. Acta 1592, 51–62 (2002).
Mohammed, M. R., El-Bahkery, A. M. & Shedid, S. M. The influence of different gamma-Irradiation patterns on factors that may affect cell cycle progression in male rats. Dose Response 20, 15593258221117898 (2022).
Pilla-Moffett, D., Barber, M. F., Taylor, G. A. & Coers, J. Interferon-inducible GTPases in host resistance, inflammation and disease. J. Mol. Biol. 428, 3495–3513 (2016).
Kozumi, K. et al. Transcriptomics Identify Thrombospondin-2 as a Biomarker for NASH and Advanced Liver Fibrosis. Hepatology 74, 2452–2466 (2021).
Kimura, T. et al. Thrombospondin 2 is a key determinant of fibrogenesis in non-alcoholic fatty liver disease. Liver Int. 44, 483–496 (2024).
Zhou, X. et al. Thrombospondin 2 is a novel biomarker of essential hypertension and associated with nocturnal Na(+) excretion and insulin resistance. Clin. Exp. Hypertens. 45, 2276029 (2023).
Zhong, W. et al. Activation of the MAPK11/12/13/14 (p38 MAPK) pathway regulates the transcription of autophagy genes in response to oxidative stress induced by a novel copper complex in HeLa cells. Autophagy 10, 1285–1300 (2014).
Tajsharghi, H. et al. Myosin storage myopathy associated with a heterozygous missense mutation in MYH7. Ann. Neurol. 54, 494–500 (2003).
Bouzeghrane, F., Reinhardt, D. P., Reudelhuber, T. L. & Thibault, G. Enhanced expression of fibrillin-1, a constituent of the myocardial extracellular matrix in fibrosis. Am. J. Physiol. Heart Circ. Physiol. 289, H982–H991 (2005).
Adamo, C. S., Zuk, A. V. & Sengle, G. The fibrillin microfibril/elastic fibre network: A critical extracellular supramolecular scaffold to balance skin homoeostasis. Exp. Dermatol. 30, 25–37 (2021).
Betrie, A. H. et al. Evidence of a cardiovascular function for microtubule-associated protein Tau. J. Alzheimers Dis. 56, 849–860 (2017).
Mitra, S. et al. Targeting Estrogen signaling in the radiation-induced neurodegeneration: a possible role of Phytoestrogens. Curr. Neuropharmacol. 21, 353–379 (2023).
Mirand, E. A., Hoffman, J. G., Reinhard, M. C. & Goltz, H. L. Sex hormones as protective agents against radiation mortality in mice. Proc. Soc. Exp. Biol. Med 86, 24–27 (1954).
Shedid, S. M., Abdel-Aziz, N., Algeda, F. R. & Saada, H. N. The mitigating effect of melatonin against radiation-induced inflammation and disturbance of reproductive hormones in female Albino Rats. Dose Response 23, 15593258251323796 (2025).
Zhou, Q. et al. Dysregulated IER3 expression is associated with enhanced Apoptosis in Titin-Based Dilated Cardiomyopathy. Int. J. Mol. Sci. 18, https://doi.org/10.3390/ijms18040723 (2017).
Zhang, R., Lahens, N. F., Ballance, H. I., Hughes, M. E. & Hogenesch, J. B. A circadian gene expression atlas in mammals: implications for biology and medicine. Proc. Natl. Acad. Sci. USA 111, 16219–16224 (2014).
Mukohda, M. et al. RhoBTB1 protects against hypertension and arterial stiffness by restraining phosphodiesterase 5 activity. J. Clin. Invest. 129, 2318–2332 (2019).
Torrealba, N. et al. Herpud1 negatively regulates pathological cardiac hypertrophy by inducing IP3 receptor degradation. Sci. Rep. 7, 13402 (2017).
Look, A. R. G. Prospective association of GLUL rs10911021 with cardiovascular morbidity and mortality among individuals with Type 2 Diabetes: The Look AHEAD Study. Diabetes 65, 297–302 (2016).
Bostrom, P. et al. C/EBPbeta controls exercise-induced cardiac growth and protects against pathological cardiac remodeling. Cell 143, 1072–1083 (2010).
Kumagai, T. et al. The proto-oncogene Bc16 inhibits apoptotic cell death in differentiation-induced mouse myogenic cells. Oncogene 18, 467–475 (1999).
Ni, J., Wu, Q. Q., Liao, H. H., Fan, D. & Tang, Q. Z. Bcl6 Suppresses cardiac fibroblast activation and function via directly binding to Smad4. Curr. Med. Sci. 39, 534–540 (2019).
Kaur, S. et al. RhoJ/TCL regulates endothelial motility and tube formation and modulates actomyosin contractility and focal adhesion numbers. Arterioscler. Thromb. Vasc. Biol. 31, 657–664 (2011).
Leszczynska, K., Kaur, S., Wilson, E., Bicknell, R. & Heath, V. L. The role of RhoJ in endothelial cell biology and angiogenesis. Biochem Soc. Trans. 39, 1606–1611 (2011).
Chen, B. R. et al. MNK2-eIF4E axis promotes cardiac repair in the infarcted mouse heart by activating cyclin D1. J. Mol. Cell Cardiol. 166, 91–106 (2022).
Brojakowska, A. et al. Lifetime evaluation of left ventricular structure and function in male C57BL/6J mice after gamma and space-type radiation exposure. Int. J. Mol. Sci. 24, https://doi.org/10.3390/ijms24065451 (2023).
Sigurgeirsson, B., Emanuelsson, O. & Lundeberg, J. Sequencing degraded RNA addressed by 3’ tag counting. PLoS One 9, e91851 (2014).
Putri, G. H., Anders, S., Pyl, P. T., Pimanda, J. E. & Zanini, F. Analysing high-throughput sequencing data in Python with HTSeq 2.0. Bioinformatics 38, 2943–2945 (2022).
Love, M. I., Huber, W. & Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550 (2014).
Chen, E. Y. et al. Enrichr: interactive and collaborative HTML5 gene list enrichment analysis tool. BMC Bioinforma. 14, 128 (2013).
Acknowledgements
The authors would like to acknowledge the support of Peter Guida, MaryAnn Petry, and their staff on the NASA support team at the Biology Department and BLAF animal facility at Brookhaven National Laboratory for their help with our longitudinal mouse lifetime studies. We would also like to acknowledge Adam Rusek and the NSRL operations team for their support in our particle radiation studies. This work was funded by the NASA Human Research Program, grant No: 80NSSC19K1079 (formerly, 80NSSC18K0921) and grant No: 80NSSC21K0549 (PI - Kenneth Walsh, DAG - subcontract) to D.A.G. and the ADVANCE Research Grant funded by the Foundation for Armenian Science and Technology and Research Grant 25FAST-1F004 funded by the Higher Education and Science Committee of the Ministry of Education, Science, Culture and Sport of the Republic of Armenia. This study was also supported by the National Institutes of Health/National Heart, Lung, and Blood Institute (NIH/NHLBI) under grants K01HL159038-01A1 and R25HL146166 (to MB), the American Heart Association Career Development Award (24CDA1269532 to MB), and the American Thoracic Society Research Program (Grant No. 23-24U1 to MB). This work was partially supported by American Heart Association Transformational Project Award 23TPA1140823, Innovative Project Award 23IPA105444, and National Institute of Health R01 grant HL164794 to V.N.S.G. The funders had no role in the data collection and analysis, the decision to publish, or the manuscript preparation.
Author information
Authors and Affiliations
Contributions
Conceptualization: D.A.G., A.A.; methodology: S.H., S.D., G.K., T.S., A.B., A.S., and V.N.S.G.; software: S.H., S.S., L.H. and S.D.; validation: A.B., M.B., A.K.R., and M.K.K.; formal analysis: A.B., M.B., S.H., T.S., A.S., G.K., and A.K.R.; investigation: A.B., M.B., M.K.K., S.Z., R.Z., S.H., S.D., A.S., A.A., V.N.S.G. and D.A.G.; resources: V.N.S.G., L.H., R.K. and D.A.G.; data curation: A.B., A.A. and D.A.G.; original draft preparation: R.Z., S.H., A.A. and D.A.G.; reviewing and editing: A.S., S.H., S.D., G.K., T.S., A.B., M.B., V.N.S.G., R.Z., A.A. and D.A.G.; visualization; S.H., S.D., T.S., G.K., A.S., A.A., and D.A.G.; supervision: A.A. and D.A.G.; project administration: A.A. and D.A.G.; funding acquisition: A.A. and D.A.G. All authors have read and agreed to the published version of the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Zakharyan, R., Hakobyan, S., Brojakowska, A. et al. The effects of space radiation on the transcriptome of heart right ventricle tissue. npj Microgravity 11, 46 (2025). https://doi.org/10.1038/s41526-025-00506-8
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41526-025-00506-8
