
Abstract
In this prospective cohort study, we analysed data from 502,364 participants (ages 40–69) in the UK Biobank, with follow-up until 2024. Logistic and Cox regression analysis identified generalized anxiety disorder (GAD) and obsessive-compulsive disorder (OCD) as independent risk factors for Parkinson’s disease (PD), with post-traumatic stress disorder (PTSD) patients under 71 also at increased risk. Panic disorder (PAD) showed no association with PD. Further analysis of anxiolytic drug use revealed that selective serotonin reuptake inhibitors (SSRIs), benzodiazepines (BDZs), medium-to-high frequency use of tricyclic antidepressants (TCAs) and serotonin norepinephrine reuptake inhibitors (SNRIs) were linked to PD incidence, while low-frequency use of TCAs and SNRIs was not. Mediation analysis indicated that GAD influenced PD risk through the thalamus, brainstem, and left putamen, while OCD and PTSD affected PD risk via brain regions including the angular gyrus, thalamus, and postcentral gyrus. These findings provide novel insights into PD mechanisms and potential therapeutic targets.
Similar content being viewed by others

Introduction
Anxiety disorders rank as the second most prevalent mental health conditions, trailing only depression, and contribute significantly to the global disease burden, accounting for 3.3% of worldwide morbidity and costing Europe ~€74 billion annually1. Anxiety disorders encompass generalised anxiety disorder (GAD, 6.2% lifetime prevalence), phobic anxiety disorders (PAD, 2.6% lifetime prevalence), obsessive-compulsive disorder (OCD, 1.3% lifetime prevalence), and post-traumatic stress disorder (PTSD, 8% lifetime prevalence)2,3. GAD is the most prevalent, particularly among women. PAD involves irrational fears of specific triggers, while OCD and PTSD, despite recent classification changes in the DSM-5, are often studied alongside traditional anxiety disorders due to their shared features4,5,6. Anxiety disorders are linked to higher risks of depression, suicidal behaviour, substance abuse, and social and occupational dysfunction, yet treatment rates remain alarmingly low globally, regardless of economic context7,8.
Recent neuroimaging and animal studies suggest that anxiety disorders may disrupt functional brain circuits involved in emotion regulation and sensorimotor control. For example, resting-state functional MRI (rsfMRI) data from two large multicenter trials involving 439 anxiety patients and 105 controls revealed increased thalamic connectivity across multiple brain regions9. In rodent models of stress-induced anxiety, glutamatergic neurons in the subthalamic nucleus (STN) exhibited enhanced excitatory projections to the lateral parabrachial nucleus (LPB)10. Studies on patients with comorbid anxiety and Parkinson’s disease (PD) have reported elevated medial prefrontal cortex (mPFC) GABA levels and enhanced amygdala-mPFC circuit connectivity11,12. These findings suggest that both glutamatergic hyperexcitability and GABAergic dysfunction may contribute to the pathophysiology of PD, possibly by increasing the vulnerability of key brain regions to neurodegeneration.
First-line treatments for anxiety include selective serotonin reuptake inhibitors (SSRIs) and serotonin-norepinephrine reuptake inhibitors (SNRIs), which increase synaptic monoamine levels and are generally considered safe for long-term use13,14. Benzodiazepines (BDZs) are also widely used for short-term symptom relief, but their long-term use is associated with significant risks, particularly in older adults and individuals with neurodegenerative conditions like PD15,16. Emerging evidence also suggests that untreated anxiety may exacerbate neuropsychiatric symptoms in PD, and genome-wide analyses have identified shared genetic loci between the two disorders17.
Moreover, long-term use of BDZs and certain tricyclic antidepressants (TCAs) has been associated with adverse neurological effects, including dopaminergic suppression, changes in receptor plasticity, and increased neuroinflammation18,19. These mechanisms may contribute to the progression of PD, raising important questions about the role of anxiety treatment in modifying disease risk.
Taken together, we propose an integrative neurobiological model in which anxiety disorders may elevate the risk of PD through both direct and indirect mechanisms that converge on shared pathophysiological pathways. Directly, chronic anxiety is associated with sustained alterations in functional connectivity and neurotransmitter balance-particularly glutamatergic hyperexcitability and GABAergic dysfunction-in brain regions central to PD pathogenesis, such as the thalamus, striatum, and brainstem. These neural changes may render these regions more susceptible to neurodegeneration. Indirectly, long-term pharmacological treatment of anxiety-especially with BDZs and certain TCAs-may exacerbate PD-related neurobiological vulnerability via dopaminergic and GABAergic dysregulation. For instance, chronic BDZs exposure has been linked to changes in receptor plasticity and neuroinflammatory processes that are also implicated in PD. We hypothesise that these direct and treatment-related effects are not independent, but cumulative and interactive, contributing jointly to regional brain volume loss, particularly in subcortical and frontal regions. This brain structural atrophy may mediate the increased PD risk observed in individuals with prolonged anxiety and/or sustained anxiolytic use.
While previous studies have primarily reported correlational associations, our model provides a mechanistic framework that connects these findings via biologically plausible pathways, informed by human imaging, animal models, and pharmacological evidence. This transition from observed correlations to a testable causal hypothesis is necessary for future targeted investigation. By proposing a unified neuropsychiatric pathway linking anxiety disorders, pharmacotherapy, and PD, we highlight the importance of early recognition, cautious long-term medication management, and the need for prospective research to validate these mechanisms.
Given the multifactorial nature of both anxiety disorders and PD, it is essential to account for potential confounding factors such as lifestyle, sociodemographic variables, and the dosage, duration, and frequency of anxiolytic use. To robustly test our proposed neurobiological model, we leveraged the UK Biobank (UKB), a comprehensive population-based cohort study with over 500,000 participants, to explore the prospective relationship between anxiety disorders and PD risk20,21. By integrating detailed sociodemographic, behavioural, and health data with primary care records, the research aimed to clarify the potential role of anxiolytic medications in PD pathogenesis, offering insights into the broader implications of anxiety treatment on neurodegenerative disease outcomes.
Results
Population baseline characteristics
During the 18-year follow-up period, 37,231 participants were diagnosed with anxiety disorders. Among them, 34,146 had GAD, 3775 had PAD, 810 had OCD, and 472 had PTSD, with 1906 participants experiencing at least two types of anxiety disorders (Supplementary Fig. 1, Table 1). In addition, 70,180 participants were prescribed anxiolytic medications: 36,707 were prescribed SSRIs (1–4 times: 11,520; 5–10 times: 6854; >10 times: 18,333), 27,004 were prescribed BDZs (1–4 times: 22,610; 5–10 times: 2338; >10 times: 2056), 36,641 were prescribed TCAs (1–4 times: 22,763; 5–10 times: 4660; >10 times: 9,218), and 5766 were prescribed SNRIs (1–4 times: 2420; 5–10 times: 944; >10 times: 2402), with 1906 participants using at least two different classes of anxiolytic drugs (Supplementary Fig. 1, Table 2). Population characteristics analysis indicated that older age, being male, white ethnicity, lower education level, history of smoking and excessive alcohol intake, BMI over 25, increased TDI index, higher PRS, and a history of hypertension, depression, and sleep disorders were associated with a higher PD risk (Table 1, Supplementary Table 1).
Anxiety disorders and PD
Univariate logistic regression analysis showed that GAD (OR = 2.331, 95% CI: 2.144–2.530, p < 0.001), PAD (OR = 1.361, 95% CI: 1.011–1.785, p = 0.033), OCD (OR = 3.162, 95% CI: 2.046–4.889, p < 0.001), and PTSD (OR = 2.235, 95% CI: 1.113–3.956, p = 0.012) were all risk factors for PD. After adjusting for covariates, multivariate logistic regression analysis indicated that GAD (OR = 1.762, 95% CI: 1.604–1.934, p < 0.001) and OCD (OR = 2.304, 95% CI: 1.471–3.439, p < 0.001) were associated with an increased risk of PD. In contrast, PAD (OR = 1.052, 95% CI: 0.778–1.388, p = 0.730) and PTSD (OR = 1.678, 95% CI: 0.869–3.053, p = 0.119) were not significantly associated with PD incidence (Fig. 1). PSM analysis confirmed that GAD and OCD are risk factors for PD, while PTSD and PAD are not (Supplementary Tables 2 and 3).

GAD generalised anxiety disorder, PAD phobic anxiety disorders, OCD obsessive-compulsive disorder, PTSD post-traumatic stress disorder. Model 0 includes four types of anxiety disorders (GAD, PAD, OCD, and PTSD). Model 1 = Model 0 + age + sex + ethnicity + education; Model 2 = Model 1 + smoking status + alcohol intake + BMI + TDI + coffee intake; Model 3 = Model 2 + PRS + hypertension + migraine + depression + sleep disorder.
Further subgroup analysis showed that, after controlling for covariates, PTSD patients younger than 71 years (OR = 4.119, 95% CI: 1.431–9.341, p = 0.003) and female PTSD patients (OR = 3.699, 95% CI: 1.428–7.859, p = 0.002) were at increased risk of PD. However, OCD patients with a university education were not associated with an increased risk of PD (Supplementary Table 4).
In the Cox regression analysis, a total of 18,419 individuals with anxiety disorders were included, comprising 16,862 with GAD, 585 with OCD, 135 with PTSD, and 1474 with PAD, with 612 individuals diagnosed with at least one of these conditions. Consistent with the results of the logistic regression analysis, after adjusting for covariates, both GAD (HR = 1.317, 95% CI: 1.089–1.593, p = 0.0045) and OCD (HR = 3.164, 95% CI: 1.580–6.336, p = 0.0011) were associated with an increased risk of developing PD, further supporting the notion that GAD and OCD may serve as independent risk factors for PD. Subgroup analyses further revealed that individuals younger than 71 years with PTSD had a significantly elevated risk of PD (HR = 7.665, 95% CI: 1.070–54.889, p = 0.043), whereas OCD in individuals with a university-level education was not significantly associated with PD incidence (HR = 2.744, 95% CI: 0.684–11.001, p = 0.154) (Supplementary Table 5).
Anxiolytic drugs and PD
Univariate logistic regression analysis indicated that low-frequency (0–4 times), medium-frequency (5–10 times), and high-frequency (>10 times) use of SSRIs, BDZs, and TCAs were associated with an increased risk of PD (all OR values > 1 and p < 0.05). Similarly, medium- and high-frequency use of SNRIs was associated with PD risk, whereas low-frequency use of SNRIs (OR = 0.312, 95% CI: 0.215–0.442, p < 0.001) was not associated with PD incidence (Fig. 2).

a selective serotonin reuptake inhibitors; b benzodiazepines; c tricyclic antidepressants; d serotonin norepinephrine reuptake inhibitors. Model 0 = four types of anxiolytic drugs (SSRIs, BDZs, TCAs, and SNRIs). Model 1 = Model 0 + age + sex + ethnicity + education; Model 2 = Model 1 + smoking status + alcohol intake + BMI + TDI + coffee intake; Model 3 = Model 2 + PRS + hypertension + migraine + depression + sleep disorder.
After adjusting for age, sex, education, BMI, smoking status, alcohol intake, hypertension, coffee intake, PRS, migraines, depression, and sleep disorders, multivariate logistic regression analysis showed that SSRIs use was associated with PD incidence (all OR values > 1 and p < 0.001) (Fig. 2a). Similarly, BDZs use was identified as a risk factor for PD (all OR values > 1 and p < 0.001) (Fig. 2b). Medium- and high-frequency use of TCAs were associated with PD risk (all OR values > 1 and p < 0.001), whereas low-frequency use of TCAs was not significantly associated with PD incidence (OR = 1.075, 95% CI: 0.931–1.236, p = 0.318) (Fig. 2c). Low-frequency (OR = 1.171, 95% CI: 0.796–1.658, p = 0.398) and medium-frequency (OR = 1.597, 95% CI: 0.938–2.534, p = 0.063) use of SNRIs were not associated with PD incidence, but high-frequency use of SNRIs (OR = 1.769, 95% CI: 1.321–2.324, p < 0.001) significantly increased PD risk (Fig. 2d). PSM analysis further confirmed that SSRI and BDZs use were risk factors for PD (all OR values > 1 and p < 0.001). Medium- and high-frequency use of TCAs and SNRIs were associated with PD incidence (all OR values > 1 and p < 0.05), whereas low-frequency use of TCAs (OR = 1.134, 95% CI: 0.972–1.318, p = 0.105) and SNRIs (OR = 1.402, 95% CI: 0.924–2.066, p = 0.099) was not associated with PD incidence (Supplementary Tables 6 and 7).
Subgroup analysis revealed that medium-frequency SNRIs use in university-educated individuals was associated with PD risk (OR = 1.727, 95% CI: 0.635–2.307, p = 0.036). In individuals younger than 71 years, high-frequency SSRIs, high-frequency SNRIs, low-frequency BDZs, and medium-frequency use of all four medication types were not associated with PD incidence (all OR values > 1 and p < 0.05). Additionally, medium-frequency TCAs and BDZs use in males was not associated with PD incidence (all OR values > 1 and p < 0.05) (Supplementary Tables 8–11).
Association between anxiolytic drug use and PD incidence in anxiety disorder patients
Given that the aforementioned medications are widely used for conditions other than anxiety disorders, we aimed to determine whether anxiolytic treatment in anxiety disorder patients specifically impacts PD risk. Excluding medication use for other conditions, we analysed 21,788 anxiety patients who used anxiolytic drugs and 171,734 non-anxiety patients who used anxiolytic drugs. After adjusting for confounders, we found that high-frequency SNRI use in anxiety disorder patients was associated with an increased risk of PD (OR = 1.929, 95% CI: 1.311–2.762, p = 0.001). Among non-anxiety disorder patients, SSRI use was identified as a risk factor for PD, as were medium-frequency BDZ use, medium- or high-frequency TCAs use, and low-frequency SNRIs use (all OR values > 1, p < 0.05). Additionally, low- and high-frequency BDZs use was associated with PD incidence, regardless of anxiety status (Supplementary Table 12).
Regional brain volumes mediate the relationship between anxiety disorders and PD
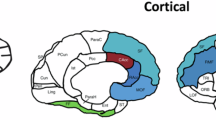
The extensive neural network connectivity between brain structures significantly influences various symptoms of diseases like PD, with anxiety potentially associated with changes in large-scale network connectivity. Exploring the interactions between these brain regions on the basis of anxiety disorders is therefore critical for understanding PD’s comprehensive impact. In our study involving 43,701 participants, we identified nine regional brain volumes that could potentially mediate the relationship between anxiety disorders and PD after adjusting for covariates. Specifically, GAD may increase the risk of PD through its impact on the volumes of the left thalamus, right thalamus, brain stem, and left putamen. OCD elevates PD risk via the left angular gyrus and right thalamus, while PTSD increases PD risk through the left postcentral gyrus and left thalamus but decreases it through the Crus II cerebellum vermis (Fig. 3a, b, Supplementary Table 13).

a β1*β2 the indirect effect, β the total effect, β’ the direct effect. MEP mediation effect proportion, GAD generalised anxiety disorder, OCD obsessive-compulsive disorder, PTSD post-traumatic stress disorder, SSRIs selective serotonin reuptake inhibitors, BDZs benzodiazepines, SNRIs serotonin norepinephrine reuptake inhibitors. b Visualisation of regional brain volumes with mediation effects. L left, R right. The structural localisation of positive brain volume clusters was conducted using the Automated Anatomical Labelling of Activations in SPM with a Macroscopic Anatomical Parcellation of the MNI MRI Single-Subject Brain (AAL), available online (https://neurovault.org/images/14257/), and the results were visualised using the Brain ViewNet system (BrainView, USA).
Regional brain volumes mediate the relationship between anxiolytic drug use and PD
In a cohort of 19,953 participants, our analysis, which controlled for covariates, revealed five regional brain volumes that may mediate the association between anxiolytic drug use and PD. Specifically, SSRIs increase PD risk through their impact on the left caudate volume, but reduce PD risk through the right IX cerebellum and right VIIIb cerebellum. BDZs decrease PD risk only through the right IX cerebellum, while SNRIs increase PD risk via the left temporal pole (Fig. 3a, b, Supplementary Table 13).
Discussion
This study indicates that GAD and OCD may be independent risk factors for PD, particularly among OCD patients without university education. PTSD patients under 71 may also be at increased PD risk, whereas PAD showed no significant association with PD incidence. SSRIs and BDZs are linked to PD risk, with medium- to high-frequency use of TCAs and high-frequency use of SNRIs also associated with increased risk. Notably, in non-anxiety patients, medium-frequency SNRI use is linked to PD. Eleven brain regions were identified as potential mediators in these relationships. GAD may increase PD risk through changes in the thalamus, brainstem, and left putamen, while OCD is associated with risk via the left angular gyrus and right thalamus. PTSD affects PD risk through the left postcentral gyrus and thalamus. SSRIs are linked to increased PD risk through the left caudate but decreased risk via the right IX and VIIIb cerebellum. BDZs decrease PD risk solely through the right IX cerebellum, whereas the left temporal pole increases PD risk.
Our study proposes a model in which anxiety-related neural circuit alterations-particularly involving the thalamus, putamen, and brainstem-create a neurobiological environment conducive to dopaminergic neuron vulnerability. The chronic use of anxiolytic medications, depending on class and usage frequency, may further influence this risk, either compounding excitotoxic stress (e.g. via TCAs or BDZs) or potentially mitigating it (e.g. via SSRIs at appropriate doses). Thus, anxiety disorders and their treatments act through partially overlapping neural pathways, providing a conceptual bridge between observed neuroimaging findings and the hypothesised pathogenesis of PD.
Anxiety disorders can be an appropriate response to stress, but they become pathological when anxiety is disabling and difficult to control. Research predominantly focuses on anxiety disorders as psychiatric comorbidities of PD. PD neuropathology likely originates in regions such as the olfactory bulb and lower brainstem, progressively involving the amygdala and hippocampus, leading to cognitive decline and emotional disturbances through disrupted dopamine, serotonin, and norepinephrine systems22,23. Retrospective studies suggest that anxiety symptoms may precede PD motor symptoms by up to 20 years24. Our findings indicate that GAD is directly associated with PD incidence, possibly due to abnormal activation and structural changes in the volumes of the amygdala, prefrontal cortex, thalamus, brainstem, and left putamen24. These changes may disrupt dopaminergic pathways, contributing to PD onset25. Therefore, we hypothesised that the long-term progression of GAD may lead to structural and functional alterations in the brain, which in turn could contribute to the pathogenesis of PD.
Similarly, OCD is a neurobehavioral disorder, with no direct evidence linking it to PD. However, retrospective studies have observed lifetime OCD in some patients before PD onset26. Our findings suggest that OCD may independently increase PD risk, particularly in those without college education. PD is characterised by dysfunction in the fronto-basal ganglia circuit, which is also implicated in OCD. In OCD, reduced top-down control by the prefrontal cortex and altered volumes in the left angular gyrus and right thalamus may disrupt basal ganglia activity, impairing dopamine regulation and potentially contributing to PD development27,28. Clinical management of OCD should thus include vigilance for PD symptoms, especially in patients lacking university education.
Subgroup analysis revealed that PTSD patients under 71 years of age may have an increased PD risk, which contrasts with studies showing a higher PD risk in older males with PTSD29. Some studies have found associations between PTSD and a milder PD motor phenotype, characterised by more neuropsychiatric and sleep disorder symptoms and fewer autonomic symptoms30. The discrepancy may result from small sample sizes. PTSD may increase PD risk via the left postcentral gyrus and thalamus, while the Crus II cerebellum vermis may mitigate this risk. Further research should explore these associations with larger PTSD cohorts. Additionally, our analysis confirmed that PAD is likely unrelated to PD, supporting the limited current research.
GAD, OCD, and PTSD may contribute to PD risk via thalamus-mediated mechanisms. Often referred to as a 'miniature cortex', the thalamus consists of multiple nuclei, many of which exhibit abnormal glutamatergic projections under conditions of negative affect such as anxiety. For instance, anxiety model mice have demonstrated enhanced glutamatergic projections from the prelimbic cortex (PL) to the mediodorsal thalamic nucleus (MD)31. Similarly, anxiety can increase the excitability of projections from glutamatergic neurons in the paraventricular thalamus (PVT) to the bed nucleus of the stria terminalis (BNST)32, while the inhibition of cerebellar nucleus glutamatergic neurons (CNGlu) projecting to the zona incerta (ZI) markedly reduces anxiety-like behaviours33. These findings suggest that anxiety disorders increase glutamatergic projections from thalamic nuclei, promoting hyperactivation and increased excitability of these pathways. Studies have also reported volumetric alterations in key brain regions in GAD patients, including reduced hypothalamic volume and increased amygdala and prefrontal cortical grey matter thickness24,34. Neuroimaging investigations of individuals with comorbid anxiety and PD have revealed enhanced connectivity within the amygdala–medial prefrontal cortex (mPFC) circuit, alongside increased GABA levels in the mPFC, underscoring the amygdala’s critical role in modulating anxiety and PD risk in an Italian cohort11,12. The current study is based on data from the UKB, which reflects a predominantly British cohort and single-centre design, thus introducing potential sample bias. Future multiethnic, multicentre studies are warranted to further explore the role of anxiety-related neurocircuitry, particularly involving the amygdala, in PD risk. Collectively, these data suggest that anxiety disorders may modulate PD risk through altered neural projections that enhance excitatory glutamatergic signalling and/or increase inhibitory GABAergic activity, both of which may contribute to the neuropathological processes underlying PD.
SSRIs work by blocking the reuptake of serotonin in neurons, making more serotonin available in the synaptic cleft, thereby enhancing its transmission in the brain. This mechanism helps regulate mood, alleviate anxiety symptoms, and improve overall psychological well-being35. SSRIs are considered first-line treatments for anxiety, with current adult practice guidelines recommending a slow titration to higher doses. Although this approach is widely accepted, it is based on clinical experience rather than robust clinical trial data. Meta-analyses have shown that SSRIs are effective in treating anxiety disorders, with higher doses within the therapeutic range providing greater benefits13. We confirmed that SSRI use in anxiety patients was not associated with an increased risk of PD, which suggests that long-term SSRI use within the therapeutic range may be safe for anxiety disorder patients. However, SSRIs are also used to treat other conditions, such as depression, bulimia nervosa, chronic pain, fibromyalgia, premenstrual syndrome (PMS), and premenstrual dysphoric disorder (PMDD)35,36,37. Our research indicates that SSRI use in non-anxiety disorder patients may increase the risk of PD, possibly due to serotonin system dysregulation leading to neuroinflammation, a critical factor in PD’s neurodegenerative process. Neuroinflammation can accelerate the loss of dopaminergic neurons, contributing to PD development35,38,39. Our findings indicate that SSRIs may increase the risk of PD through their impact on the volume of the left caudate, while potentially reducing PD risk by affecting the right IX and VIIIb cerebellum. This suggests that SSRIs may promote neuroplasticity, reduce inflammation, and improve mood through compensatory or adaptive mechanisms, which in turn influence cerebellar volume and potentially decrease PD risk. However, these compensatory mechanisms are limited. This study suggests that PD may be a neurological side effect of SSRIs, and further research is needed to elucidate the underlying molecular mechanisms.
SNRIs, also used as first-line treatments for anxiety disorders, regulate both serotonin and norepinephrine levels. However, high doses of SNRIs do not provide additional anti-anxiety benefits. Arai’s team reported the first clinical case of milnacipran (an SNRI) potentially linked to PD, suggesting that SNRIs could cause severe PD13,40. Our analysis of UKB data also found that high-frequency SNRI use for anxiety disorders might increase PD risk by affecting the left temporal pole. Neuroimaging studies have shown that changes in the volume of the temporal pole are associated with alterations in the structure and function of the basal ganglia. Therefore, we propose that long-term SNRI use may lead to damage to the dopaminergic system, potentially due to increased dopamine D2 receptor density in the striatum and infarction of the basal ganglia, ultimately contributing to PD pathology41.
BDZs, commonly used in clinical practice, bind to the ɑ and γ subunits of GABA receptors. BDZs produce hypnotic effects by binding to the ɑ1 subunit and anxiolytic effects by binding to the ɑ2 subunit. They also inhibit adenosine reuptake, exerting anxiolytic, anticonvulsant, and muscle relaxant effects42. The adverse effects of BDZs are controversial, with excessive use linked to Alzheimer’s disease (AD) due to their impact on GABA receptor activity and excitatory synaptic function, which can lead to neuronal dysfunction. BDZs may also influence neuroinflammatory responses in ways that lead to neuronal dysfunction43,44. Few studies have explored the association between BDZ use and PD, but our research suggests that there is an association between BDZ use and PD risk. However, although BDZs may influence PD risk through a reduction in right lobule IX of the cerebellar volume, the effect size is minimal. While the statistical significance suggests the presence of this indirect pathway, its clinical or biological relevance may be limited-particularly considering the potential compensatory or adaptive changes within the cerebellum that should not be overlooked. This finding might indicate that the primary impact of BDZs on PD is not through brain structure but through other pathways or mechanisms. Given BDZs’ substantial impact on neuronal function, they might influence ɑ-synuclein, a key protein in PD pathogenesis, through GABA receptors, warranting further molecular investigation. Retrospective analyses indicate that BDZ use is more prevalent among women and increases with age, which could explain our subgroup analysis findings showing an association between BDZ use and PD in women and older adults19.
While no direct evidence links TCAs to PD, their anticholinergic effects could lead to Parkinsonian symptoms. TCA use can lead to drug-induced Parkinsonism (DIP), which resembles PD but is typically reversible upon discontinuation45. Our analysis indicates that medium-to high-frequency TCA use, particularly in older women, may increase PD risk, potentially due to slower drug metabolism in this population, which leads to prolonged exposure and heightened risk.
Anxiety disorders are under-recognised, and the potential neurological side effects of anxiolytics may be underestimated. Early detection and further research are crucial for optimising treatment and understanding the underlying mechanisms.
Therefore, our findings support that the relationship between anxiety disorders, pharmacological treatment, and PD risk may be explained by an integrative neurobiological framework involving two interacting pathways. The first is a direct neural pathway, whereby chronic anxiety leads to sustained alterations in brain connectivity and neurotransmitter balance, particularly glutamatergic hyperexcitability and GABAergic dysfunction, in regions implicated in PD pathogenesis such as the thalamus, striatum and brainstem. These changes may increase the vulnerability of these regions to neurodegeneration. The second is an indirect pharmacological pathway, in which long-term use of anxiolytic drugs, especially BDZs and certain TCAs, may contribute to neurotoxicity through dopaminergic suppression, altered receptor plasticity and neuroinflammatory processes. These two pathways may act synergistically and cumulatively, accelerating structural brain changes, particularly in subcortical and frontal regions. This dual-pathway model provides a biologically grounded account of the observed associations, establishing a conceptual foundation for future longitudinal and mechanistic studies. It advances our understanding of the neuropsychiatric precursors of PD and highlights candidate targets for early intervention.
However, this study has several limitations. Although extensive adjustments were made for demographic and clinical confounders, residual confounding due to unmeasured factors, such as lifestyle behaviours, genetic predisposition, and environmental exposures, cannot be fully excluded. The lack of detailed clinical information on anxiety disorder severity, frequency, and duration in the UKB restricts our ability to explore dose–response relationships. The observational nature of the study, despite its prospective design, limits the ability to draw causal inferences. Additionally, potential selection bias may exist, as UKB participants tend to be healthier and more socioeconomically advantaged than the general population, which may affect generalisability. Although PSM and multivariable Cox models were applied to control for key covariates, residual heterogeneity in age, sex, and comorbidities may remain. Furthermore, medication data were based on self-reports and prescription records, which could lead to exposure misclassification or incomplete data capture. Future large-scale longitudinal studies with more comprehensive clinical characterisation, extended follow-up, and more diverse populations are needed to validate and expand on our findings.
Methods
Participants
This large-scale prospective cohort study utilised data from the UK Biobank, which recruited 502,364 participants aged 40–69 between 2006 and 2010, collecting extensive phenotypic and genotypic data. Since recruitment, participants have been followed for clinical outcomes, including PD, through hospital inpatient records, death certificates, and primary care records. Detailed descriptions of the UKB data are provided elsewhere. All participants provided written informed consent, and the UKB received ethical approval from the North West Multi-centre Research Ethics Committee.
In examining the relationship between anxiety disorders and PD, participants with missing baseline covariate data (n = 29,406) and those lost to follow-up or deceased (n = 28,404) were excluded, leaving 432,554 participants for analysis. Of these, 21,788 individuals were diagnosed with anxiety disorders. We also analysed the relationship between anxiolytic drug use and PD among 193,522 anxiolytic drug users.
Assessment of outcomes
The primary outcome was the incidence of PD, diagnosed through hospital inpatient records, Scottish Morbidity Records, and the Welsh Patient Episode Database, defined according to the International Classification of Diseases, 10th Revision (ICD-10), with Parkinson’s disease coded as G20. Follow-up began at the initial assessment and continued until the earliest date of PD diagnosis.
Anxiety disorders
Anxiety disorders were classified according to ICD-10 definitions, including GAD (F41.1), PAD (F40), OCD (F42), and PTSD (F43.1). Diagnoses were obtained through hospital inpatient records, Scottish Morbidity Records, and the Welsh Patient Episode Database.
Anxiolytic drugs
GP prescription records (1062) from the UK Biobank were analysed, with 193,522 individuals remaining after those with missing data were excluded. Anxiolytic drugs were categorised using the Anatomical Therapeutic Chemical (ATC) classification system, coded as N05, and grouped into four classes: (1) SSRIs: citalopram, escitalopram, fluoxetine, fluvoxamine, paroxetine, sertraline, and vilazodone; (2) BDZs: alprazolam, chlordiazepoxide, clobazam, clonazepam, clorazepate, diazepam, halazepam, lorazepam, oxazepam, and prazepam; (3) TCAs: amitriptyline, clomipramine, desipramine, doxepin, imipramine, maprotiline, nortriptyline, protriptyline, trimipramine, and amoxapine; and (4) SNRIs: desvenlafaxine, duloxetine, levomilnacipran, milnacipran, and venlafaxine.
Assessment of covariates
Baseline questionnaires provided data on sociodemographic factors, physical measurements, and medical history. Covariates included age (years), sex (male or female), ethnicity (white or non-white), education level (college or university degree vs. no college or university degree), smoking status (never, former, or current smoker), alcohol consumption frequency (never or special occasions only, 1–3 times per month, 1–2 times per week, 3–4 times per week, or daily/almost daily), body mass index (BMI, kg/m²), polygenic risk score (PRS), Townsend deprivation index (TDI), coffee intake (none, 0–2 cups per day, 2–4 cups per day, or more than 4 cups per day), hypertension (yes or no), diabetes (yes or no), migraine (yes or no), depression (yes or no), and sleep disorder (yes or no). Trained nurses measured height and weight at baseline assessment centres, and BMI was calculated as weight in kilograms divided by height in metres squared. The PRS was derived by comparing individual genotypes to known PD-associated genotypes to predict the probability of developing PD. Hypertension, migraine, depression, and sleep disorders were determined based on self-reported information and medical records.
Anxiety disorders and Parkinson’s disease
To assess the relationship between anxiety disorders and the risk of PD, we employed logistic regression analysis, a statistical method used to estimate the probability of a specific outcome given a set of predictor variables46. In this study, logistic regression was used to evaluate the association between anxiety disorders and PD incidence, controlling for potential confounders. We constructed four models with varying combinations of covariates: Model 0 included the four types of anxiety disorders (GAD, PAD, OCD, and PTSD); Model 1 was Model 0 + age + sex + ethnicity + education; Model 2 was Model 1 + smoking status + alcohol intake + BMI + TDI + coffee intake; and Model 3 was Model 2 + PRS + hypertension + migraine + depression + sleep disorder.
Sensitivity analysis of anxiety disorders and Parkinson’s disease
To ensure the robustness of our findings, we performed a sensitivity analysis using propensity score matching (PSM). PSM is a technique commonly used in observational studies to reduce bias and confounding, thereby clarifying the relationship between exposure and outcome47. By controlling for variables such as age, sex, ethnicity, education, smoking status, alcohol intake, BMI, TDI, coffee intake, hypertension, migraines, depression, and sleep disorders, we further elucidated the relationship between anxiety disorders and PD.
Subgroup analysis of anxiety disorders and Parkinson’s disease
We also performed subgroup analyses based on age, sex, and education level. Participants were divided into two age groups using the median value of 70 years (>71 years and ≤70 years). We then assessed the relationship between anxiety disorders and PD incidence within these age groups, as well as between males and females, and those with and without college education.
Cox proportional hazard model analysis
To further validate our findings and clarify the risk association between anxiety disorders and PD, we employed a Cox proportional hazards model incorporating a minimum latency period, wherein only PD events occurring at least five years after baseline were considered. At baseline (n = 502,364), participants with missing covariate data (n = 29,406), those diagnosed with PD before follow-up, and those who developed PD within five years of baseline (n = 28,404) were excluded. The final analytic sample comprised 472,541 individuals, of whom 2815 developed PD during follow-up. Individuals diagnosed with anxiety disorders prior to baseline were classified as having anxiety, while those diagnosed after baseline were considered non-anxious at baseline.
Anti-anxiety drugs and Parkinson’s disease
We conducted a cross-sectional analysis to explore the association between anxiolytic drug use and PD risk. Clinical management of anxiety disorders often involves various anxiolytic drugs, with treatment outcomes typically observed within 1–4 weeks and optimal results after 8–12 weeks or longer. Maintenance therapy generally extends for 1–2 years. We categorised medication use by frequency: no medication use (diagnosed but untreated), 1–4 times, 5–10 times, and more than 10 times. The analysis employed four models similar to those used for anxiety disorder analysis. Model 0 included four types of anxiolytic drugs (SSRIs, BDZs, TCAs, and SNRIs); Model 1 was Model 0 + age + sex + ethnicity + education; Model 2 was Model 1 + smoking status + alcohol intake + BMI + TDI + coffee intake; Model 3 was Model 2 + PRS + hypertension + migraine + depression + sleep disorder.
Sensitivity and subgroup analysis of anti-anxiety drugs and Parkinson’s disease
Sensitivity analysis using PSM controlled for age, sex, ethnicity, education, smoking status, alcohol intake, BMI, TDI, coffee intake, hypertension, migraines, depression, and sleep disorders. Subgroup analyses were also performed by age, sex, and education level to better understand the impact of anxiolytic drugs on PD incidence.
Mediation analysis
To further investigate the neuroanatomical correlates of anxiety disorders and anxiolytic medication use in PD, we examined 153 image-derived phenotypes (IDPs) obtained from T1-weighted MPRAGE data, preprocessed using the UK Biobank imaging pipeline. These included 139 regional grey matter volume estimates derived using FMRIB’s Automated Segmentation Tool (FAST) and 14 subcortical volume measures obtained using FMRIB’s Integrated Registration and Segmentation Tool (FIRST)48,49. All MRI data were acquired on a Siemens Skyra 3T scanner equipped with a 32-channel RF head coil at the UKB imaging centre in Stockport, with acquisition parameters set to a resolution of 1 × 1 × 1 mm³, a field of view of 208 × 256 × 256 matrix, and a total duration of ~5 min. Grey matter volume (GMV) estimates were generated and quality-controlled by the UKB imaging team, and made available as IDPs to approved researchers. Comprehensive details regarding imaging acquisition and processing are documented in the UKB imaging protocol (https://biobank.ctsu.ox.ac.uk/crystal/docs/brain_mri.pdf). Based on the imaging visits (Instance 2) of 46,831 participants (n = 25,023), we identified 102 individuals with a confirmed diagnosis of PD and complete imaging data, who were subsequently included in the neuroimaging analyses. Extreme outliers, defined as values exceeding ±4 standard deviations from the group mean, were excluded from the final imaging dataset to minimise the impact of data artefacts.
Data availability
The authors declare that the data supporting the findings of this study are available within the paper and its supplementary information files, all relevant data of the present paper are available from the corresponding author on reasonable request.
Code availability
The underlying code for this study is available in the git repository: https://github.com/tenayatherapeutics/Genetic-Survival-Analysis-in-UKB and https://github.com/yochaiedlitz/T2DM_UKB_predictions. The R version used is version 4.2.
References
Penninx, B. W. J. H., Pine, D. S., Holmes, E. A. & Reif, A. Anxiety disorders. Lancet 397, 914–927 (2021).
Szuhany, K. L. & Simon, N. M. Anxiety disorders: a review. JAMA 328, 2431–2445 (2022).
Kendler, K. S., Abrahamsson, L., Ohlsson, H., Sundquist, J. & Sundquist, K. Obsessive-compulsive disorder and its cross-generational familial association with anxiety disorders in a national Swedish extended adoption study. JAMA Psychiatry 80, 314–322 (2023).
DeMartini, J., Patel, G. & Fancher, T. L. Generalized anxiety disorder. Ann. Intern. Med. 170, ITC49–ITC64 (2019).
Bagatska, N. et al. Cytogenetic characteristic the patients of both sexes with phobic-anxiety disorders. Eur. Psychiatry 41, S306–S306 (2017).
Kalin, N. H. Novel insights into pathological anxiety and anxiety-related disorders. Am. J. Psychiatry 177, 187–189 (2020).
Khan, A., Faucett, J., Morrison, S. & Brown, W. A. Comparative mortality risk in adult patients with schizophrenia, depression, bipolar disorder, anxiety disorders, and attention-deficit/hyperactivity disorder participating in psychopharmacology clinical trials. JAMA Psychiatry 70, 1091–1099 (2013).
Erlangsen, A. et al. Association between spousal suicide and mental, physical, and social health outcomes: a longitudinal and nationwide register-based study. JAMA Psychiatry 74, 456–464 (2017).
Langhammer, T. et al. Resting-state functional connectivity in anxiety disorders: a multicenter fMRI study. Mol. Psychiatry 30, 1548–1557 (2025).
Jia, T., Chen, J., Wang, Y. -d, Xiao, C. & Zhou, C. -y A subthalamo-parabrachial glutamatergic pathway is involved in stress-induced self-grooming in mice. Acta Pharmacol. Sin. 44, 2169–2183 (2023).
Delli Pizzi, S. et al. Altered medial prefrontal connectivity in Parkinson’s disease patients with somatic symptoms. Mov. Disord. 37, 2226–2235 (2022).
Delli Pizzi, S. et al. High gamma-aminobutyric acid content within the medial prefrontal cortex is a functional signature of somatic symptoms disorder in patients with Parkinson’s disease. Mov Disord. 35, 2184–2192 (2020).
Jakubovski, E., Johnson, J. A., Nasir, M., Müller-Vahl, K. & Bloch, M. H. Systematic review and meta-analysis: dose–response curve of SSRIs and SNRIs in anxiety disorders. Depress. Anxiety 36, 198–212 (2019).
Bandelow, B. Current and novel psychopharmacological drugs for anxiety disorders. Anxiety disorders: rethinking and understanding recent discoveries. Adv. Exp. Med. Biol 1191, 347–365 (2020).
Baldwin, D. S. Clinical management of withdrawal from benzodiazepine anxiolytic and hypnotic medications. Addiction 117, 1472–1482 (2022).
Bazo-Alvarez, J. C., Nimmons, D., Walters, K., Petersen, I. & Schrag, A. Risk of Parkinson’s disease in people aged≥ 50 years with new-onset anxiety: a retrospective cohort study in UK primary care. Br. J. Gen. Pract. 74, e482–e488 (2024).
Li, W. et al. Genome-wide meta-analysis, functional genomics and integrative analyses implicate new risk genes and therapeutic targets for anxiety disorders. Nat. Hum. Behav. 8, 361–379 (2024).
Pena, E. et al. Antidepressants in Parkinson’s disease. Recommendations by the movement disorder study group of the Neurological Association of Madrid. Neurología 33, 395–402 (2018).
Olfson, M., King, M. & Schoenbaum, M. Benzodiazepine use in the United States. JAMA Psychiatry 72, 136–142 (2015).
Feng, Y. et al. Air pollution, greenspace exposure and risk of Parkinson’s disease: a prospective study of 441,462 participants. J. Neurol.271, 5233–5245 (2024).
Wang, Z.-Y et al. Association between irritable bowel syndrome and Parkinson’s disease by Cohort study and Mendelian randomization analysis. npj Parkinson’s Dis. 10, 70 (2024).
Khatri, D. K., Choudhary, M., Sood, A. & Singh, S. B. Anxiety: An ignored aspect of Parkinson’sś disease lacking attention. Biomed. Pharmacother. 131, 110776 (2020).
Dickson, D. W. Parkinson’s disease and Parkinsonism: neuropathology. Cold Spring Harb. Perspect. Med. 2, a009258 (2012).
Harrewijn, A. et al. Cortical and subcortical brain structure in generalized anxiety disorder: findings from 28 research sites in the ENIGMA-anxiety working group. Transl. Psychiatry 11, 502 (2021).
Brotchie, J. & Fitzer-Attas, C. Mechanisms compensating for dopamine loss in early Parkinson's disease. Neurology 72, S32–S38 (2009).
Kim, T. et al. Neurocomputational model of compulsivity: deviating from an uncertain goal-directed system. Brain 147, 2230–2244 (2024).
McGregor, M. M. & Nelson, A. B. Circuit mechanisms of Parkinson’s disease. Neuron 101, 1042–1056 (2019).
Fineberg, N., Chamberlain, S., Hollander, E., Boulougouris, V. & Robbins, T. Translational approaches to obsessive-compulsive disorder: from animal models to clinical treatment. Br. J. Pharmacol. 164, 1044–1061 (2011).
Barer, Y., Chodick, G., Chodick, N. G. & Gurevich, T. Risk of Parkinson disease among adults with vs without posttraumatic stress disorder. JAMA Netw. Open 5, e2225445 (2022).
Syed, G., Cheung, E., Saleh, Y., Pandey, D. & Barton, B. Influence of Co-Morbid PTSD in Parkinson’s Disease on motor and non-motor symptoms (P4-11.003). Neurology 98, 632 (2022).
Zhang, S.-R. et al. A prelimbic cortex-thalamus circuit bidirectionally regulates innate and stress-induced anxiety-like behavior. J. Neurosci. 44, e2103232024 (2024).
Zhao, D. et al. The altered sensitivity of acute stress induced anxiety-related behaviors by modulating insular cortex-paraventricular thalamus-bed nucleus of the stria terminalis neural circuit. Neurobiol. Dis. 174, 105890 (2022).
Zhao, Y. et al. Dual and plasticity-dependent regulation of cerebello-zona incerta circuits on anxiety-like behaviors. Nat. Commun. 16, 3339 (2025).
Terlevic, R. et al. Decreased hypothalamus volumes in generalized anxiety disorder but not in panic disorder. J. Affect. Disord. 146, 390–394 (2013).
Murphy, S. E. et al. The knowns and unknowns of SSRI treatment in young people with depression and anxiety: efficacy, predictors, and mechanisms of action. Lancet Psychiatry 8, 824–835 (2021).
Jannini, T. B. et al. Off-label uses of selective serotonin reuptake inhibitors (SSRIs). Curr. Neuropharmacol. 20, 693 (2022).
Halbreich, U., Borenstein, J., Pearlstein, T. & Kahn, L. S. The prevalence, impairment, impact, and burden of premenstrual dysphoric disorder (PMS/PMDD). Psychoneuroendocrinology 28, 1–23 (2003).
Howerton, A. R., Roland, A. V. & Bale, T. L. Dorsal raphe neuroinflammation promotes dramatic behavioral stress dysregulation. J. Neurosci. 34, 7113–7123 (2014).
Wang, Q., Liu, Y. & Zhou, J. Neuroinflammation in Parkinson’s disease and its potential as therapeutic target. Transl. Neurodegener. 4, 1–9 (2015).
Arai, M. Parkinsonism associated with a serotonin and noradrenaline reuptake inhibitor, milnacipran. J. Neurol. Neurosurg. Psychiatry 74, 137–138 (2003).
Ang, Y.-S. et al. A multi-pronged investigation of option generation using depression, PET and modafinil. Brain 145, 1854–1865 (2022).
Sieghart, W. & Savić, M. M. International union of basic and clinical pharmacology. CVI: GABAA receptor subtype-and function-selective ligands: key issues in translation to humans. Pharmacol. Rev. 70, 836–878 (2018).
Al-Kuraishy, H. M., Al-Gareeb, A. I., Saad, H. M. & Batiha, G. E.-S. Benzodiazepines in Alzheimer’s disease: beneficial or detrimental effects. Inflammopharmacology 31, 221–230 (2023).
Emmanouilidou, E. et al. GABA transmission via ATP-dependent K+ channels regulates α-synuclein secretion in mouse striatum. Brain 139, 871–890 (2016).
Lee, M. -y, Hong, S., Kim, N., Shin, K. S. & Kang, S. J. Tricyclic antidepressants amitriptyline and desipramine induced neurotoxicity associated with Parkinson’s disease. Mol. Cells 38, 732–738 (2015).
Schober, P. & Vetter, T. R. Logistic regression in medical research. Anesth. Anal.132, 365–366 (2021).
Morgan, C. J. Reducing bias using propensity score matching. J. Nucl. Cardiol. 25, 404–406 (2018).
Jiang, R. et al. Associations between grip strength, brain structure, and mental health in >40,000 participants from the UK biobank. BMC Med. 20, 286 (2022).
Bhatt, R. R. et al. Mapping brain structure variability in chronic pain: the role of widespreadness and pain type and its mediating relationship with suicide attempt. Biol. Psychiatry 95, 473–481 (2024).
Acknowledgements
This study was conducted the under application number 104811 for UK Biobank Resource. We would like to express our sincere gratitude to all associated researchers of UK Biobank. Our research was supported by the National Natural Science Foundation of China (Nos. 82171247 and No.82371433), and Scientific Research and Innovation Team of The First Affiliated Hospital of Zhengzhou University (No. ZYCXTD2023011).
Author information
Authors and Affiliations
Contributions
All authors made great contributions and have given permission to submit this manuscript. H.X.Y. was responsible for writing the article and designing the methodology. W.Z.Y. and F.Y.M. completed the code organisation, while L.M.J. and H.C.W. primarily handled data downloading and collection. L.Y.Y., Z.C.Y., and Y.X.H. managed and summarised the tables and figures. M.D.R. and W.Y.Y. were responsible for formatting the regional brain volume data. L.S.J. corrected the statistical analysis. Q.S.S., S.Y.M., M.C.Y., S.S.L., and X.Y.M. were in charge of the article's layout and formatting. S.C.H. contributed to the conception of this research, polished the writing, improved the logical coherence of the article, and ensured the final manuscript's submission. All authors collaborated to complete the discussion. Thanks to all authors for their efforts and support.
Corresponding author
Ethics declarations
Competing interests
All the authors declare that we have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Hao, X., Wang, Z., Feng, Y. et al. Association between anxiety disorder, anxiolytic drugs, and risk of incident Parkinson’s disease. npj Parkinsons Dis. 11, 252 (2025). https://doi.org/10.1038/s41531-025-01104-x
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41531-025-01104-x
