
Abstract
Ageing is the primary risk factor for Parkinson’s disease, yet the intricate interplay between these processes remains ambiguous. This position paper, a collaborative output from the PD-AGE consortium, addresses the urgent need for standardising methods in in vitro modelling. A panel of international experts recommends human induced pluripotent stem cell (iPSC)-derived models, with chemically induced ageing methods, such as the SLO cocktail, as a robust system. Furthermore, the consortium highlights the value of direct and semi-direct reprogramming for retaining donor-specific ageing phenotypes. The paper also outlines a prioritised panel of measurable parameters, categorised into senescence, inflammaging, omics profiling, and mitochondrial dysfunction, providing a consistent framework to enhance research reproducibility, investigating the nexus of ageing and Parkinson’s. In addition, we provide links to SOPs (https://doi.org/10.5281/zenodo.15056603) [1] to measure the key measurable ageing parameters outlined in this review to facilitate consistency and reproducibility within the field.
Similar content being viewed by others

Introduction
Parkinson’s disease epidemiology
Parkinson’s disease (PD) affects 0.3% of the global population, 1% of the over-60s and 5% of the over-80s population – this reflects the fact that ageing is the chief risk factor of PD1,2. PD is best characterised by its motor symptoms: a resting tremor, postural instability, rigidity and bradykinesia; resulting from the neurodegeneration of dopaminergic (DA) neurons in the Substantia Nigra pars compacta (SNpc)3. Non-motor symptoms, which are less well characterised, include: impaired REM sleep, cognitive dysfunction, depression and anxiety are more associated with neurodegeneration of non-dopaminergic neuronal populations4,5. A main pathological hallmark of PD is the aggregation of misfolded α-synuclein (aSyn), which is incorporated within Lewy Body structures. The exact role of Lewy bodies in contributing to, or otherwise ameliorating, PD pathogenesis caused by toxic soluble a-Synspecies, remains unresolved6. DA neurons are the most affected neuronal population in PD, but progressive loss of this subtype in the SNpc is also shown to occur in normal ageing7,8,9,10,11,12. This age-associated decline in DA neurons has been shown by a reduction in tyrosine hydroxylase (TH) staining in the SNpc of healthy, aged non-human primates1,13. This also suggests that though there are shared mechanisms of neurodegeneration, particularly of DA neurons in PD and ageing, they appear to be more pronounced in PD6,14,
Parkinson’s disease and ageing
There are multiple shared mechanisms in ageing and the pathogenesis of PD, including dysregulated autophagy, genomic instability, telomere attrition, impaired proteostasis, senescence, epigenetic modulation, inflammation, impaired intercellular communication, nutrient sensing, microbiota and mitochondrial dysfunction15,16. Of these mechanisms, mitochondrial dysfunction, dysregulated proteostasis, inflammation and cellular senescence show the greatest degree of overlap between PD and ageing14. However, the molecular intricacies underlying these two distinct processes remain unclear.
Mitochondrial dysfunction
Mitochondrial dysfunction in PD is linked to reduced electron transport chain complex I activity, which has been observed in PD patient nigral tissue homogenate17 and PD patient-derived fibroblasts18,19. Mitochondrial dysfunction is also implicated in PD through an increase of somatic mitochondrial DNA (mtDNA) deletions20 and point mutations21, in the SNpc of PD patients, resulting in impaired oxidative phosphorylation. MtDNA deletions are also characteristic of pathological ageing within highly metabolic cells, such as SNpc DA neurons22. Reactive oxygen species (ROS) are implicated in mtDNA deletion formation. DA neurons are at a higher risk to develop oxidative stress-associated damage, because ROS are generated through oxidative phosphorylation and through DA metabolism23. In addition to mutation load, mtDNA damage is independently complicit in PD pathophysiology. Leveraged against a multi-copy genome, mtDNA repair is more rudimentary compared with nuclear (nDNA) and lacks some mechanisms associated with oxidative lesion repair, such as Nucleotide Excision Repair (NER)24. Recent imaging and qPCR-based methodologies, measuring mtDNA lesion frequency, highlight elevated oxidative mtDNA damage in human post-mortem tissue25,26 Peripheral Blood Mononuclear Cells (PBMCs)27 and a human induced Pluripotent Stem Cell (hiPSC)-derived neuronal model of familial PD25. The exact role of mtDNA lesions in PD pathophysiology is not fully understood, but it has been proposed that they could impact mitochondrial homoeostasis, as evidenced by the increase in mtDNA biogenesis as a compensatory mechanism primarily in SNpc DA neurons28 or trigger an inflammatory response29,30,31.
Inflammaging
The combination of mitochondrial dysfunction, elevated ROS, and proteotoxicity, associated with overloaded protein degradation systems, are drivers of inflammation in PD and ageing32. Chronic inflammation, termed “inflammaging”, is another hallmark of ageing and neurodegeneration triggered by damage-associated molecular patterns (DAMPs), such as ROS, ATP and extracellular mtDNA. This results in the production of cytokines and further oxidative species, which directly damage cells and tissues33. Inflammatory markers, such as IL-6 and IL-8, also contribute to the senescence-associated secretory phenotype (SASP), which leads to the induction of cellular senescence34,35. Cells with a higher metabolic threshold, such as DA neurons, are most susceptible to chronic inflammation and are particularly prone to induced senescence36. PD is heterogeneous and arises from a number of genetic and environmental factors26,37. A recent study suggests that inflammaging may be specific to industrialised populations38, whilst other work suggests that inflammatory responses with age are common within species39,40. This supports and suggests both ageing and PD are heterogeneous and influenced by environmental factors in a manner that is human specific41.
Senescence
Senescence in mitotic cells is associated with irreversible cell cycle arrest in response to oncogenic stressors such as DNA damage42,43, telomere shortening44 and epigenetic perturbations45. Senescence is initiated by p16 or p21 cyclin-dependent kinase inhibitors, which trigger cell cycle arrest in response to the DNA damage response or telomere attrition46,47. Though the mechanistic basis of senescence is less well defined in neurons, relative to mitotic cells, a senescent phenotype has been reported in mouse primary Purkinje neurons through p21 in response to DNA damage and pro-inflammatory factors48,49. This p21-dependent senescence phenotype has, to our knowledge, not been investigated in PD, although a study has reported an increase of p21+ cells in the midbrain of PD patients, and that loss of SATB1, a DNA-binding protein, could induce p21-dependant cellular senescence in iPSC-derived DA neurons36. Whilst p16 levels have been shown to be elevated in PD32 and some studies have suggested telomere attrition is predictive of PD progression and severity50,51, contradictory data and lack of consensus on the role of telomere length in PD aetiology, limits the use of telomere length as a robust biomarker of PD in the context of ageing52,53,54. SASP comprises a number of factors that contribute to the senescent phenotype at the cellular level, these include growth factors, chemokines and cytokines, the latter of which can also act in paracrine fashion, spreading senescence to neighbouring cells34. The senescent phenotype is also characterised by mitochondrial-dependent ROS generation55, the accumulation of senescence-associated beta-galactosidase (SA-β-gal) in the lysosomes56,57, senescence-associated heterochromatic foci (SAHF)58 and phosphorylation of the histone protein H2AX (γH2AX) in response to double-stranded DNA breaks59. Biomarkers associated with senescence such as SA-β-gal activation, SASP induction, loss of lamin B1, γH2AX foci and oxidative stress have been observed in aged36,49,55,60,61,62,63,64,65and paraquat-induced Parkinsonian mouse models32. SA-β-gal and SASP biomarkers have also been observed in rat66,67,68,69 and non-human primate models of ageing70,71.
Disparities between Parkinson’s disease and ageing
Whilst there is clear evidence supporting the association between PD and ageing, there are notable differences in reported changes in DA neurons in Parkinsonian and aged individuals. These include: the number of neurons, levels of oxidative species, αSyn pathology, microglial activation, proteasomal and lysosomal dysfunction. This suggests that the interaction between ageing and PD pathophysiology is complex and not fully understood1,72. To better understand common and distinct mechanisms between ageing and PD, it is necessary to standardise the way in which we conduct research in both the fields of ageing and neurodegeneration. This includes recognising the most appropriate disease model(s) and selecting an appropriate panel of biomarkers to best investigate common pathways.
Cellular models of PD and ageing
Whilst animal models are an important tool to understand the mechanistic basis of ageing and PD, a key limitation of animal models is that PD is a uniquely human disease. The time taken for features of PD to manifest necessitates the use of exogenous induction of certain aspects of PD pathophysiology in animal models73,74,75,76,77 and no animal can adequately model all facets of PD simultaneously41. Cells sampled from patient peripheral tissues, such as PBMCs, allow more discrete means to assay human tissue biomarkers, but data have so far had limited reproducibility78,79. Fibroblasts can also be cultured from patients and age-matched donors and retain age-associated characteristics, although many features of the PD pathophysiology are less pronounced or not expressed in fibroblasts compared to neurons18,80,81,82. Features of PD pathology that have been successfully modelled in fibroblasts include mitochondrial dysfunction and turnover19,83,84,85,86,87,88, lysosomal dysfunction18,89,90,91,92,93 and inflammation14,35,94.
The discovery and use of “Yamanaka” transcription factors to convert human fibroblasts into hiPSCs, which can then be differentiated into neurons, provides a means to model PD and age-associated disease in a human-based system95,96,97,98. Since then, a number of cell reprogramming strategies have become available for the conversion of human fibroblasts into neural cells: the differentiation of reprogrammed hiPSCs, the differentiation of reprogrammed induced neuronal progenitor cells (iNPCs)99,100 and finally direct reprogramming from fibroblasts into neurons101,102,103 and astrocytes104.
Small molecules can be used to differentiate hiPSCs into DA neurons, which express pan-neuronal markers such as βIII-tubulin and the DA machinery, including the marker TH105,106. Subsequently, a range of hiPSC-derived neuronal models of idiopathic and familial PD have been derived from patients, including from patients harbouring SNCA, PINK1, PRKN, LRRK2 and GBA mutations80,96,107,108,109,110,111,112,113,114,115,116,117,118,119,120, as well as sporadic cases97,114. Transcription factors, such as Neurogenin 2 (NGN2), can also be used to generate a high induced neuron yield rapidly, bypassing the neuronal progenitor stage121,122. Using this approach in combination with other transcription factors or small molecules, the generation of iDAs has been possible123,124,125. A further refinement of this methodology is the use of doxycycline-induced NGN2, which improves efficiency and reduces batch heterogeneity126. A next step after the development of neuronal models was brain organoids, where midbrain organoids are of particular relevance for PD127,128,129,130,131,132. More recently, the level of complexity of 3D models have improved, with assembling of midbrain and striatal organoids to mimic the nigrostriatal pathway, as well as with the experimental induction of cellular ageing133. Like iPSC-derived neuronal differentiation, which provides the basis for differentiated organoid models131,134 cocktails of growth factors and small molecules can be used to differentiate stem cells into cells of a specific tissue type – such as midbrain neuronal populations135,136,137. Organoid systems offer the potential to model a number of disease features of PD and pathological ageing – including mitochondrial dysfunction, senescence, neuro-inflammation and omic-signatures138,139, Whilst organoids themselves are beyond the remit of this article, they do offer long term potential for modelling ageing and neurodegenerative disease. The standardisation of two-dimensional models will only serve to facilitate the development of organoid models going forward.
A notable consequence of the reprogramming process into pluripotency is the loss of cellular ageing signatures, including age-associated changes in DNA methylation patterns and histone modifications, and telomere shortening, which can affect the suitability of this approach in modelling certain aspects of cellular ageing and neurodegenerative diseases140,141. Other key age-associated features lost during rejuvenation are the progressive impairment in oxidative phosphorylation142, and the age-associated impairment in autophagy143. To overcome this limitation, researchers have developed several strategies to induce features of ageing in iPSC-derived cells. These include: long-term culturing144 and induced telomere shortening145.
By reprogramming terminally differentiated cells directly into somatic cells of another tissue, it is possible to circumvent the pluripotency stage101,102,103, a methodology termed ‘direct cell reprogramming’. Lineage-determining transcription factors can be used to reprogramme somatic cells into subtype-specific neurons, including DA neurons111,143,146.
The use of Yamanaka factors supplemented with neural transcription factors, can be used to reprogramme somatic cells into tri-potent induced neural progenitor cells (iNPCs) – which can be differentiated into neurons, astrocytes or oligodendrocytes99,100. This process of ‘semi-direct reprogramming’ differs from ‘direct reprogramming’ because somatic cells are first differentiated into progenitor cells, bypassing pluripotency, before being terminally differentiated into cells of a different lineage. This introduces an intermediate step in the differentiation process with the possibility of cryopreserving progenitor cells. The differentiation of human fibroblast-derived iNPCs into iDAs has been demonstrated for the investigation of metabolic and mitochondrial dysfunction in Parkinson’s disease18,147,148. Importantly, dermal fibroblasts harbour an endogenous heterogeneity149, which can lead to issues related to the clonal nature of iPSCs, which is not the case when using direct and semi-direct reprogramming. However, inherent inter-individual variability can impact the yield and reprogramming efficiency of directly reprogrammed cells143,150,151, or result in phenotypically immature neurons152. Overall, directly or semi-directly reprogrammed cells are less characterised than iPSC-derived cells. For all reprogramming methods, the somatic mosaicism of the starting cell type can affect the resulting cells. Dermal fibroblasts are thought to have more mosaicism than PBMC’s for example; however, a comparison of the starting cell type for reprogramming is beyond the scope of this review. Furthermore, the composition of the culture can change over time, especially with extended periods of culture. Hence, it is important to monitor the cellular makeup of the cultures on which the experimental assays have been performed on.
The PD-Age network
Accurately measuring ageing in the cellular models of PD is a complex challenge, which demands a collaborative, interdisciplinary approach. The PD-Age network fosters partnership and knowledge sharing among researchers to identify the most valid, reliable and scalable methodologies of assessment of the interplay between PD and ageing. Working group 2 emphasised the pressing necessity for robust and standardised methods to elucidate the overlapping mechanisms to establish the best practices for incorporating ageing into patient-derived cellular models used for PD research. With the aim of harmonising methodologies across studies, the group discussion was divided into two main objectives.
The PD-age network: standard operating procedures development
Firstly, the PD-Age Network developed rigorous methodological frameworks of the Standard Operating Procedures (SOPs) utilised for assessing key cellular processes involved in ageing and PD153. The group reached a consensus on the importance of methodological precision of SOPs for measuring senescence, inflammaging, telomere length detection and mitochondrial function. The group discussed which pathways should be included in the SOP list, deciding to focus on pathways that had wide applicability and would be accessible to a wide number of labs worldwide. Detailed protocols for assessment of these changes were developed cooperatively, including equipment and reagents necessary, software and step-by-step experimental procedures. This selection of cellular mechanism categories is not exhaustive but does reflect a representative cross-section of pathways that are compatible with commonly available methodologies and align with the collective expertise of the working group members. Moreover, various factors were taken into consideration when deciding on the relevant methods, including but not limited to: feasibility and scalability, sensitivity, reliability, practicability, ease of adoption and robustness across sites and levels of expertise required. These SOPs can be found at: https://doi.org/10.5281/zenodo.15056603153.
The PD-age network: choosing the reprogramming route
The second focus of this working group was to develop structured and consistent experimental frameworks for measuring PD- and age-relevant changes in vitro to minimise challenges which may be associated with it. This was done via workshops, questionnaires and literature reviews, to reach consensus on the most appropriate cellular reprogramming models and biomarkers for investigating both ageing and PD. Several measurable markers have been observed in cellular models of PD and ageing, which can largely be categorised as either inflammatory154, metabolic155, (multi)omic156 or senescent157. This part of the discussion centred on the importance of selecting the appropriate method to ensure that the cellular model captures the age-dependent vulnerabilities, which characterise PD pathology, while also recapitulating the disease phenotype. The working group undertook an in-depth comparison of iPSC (and iPSC with exogenously induced ageing phenotype), direct and semi-direct routes of reprogramming, to ensure the most appropriate method is chosen.
Here, we document the outputs from these sessions, made up of a panel of researchers with expertise in ageing and PD, to facilitate the standardisation of methodologies for the benefit of researchers of all experience levels wishing to conduct research into the impact of ageing on PD progression.
Choosing cell reprogramming route
iPSCs as a versatile tool for modelling neurodegenerative diseases
To date, many protocols have been established to differentiate iPSCs into various cell types of the brain, utilising developmental signalling cues—such as proteins, small molecules, and transcription factors—that are active during embryonic development. Although efficiency and best practices to differentiate various lineages of neuronal and glial cells have not been discussed by the panel, we recommend the book “Induced Pluripotent Stem Cells - Methods and Protocols”, for material and reagents, step-by-step protocols, and troubleshooting strategies158. Since differentiation protocols mimic natural developmental trajectories, cells differentiated from iPSCs typically resemble primary cell types more closely than those that are directly reprogrammed from fibroblasts.
Due to their pluripotent nature, iPSCs can be expanded in vitro prior to differentiation, thus resulting in a high yield of both pluripotent and differentiated cells, and the possibility to scale-up experiments to perform large-scale screens, deep phenotyping experiments or large-scale omics-related studies. As a direct consequence, iPSCs have been characterised in detail, and in some instances, their differentiation paths have been better described than the corresponding processes that control direct reprogramming. Therefore, iPSCs constitute the method of choice when setting up co-culture experiments159,160, microfluidic-based organ-on-a-chip cultures161,162,163,164,165,166,167 and 3D cultures168,169,170,171,172. The advent of efficient gene editing methods expanded iPSC’s versatility, allowing the generation of genetically modified cell lines carrying known pathological mutations or risk variants or correcting such mutations in patient-derived iPSCs, to produce isogenic control lines173. Finally, iPSC-derived precursor cells show great structural and functional integration when engrafted, and have been used in clinical trials, further consolidating their biological and translational relevance57,174,175,176.
Induction of cellular ageing in iPSC-derived cells: where do we stand
Historically, the most used methods were based on replicative stress17, ionizing gamma-ray irradiation177 or ectopic expression of progerin95, a truncated, pathogenic version of the nuclear lamina protein Lamin A. Although very efficient, those methods present caveats and limitations that weaken their relevance in the study of brain ageing in neurodegenerative diseases. For instance, replicative stress is not compatible with post-mitotic cells such as neurons, which also exhibit a high resistance to ionizing gamma-ray irradiation. While the progerin-based approach affects the nuclear envelope, it does not reproduce epigenetic reprogramming, and it has been shown to lack the full complexity of age-related epigenetic drift178. Moreover, this approach can trigger acute cellular stress, such as apoptosis and rapid senescence, which may mask or exaggerate the induced ageing and disease phenotypes.
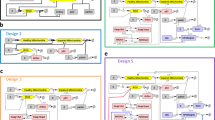
More recent studies have identified alternative methods to induce ageing in iPSC-derived models. It has been proposed that genetic inactivation of SATB1179, a transcriptional regulator whose expression is reduced in DA neurons of PD patients, could be used to investigate the drivers of ageing specifically occurring in PD as opposed to the broad ageing phenotype. RNage180, an RNA-seq-based method to calculate ageing scores, can be used to both validate and compare existing protocols and as a screening tool to identify novel strategies. When used to study gene expression profiles from cells treated with several hundreds of compounds, it showed that a few of them, including Fludarabine, could induce an increased RNage score, and cause typical markers of cellular ageing. Similarly, a CRISPR-based whole genome screening181 can also be used to identify regulators of ageing. This approach in iPSC-derived neurons helped identify the neddylation pathway as a potential regulation of ageing in Alzheimer’s disease (AD) and could be used to model late-onset phenotypes in PD models. Although promising, these techniques need to be validated in different systems, and further optimization is also required. All methods, along with a more detailed description of their specific pros and cons can be found in Fig. 1.

Each method is defined as follows: Long-term culture17, cells are maintained in culture and frequently passaged until their proliferative potential is exhausted; γ-Ray irradiation177, sublethal doses are used to induce DNA-damage and trigger senescence; Progerin95, the ectopic expression of this mutant form of lamin A is used to mimic the effect on cells of the Hutchinson-Gilford Progeria Syndrome; SATB1179, genetic downregulation of this chromatin remodeller in the context of PD leads to the activation of key senescence genes and ageing pathways; Fludarabine180, can cause signs of ageing in hiPSCs by interfering with DNA synthesis; Neddylation181 loss of function in iPSC-derived neurons leads to increased hallmarks of ageing and exacerbates neuronal loss in AD and PD neurons; Small molecules182,185 that pharmacologically target autophagy, exclusively (SBI-026965) or in combination with nuclear lamina formation and DNA repair (SLO cocktail), induce signs of ageing in iPSC derived neuronal and glial cells.
Among the discussed strategies, we identified the administration of small molecules targeting known ageing-related molecular pathways as the most relevant method to induce ageing in iPSC-derived models of PD182. This strategy has multiple advantages: it is easy to use and accessible, time- and cost-effective and very versatile, as the used compounds and their dosage can be adapted based on their relevance to the cell type of choice and the disease to be modelled. Furthermore, simultaneously targeting multiple pathways better mimics the effects of ageing on overall cell health, thus providing many features associated with ageing. To date, the most promising treatment is the SLO cocktail, which combines three molecules, SBI-0206965, Lopinavir and O-151, that respectively target autophagy, Lamin A biogenesis and DNA glycosylase and together base excision repair183,184. Defective autophagosomes lead to impaired mitochondrial clearance and increased oxidative stress, whereas DNA glycosylase and Lamin A biosynthesis impairment affect nuclear architecture and lead to DNA damage accumulation. Although it has not yet been tested on DA neurons, the SLO cocktail has been successfully applied to age iPSC-derived cortical neurons182, and human microglia185, suggesting the method has the potential to be used with many other PD-relevant cell types. As such, the working group recommends the SLO cocktail treatment as the preferred method to age iPSC-derived brain cells.
Preserving the ageing signature with semi-direct and direct reprogramming
Although our working group established that iPSC-derived models with accelerated ageing should be the system of choice to study the effects of ageing on PD, there are a few instances where preserving the ageing signature of the donor should be preferred. Neurons and astrocytes directly or semi-directly reprogrammed from patient-derived skin fibroblasts maintain the ageing signature of the donor143,186,187.
Directly reprogrammed induced neurons (iNs) retain their age-associated epigenetic and transcriptomic signatures, Oxidative Phosphorylation (OXPHOS) and autophagy impairment143,187,188,189,190, DNA damage143,187 and expression of mature TAU isoforms143,191. A select number of studies using directly or semi-directly reprogrammed iNs have successfully modelled mitochondrial and lysosomal dysfunction associated with ageing or neurodegenerative disease, including PD18,142,143,148,187,188,192,193.
By replicating the exact ageing profile in a patient-specific matter, these strategies represent a powerful tool to study disease mechanisms in an ageing context. However, direct reprogramming methods were not selected as the preferred reprogramming route because of the current challenges associated with their use. Since iNs become post-mitotic early in the conversion process194, any study necessitating a high neuronal yield requires extensive expansion of fibroblasts, which can lead to replicative senescence or metabolic changes in parental cells impacting on the reprogramming efficiency and the generated cell product195. However, semi-reprogramming methods effectively overcome yield limitations, enabling the production of large quantities of neurons148. Heterogeneity between batches14,143 and lack of protocol standardisation are also prominent limitations of these models. A detailed list of advantages and disadvantages of direct- and semi-direct reprogramming is reported in Fig. 2.

Advantages and disadvantages of direct and semi-direct reprogramming in the study of ageing in PD.
A feature of PD pathology and ageing not successfully modelled by any of the cell reprogramming methods described so far, is the interaction between distinct cell types within the brain. This could potentially be achieved by the co-culture of different reprogrammed cell types, but success in this area has thus far been limited104,163,186,196,197,198,199.
In conclusion, iPSC-derived models, and direct and semi-direct reprogramming all present advantages and disadvantages (Fig. 1). While direct and semi-direct reprogramming is generally more time and cost-effective and present the clear advantage of retaining the donor ageing signature, iPSC-based models are overall more standardised, high-throughput and versatile. Thus, selecting the appropriate method should be driven by the specific objectives and needs of the study at hand.
Assessing ageing in cellular models
Selection and prioritisation of ageing assays
To comprehensively evaluate assays that can be used to evaluate cellular age in in vitro models of PD, the working group identified commonly used assays across four key areas: senescence and inflammaging, omics profiling and mitochondrial function. These key areas were prioritised because they have been clearly linked to ageing16,200,201 but, except for mitochondrial function, remain distinct from the aetiology of heritable PD202. Subsequently, the collective expertise of the working group members was surveyed to establish a prioritised list of assays that are robust and can be used to validate ageing phenotypes in cellular models. Figure 3 summarises the selected tests and their corresponding functionalities.

Each criteria is defined as follows: time effective, how quickly the method produces the cell type of interest; cost effective, relative expense of the approach; easy to implement, overall complexity of the method; protocol standardization, availability of standard procedures; quality controlled parental cells, ability to maintain high-quality cells without unwanted mutations or inconsistencies, and accessibility of quality control assays; retention of donor epigenetic age, whether the method preserves age related epigenetic modifications; high yield, efficiency of producing a large number of viable cells; compatibility with high throughput screening, assesses if the method can be used for large-scale drug and guide screening and automated testing; compatibility with parallel cell line handling, easiness to process multiple cell lines at the same time; derived cell type identity and functionality, whether the produced cells accurately resemble there in vivo counterpart; versatility, ability to produce a variety of different cell types; compatibility with gene editing, how well the method supports genome editing techniques; availability of patient-derived cell lines, assesses the availability of cell lines derived from human patients, as well as centralised cell banks and depositories; compatibility with rejuvenation studies, whether the method is suited to test strategies to reverse cellular ageing. The + represents if the reprogramming method has this criterion, with more + the better. – represents the method does not have that criterion.
To further support researchers looking to perform these assays in their own laboratories, we have generated a standardised web platform to share protocols (SOPs) for key assays. Standardisation will help ensure consistency and reproducibility across research groups and provide guidance when there are multiple different methods that can be used to measure an age-related change The web platform can be found here: https://doi.org/10.5281/zenodo.15056603153.
Senescence and Inflammaging
Cellular senescence phenotypes are highly heterogeneous and vary based on both cell type and the senescence-initiating stimulus. As such, there is no single assay that can be used to define senescence; rather a combinatorial approach should be used with careful consideration paid to cell type (Fig. 4). Notably, neurons require a tailored approach as they are post-mitotic and therefore it is not appropriate to measure senescence using assays that are directly tied to replication potential. Following discussion by the working group, we recommend prioritising SA-β-Gal (fluorescent probe), γH2AX (immunocytochemistry) and SASP (ELISA) when establishing assays to measure senescence and DNA damage. The group recognised this is not an exhaustive list and additional assays that can be used to further strengthen evidence of senescence are outlined in Fig. 4 and include p16 and p21and loss of HMGB1 and Lamin B1.

SOPs for the methods in bold can be found at https://doi.org/10.5281/zenodo.15056603153.
Inflammaging is defined as an increase in proinflammatory cytokines as individuals age. Proinflammatory cytokines are also components of SASP. As outlined in Fig. 4, key SASP factors have not been well described across neuronal and glial types and further characterisation and validation are necessary. While IL-6, IL-8, and IL-1β are likely to be relevant, gene expression studies and multiplex cytokine arrays should be used to establish cell type and stimuli-specific profiles of cell types of the brain.
Omics
Omics-related technologies represent fast-moving and evolving tools to measure ageing. DNA methylation clocks are the most established ageing clocks and the recently published Universal ageing clock203 represents an important step forward in using this technology to measure age in cultured cells such as neurons, and glia. We would point researchers wanting to use this tool to the consortia website (https://clockfoundation.org) for further information. Rapid progress is also being made to develop transcriptomic, proteomic and metabolomic-based clocks. Researchers should stay updated with evolving technologies and remain open to the limitations and context-specific applications of these omics approaches.
Mitochondria in ageing
Mitochondria have critical roles in both ageing and PD. To measure mitochondrial function, it is recommended that the following assays be prioritised by researchers: mtDNA damage Detection, Mitochondrial Morphology Analysis, and Mitochondrial Respiration. However, mitochondrial dysfunction is a key pathology in PD and all the suggested assays have also been used to study PD in the absence of age. Therefore, results should be interpreted carefully, and controls included that allow the impact of the disease model and age to be distinguished.
Key findings and future directions
Age is the single most important risk factor for PD, but the complexity of the interplay between ageing and PD is yet to be fully determined. The various available in vitro models for this investigation provide a distinct set of advantages and disadvantages. By discussing these properties, the PD-Age network identified an urgent need for methodological rigour to strengthen the understanding of common mechanisms behind ageing and PD.
Out of the wide range of overlapping mechanisms implicated in both ageing and PD studies, this consortium has prioritised protocols utilised for the investigation of senescence, inflammaging, omics profiling and mitochondrial dysfunction (https://doi.org/10.5281/zenodo.15056603153). Therefore, the standardisation of the in vitro techniques utilised to investigate the underlying pathways will not only reinforce individual studies within this field but also provide a robust framework to minimise variability and improve reproducibility.
While the current most preferred approach to studying PD-related changes, the iPSC cell reprogramming route, is well characterised, the benefits of other in vitro models conserving the epigenetic signature of the donor should be considered. This versatile tool is a method choice of a majority of the in vitro studies into neurodegeneration. With multiple techniques of induction of the ageing features into the iPSC-derived cells, further standardisation is necessary to increase the ability to compare findings across studies. On the contrary, the less characterised practice of obtaining cultured cells carrying the original ageing profile of the donor, have been shown to be a powerful tool for investigating age-related diseases. Despite the advantages of utilising the direct and semi-direct reprogramming approaches of generating cells and maintaining the biological background of the individual biopsy donor, robustness is necessary in the developing procedures. Uniform and efficient protocols will facilitate greater consistency and more accurate comparisons across studies from various institutions.
However, there are multiple outstanding questions future research should continue to explore to advance the ageing research in PD. One of the crucial challenges facing this field is distinguishing the age-specific effects from the PD-specific effects in vitro. This issue, especially vital in distinguishing mitochondrial characteristics and their age- and PD-specific changes will require further elucidation. Secondly, modelling disease progression in the context of ageing still requires refinement, as the current cellular models are unable to fully capture the gradual progression of the disease. Therefore, while the outlined practices may create a solid foundation for ageing and PD studies, avenues such as multi-cellular models, time-lapse investigations and incorporation of risk factors will be critical in the general standardisation across the field.
While the harmonisation of research practices is essential, its challenges should also be considered. For the research community to be able to draw meaningful conclusions achieved from standardised frameworks, the heterogeneity of research settings must be reviewed. Moreover, a key factor to evaluate during this process is the high level of complexity of PD and ageing, their various pathways of pathophysiology and the variability in their presentation across individuals. To overcome this, the process must be adaptable and constantly updated. Also, while great effort was implicated in the selection of methods for measuring the chosen parameters to include the most common laboratory equipment, the differences in technology and resource access may decelerate the standardisation process across regions. Therefore, the PD-Age network emphasises the importance of international partnership and technological unification as indispensable means for establishing these common protocols.
Data availability
No datasets were generated or analysed during the current study.
References
Collier, T. J., Kanaan, N. M. & Kordower, J. H. Ageing as a primary risk factor for Parkinson’s disease: evidence from studies of non-human primates. Nat. Rev. Neurosci. 12, 359–366 (2011).
Driver, J. A. et al. Incidence and remaining lifetime risk of Parkinson disease in advanced age. Neurology 72, 432–438 (2009).
Balestrino, R. & Schapira, A. H. Parkinson disease. Eur. J. Neurol. 27, 27–42 (2020).
Bury, A. G. et al. Mitochondrial DNA changes in pedunculopontine cholinergic neurons in Parkinson disease. Ann. Neurol. 82, 1016–1021 (2017).
Poewe, W. Non-motor symptoms in Parkinson’s disease. Eur. J. Neurol. 15, 14–20 (2008).
Buchman, A. S. et al. Nigral pathology and Parkinsonian signs in elders without Parkinson disease. Ann. Neurol. 71, 258–266 (2012).
Double, K. et al. The comparative biology of neuromelanin and lipofuscin in the human brain. Cell. Mol. Life Sci. 65, 1669–1682 (2008).
Fedorow, H. et al. Neuromelanin in human dopamine neurons: comparison with peripheral melanins and relevance to Parkinson’s disease. Prog. Neurobiol. 75, 109–124 (2005).
McGeer, P. L. et al. Rate of cell death in Parkinsonism indicates an active neuropathological process. Ann. Neurol. J. Am. Neurol. Assoc. Child Neurol. Soc. 24, 574–576 (1988).
Ma, S. Y. et al. Unbiased morphometrical measurements show loss of pigmented nigral neurones with ageing. Neuropathol. Appl. Neurobiol. 25, 394–399 (1999).
Rudow, G. et al. Morphometry of the human substantia nigra in ageing and Parkinson’s disease. Acta Neuropathol.115, 461–470 (2008).
Stark, A. & Pakkenberg, B. Histological changes of the dopaminergic nigrostriatal system in aging. Cell Tissue Res. 318, 81–92 (2004).
Kanaan, N. M., Kordower, J. H. & Collier, T. J. Age-related accumulation of Marinesco bodies and lipofuscin in rhesus monkey midbrain dopamine neurons: relevance to selective neuronal vulnerability. J. Comp. Neurol. 502, 683–700 (2007).
Rocha, E. et al. Aging, Parkinson’s disease, and models: what are the challenges?. Aging Biol. 1, e20230010 (2023).
López-Otín, C. et al. The hallmarks of aging. Cell 153, 1194–1217 (2013).
Odawara, A. et al. Physiological maturation and drug responses of human induced pluripotent stem cell-derived cortical neuronal networks in long-term culture. Sci. Rep. 6, 26181 (2016).
Schapira, A. et al. Mitochondrial complex I deficiency in Parkinson’s disease. J. Neurochem. 54, 823–827 (1990).
Carling, P. J. et al. Deep phenotyping of peripheral tissue facilitates mechanistic disease stratification in sporadic Parkinson’s disease. Prog. Neurobiol. 187, 101772 (2020).
Mortiboys, H. et al. Mitochondrial function and morphology are impaired in parkin-mutant fibroblasts. Ann. Neurol.:J. Am. Neurol. Assoc. Child Neurol. Soc. 64, 555–565 (2008).
Bender, A. et al. High levels of mitochondrial DNA deletions in substantia nigra neurons in aging and Parkinson disease. Nat. Genet. 38, 515–517 (2006).
Coxhead, J. et al. Somatic mtDNA variation is an important component of Parkinson’s disease. Neurobiol. Aging 38, 217.e1 (2016).
Bender, A. et al. Dopaminergic midbrain neurons are the prime target for mitochondrial DNA deletions. J. Neurol. 255, 1231–1235 (2008).
Krishnan, K. J. et al. What causes mitochondrial DNA deletions in human cells?. Nat. Genet. 40, 275–279 (2008).
Alexeyev, M. et al. The maintenance of mitochondrial DNA integrity—critical analysis and update. Cold Spring Harb. Perspect. Biol. 5, a012641 (2013).
Sanders, L. H. et al. Mitochondrial DNA damage: molecular marker of vulnerable nigral neurons in Parkinson’s disease. Neurobiol. Dis. 70, 214–223 (2014).
Greenland, J. C., Williams-Gray, C. H. & Barker, R. A. The clinical heterogeneity of Parkinson’s disease and its therapeutic implications. Eur. J. Neurosci. 49, 328–338 (2019).
Qi, R. et al. A blood-based marker of mitochondrial DNA damage in Parkinson’s disease. Sci. Transl. Med. 15, eabo1557 (2023).
Dölle, C. et al. Defective mitochondrial DNA homeostasis in the substantia nigra in Parkinson disease. Nat. Commun. 7, 13548 (2016).
Tresse, E. et al. Mitochondrial DNA damage triggers spread of Parkinson’s disease-like pathology. Mol. Psychiatry 28, 4902–4914 (2023).
Sliter, D. A. et al. Retraction Note: Parkin and PINK1 mitigate STING-induced inflammation. Nature 644, 1116 (2025).
Wilkins, H. M. et al. Mitochondria-derived damage-associated molecular patterns in neurodegeneration. Front. Immunol. 8, 508 (2017).
Chinta, S. J. et al. Cellular senescence is induced by the environmental neurotoxin paraquat and contributes to neuropathology linked to Parkinson’s disease. Cell Rep. 22, 930–940 (2018).
Franceschi, C. & Campisi, J. Chronic inflammation (inflammaging) and its potential contribution to age-associated diseases. J. Gerontol. Ser. A: Biomed. Sci. Med. Sci. 69, S4–S9 (2014).
Acosta, J. C. et al. A complex secretory program orchestrated by the inflammasome controls paracrine senescence. Nat. cell Biol. 15, 978–990 (2013).
Coppé, J.-P. et al. Senescence-associated secretory phenotypes reveal cell-nonautonomous functions of oncogenic RAS and the p53 tumor suppressor. PLoS Biol. 6, e301 (2008).
Riessland, M. et al. Loss of SATB1 induces p21-dependent cellular senescence in post-mitotic dopaminergic neurons. cell stem cell 25, 514–530.e8 (2019).
Payne, T. et al. Multimodal assessment of mitochondrial function in Parkinson’s disease. Brain 147, 267–280 (2024).
Franck, M. et al. Nonuniversality of inflammaging across human populations. Nature Aging, 5, 1471–1480 (2025).
Negrey, J. D. et al. Urinary neopterin of wild chimpanzees indicates that cell-mediated immune activity varies by age, sex, and female reproductive status. Sci. Rep. 11, 9298 (2021).
Zhang, P. et al. Inhibition of S6K lowers age-related inflammation and increases lifespan through the endolysosomal system. Nat. Aging 4, 491–509 (2024).
Barker, R. A. & Björklund, A. Animal models of Parkinson’s disease: are they useful or not?. J. Parkinson’s. Dis. 10, 1335–1342 (2020).
Halazonetis, T. D., Gorgoulis, V. G. & Bartek, J. An oncogene-induced DNA damage model for cancer development. science 319, 1352–1355 (2008).
Serrano, M. et al. Oncogenic Ras provokes premature cell senescence associated with the accumulation of p53 and p16INK4a. Cell 88, 593–602 (1997).
Harley, C. B., Futcher, A. B. & Greider, C. W. Telomeres shorten during ageing of human fibroblasts. Nature 345, 458–460 (1990).
Dasgupta, N. et al. The role of the dynamic epigenetic landscape in senescence: orchestrating SASP expression. npj Aging 10, 48 (2024).
McConnell, B. B. et al. Inhibitors of cyclin-dependent kinases induce features of replicative senescence in early passage human diploid fibroblasts. Curr. Biol. 8, 351–354 (1998).
Rovillain, E. et al. An RNA interference screen for identifying downstream effectors of the p53 and pRB tumour suppressor pathways involved in senescence. BMC Genomics 12, 1–12 (2011).
Jurk, D. et al. Postmitotic neurons develop a p21-dependent senescence-like phenotype driven by a DNA damage response. Aging Cell 11, 996–1004 (2012).
Raffaele, S. et al. TNF production and release from microglia via extracellular vesicles: impact on brain functions. Cells 9, 2145 (2020).
Maeda, T. et al. Aging-associated alteration of telomere length and subtelomeric status in female patients with Parkinson’s disease. J. Neurogenet. 26, 245–251 (2012).
Martin-Ruiz, C. et al. Senescence and inflammatory markers for predicting clinical progression in Parkinson’s disease: the ICICLE-PD study. J Parkinsons Dis. 10, 193–206 (2020).
Asghar, M. et al. Mitochondrial biogenesis, telomere length and cellular senescence in Parkinson’s disease and Lewy body dementia. Sci. Rep. 12, 17578 (2022).
Forero, D. A. et al. Telomere length in Parkinson’s disease: A meta-analysis. Exp. Gerontol. 75, 53–55 (2016).
Hudson, G. et al. No evidence of substantia nigra telomere shortening in Parkinson’s disease. Neurobiol. Aging 32, 2107. e3–2107.e5 (2011).
Correia-Melo, C. et al. Mitochondria are required for pro-ageing features of the senescent phenotype. EMBO J. 35, 724–742 (2016).
Dimri, G. P. et al. A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proc. Natl. Acad. Sci. 92, 9363–9367 (1995).
Lee, J. et al. Induced pluripotency and spontaneous reversal of cellular aging in supercentenarian donor cells. Biochem. Biophys. Res. Commun. 525, 563–569 (2020).
Narita, M. et al. Rb-mediated heterochromatin formation and silencing of E2F target genes during cellular senescence. Cell 113, 703–716 (2003).
Noubissi, F. K. et al. Detection and quantification of γ-H2AX using a dissociation enhanced lanthanide fluorescence immunoassay. Sci. Rep. 11, 8945 (2021).
Bussian, T. J. et al. Clearance of senescent glial cells prevents tau-dependent pathology and cognitive decline. Nature 562, 578–582 (2018).
Hou, Y. et al. NAD+ supplementation reduces neuroinflammation and cell senescence in a transgenic mouse model of Alzheimer’s disease via cGAS–STING. Proc. Natl. Acad. Sci. 118, e2011226118 (2021).
Itahana, K., Campisi, J. & Dimri, G. P. Mechanisms of cellular senescence in human and mouse cells. Biogerontology 5, 1–10 (2004).
Shen, Q. -q. et al. Cell senescence induced by toxic interaction between α-synuclein and iron precedes nigral dopaminergic neuron loss in a mouse model of Parkinson’s disease. Acta Pharmacol. Sin. 45, 268–281 (2024).
Xia, M.-L. et al. Astragaloside IV inhibits astrocyte senescence: implication in Parkinson’s disease. J. Neuroinflamm. 17, 1–13 (2020).
Xu, M. et al. Senolytics improve physical function and increase lifespan in old age. Nat. Med. 24, 1246–1256 (2018).
Ishikawa, S. & Ishikawa, F. Proteostasis failure and cellular senescence in long-term cultured postmitotic rat neurons. Aging Cell 19, e13071 (2020).
Liu, X. et al. The combination of nicotinamide mononucleotide and lycopene prevents cognitive impairment and attenuates oxidative damage in D-galactose induced aging models via Keap1-Nrf2 signaling. Gene 822, 146348 (2022).
Moreno-Blas, D. et al. Cortical neurons develop a senescence-like phenotype promoted by dysfunctional autophagy. Aging 11, 6175 (2019).
Trias, E. et al. Emergence of microglia bearing senescence markers during paralysis progression in a rat model of inherited ALS. Front. Aging Neurosci. 11, 42 (2019).
Csiszar, A. et al. Age-associated proinflammatory secretory phenotype in vascular smooth muscle cells from the non-human primate Macaca mulatta: reversal by resveratrol treatment. J. Gerontol. Ser. A: Biomed. Sci. Med. Sci. 67, 811–820 (2012).
Yan, L. et al. Stem cell transplantation extends the reproductive life span of naturally aging cynomolgus monkeys. Cell Discov. 10, 111 (2024).
Fearnley, J. M. & Lees, A. J. Ageing and Parkinson’s disease: substantia nigra regional selectivity. Brain 114, 2283–2301 (1991).
Burns, R. S. et al. A primate model of Parkinsonism: selective destruction of dopaminergic neurons in the pars compacta of the substantia nigra by N-methyl-4-phenyl-1, 2, 3, 6-tetrahydropyridine. Proc. Natl. Acad. Sci. 80, 4546–4550 (1983).
Ovadia, A., Zhang, Z. & Gash, D. M. Increased susceptibility to MPTP toxicity in middle-aged rhesus monkeys. Neurobiol. aging 16, 931–937 (1995).
Ungerstedt, U. 6-Hydroxy-dopamine induced degeneration of central monoamine neurons. Eur. J. Pharmacol. 5, 107–110 (1968).
Van Den Berge, N. & Ulusoy, A. Animal models of brain-first and body-first Parkinson’s disease. Neurobiol. Dis. 163, 105599 (2022).
Visanji, N. P. et al. α-Synuclein-based animal models of Parkinson’s disease: challenges and opportunities in a new era. Trends Neurosci. 39, 750–762 (2016).
Davis, R. L. et al. Serum FGF-21, GDF-15, and blood mtDNA copy number are not biomarkers of Parkinson disease. Neurol.: Clin. Pract. 10, 40–46 (2020).
Pyle, A. et al. Reduced mitochondrial DNA copy number is a biomarker of Parkinson’s disease. Neurobiol. Aging 38, 216.e7–216. e10 (2016).
Devine, M. J. et al. Parkinson’s disease induced pluripotent stem cells with triplication of the α-synuclein locus. Nat. Commun. 2, 440 (2011).
Olesen, M. A., Villavicencio-Tejo, F. & Quintanilla, R. A. The use of fibroblasts as a valuable strategy for studying mitochondrial impairment in neurological disorders. Transl. Neurodegen.11, 36 (2022).
Yang, C. et al. Single-cell spatiotemporal analysis reveals cell fates and functions of transplanted mesenchymal stromal cells during bone repair. Stem Cell Rep. 17, 2318–2333 (2022).
Ambrosi, G. et al. Bioenergetic and proteolytic defects in fibroblasts from patients with sporadic Parkinson’s disease. Biochim. Biophys. Acta (BBA)-Mol. Basis Dis. 1842, 1385–1394 (2014).
del Hoyo, P. et al. Oxidative stress in skin fibroblasts cultures from patients with Parkinson’s disease. BMC Neurol. 10, 1–7 (2010).
Grünewald, A. et al. Mutant Parkin impairs mitochondrial function and morphology in human fibroblasts. PloS One 5, e12962 (2010).
Mytilineou, C. et al. Impaired oxidative decarboxylation of pyruvate in fibroblasts from patients with Parkinson’s disease. J. Neural Transm. -Parkinson’s. Dis. Dement. Sect. 8, 223–228 (1994).
Papkovskaia, T. D. et al. G2019S leucine-rich repeat kinase 2 causes uncoupling protein-mediated mitochondrial depolarization. Hum. Mol. Genet. 21, 4201–4213 (2012).
Rakovic, A. et al. Effect of endogenous mutant and wild-type PINK1 on Parkin in fibroblasts from Parkinson disease patients. Hum. Mol. Genet. 19, 3124–3137 (2010).
Dehay, B. et al. Loss of P-type ATPase ATP13A2/PARK9 function induces general lysosomal deficiency and leads to Parkinson disease neurodegeneration. Proc. Natl. Acad. Sci. 109, 9611–9616 (2012).
Hockey, L. N. et al. Dysregulation of lysosomal morphology by pathogenic LRRK2 is corrected by TPC2 inhibition. J. Cell Sci. 128, 232–238 (2015).
McNeill, A. et al. Ambroxol improves lysosomal biochemistry in glucocerebrosidase mutation-linked Parkinson disease cells. Brain 137, 1481–1495 (2014).
Smith, G. et al. Fibroblast biomarkers of sporadic Parkinson’s disease and LRRK2 kinase inhibition. Mol. Neurobiol. 53, 5161–5177 (2016).
Usenovic, M. et al. Deficiency of ATP13A2 leads to lysosomal dysfunction, α-synuclein accumulation, and neurotoxicity. J. Neurosci. 32, 4240–4246 (2012).
Wiley, C. D. et al. Mitochondrial dysfunction induces senescence with a distinct secretory phenotype. Cell Metab. 23, 303–314 (2016).
Miller, J. D. et al. Human iPSC-based modeling of late-onset disease via progerin-induced aging. Cell Stem Cell 13, 691–705 (2013).
Ren, Y. et al. Parkin mutations reduce the complexity of neuronal processes in iPSC-derived human neurons. Stem Cells 33, 68–78 (2015).
Soldner, F. et al. Parkinson’s disease patient-derived induced pluripotent stem cells free of viral reprogramming factors. Cell 136, 964–977 (2009).
Takahashi, K. et al. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 131, 861–872 (2007).
Kim, J. et al. Direct reprogramming of mouse fibroblasts to neural progenitors. Proc. Natl. Acad. Sci. 108, 7838–7843 (2011).
Meyer, K. et al. Direct conversion of patient fibroblasts demonstrates non-cell autonomous toxicity of astrocytes to motor neurons in familial and sporadic ALS. Proc. Natl. Acad. Sci. 111, 829–832 (2014).
Ambasudhan, R. et al. Direct reprogramming of adult human fibroblasts to functional neurons under defined conditions. Cell Stem Cell 9, 113–118 (2011).
Pang, Z. P. et al. Induction of human neuronal cells by defined transcription factors. Nature 476, 220–223 (2011).
Yoo, A. S. et al. MicroRNA-mediated conversion of human fibroblasts to neurons. Nature 476, 228–231 (2011).
Quist, E. et al. Transcription factor-based direct conversion of human fibroblasts to functional astrocytes. Stem Cell Rep. 17, 1620–1635 (2022).
Chambers, S. M. et al. Highly efficient neural conversion of human ES and iPS cells by dual inhibition of SMAD signaling. Nat. Biotechnol. 27, 275–280 (2009).
Marton, R. M. & Ioannidis, J. P. A comprehensive analysis of protocols for deriving dopaminergic neurons from human pluripotent stem cells. Stem Cells Transl. Med. 8, 366–374 (2019).
Byers, B. et al. SNCA triplication Parkinson’s patient’s iPSC-derived DA neurons accumulate α-synuclein and are susceptible to oxidative stress. PloS One 6, e26159 (2011).
Chung, S. Y. et al. Parkin and PINK1 patient iPSC-derived midbrain dopamine neurons exhibit mitochondrial dysfunction and α-synuclein accumulation. Stem Cell Rep. 7, 664–677 (2016).
Cooper, O. et al. Differentiation of human ES and Parkinson’s disease iPS cells into ventral midbrain dopaminergic neurons requires a high activity form of SHH, FGF8a and specific regionalization by retinoic acid. Mol. Cell. Neurosci. 45, 258–266 (2010).
Imaizumi, Y. et al. Mitochondrial dysfunction associated with increased oxidative stress and α-synuclein accumulation in PARK2 iPSC-derived neurons and postmortem brain tissue. Mol. Brain 5, 1–13 (2012).
Jiang, H. et al. Parkin controls dopamine utilization in human midbrain dopaminergic neurons derived from induced pluripotent stem cells. Nat. Commun. 3, 668 (2012).
Nguyen, H. N. et al. LRRK2 mutant iPSC-derived DA neurons demonstrate increased susceptibility to oxidative stress. Cell Stem Cell 8, 267–280 (2011).
Ryan, S. D. et al. Isogenic human iPSC Parkinson’s model shows nitrosative stress-induced dysfunction in MEF2-PGC1α transcription. Cell 155, 1351–1364 (2013).
Sánchez-Danés, A. et al. Disease-specific phenotypes in dopamine neurons from human iPS-based models of genetic and sporadic Parkinson’s disease. EMBO Mol. Med. 4, 380–395 (2012).
Sanders, L. H. et al. LRRK2 mutations cause mitochondrial DNA damage in iPSC-derived neural cells from Parkinson’s disease patients: reversal by gene correction. Neurobiol. Dis. 62, 381–386 (2014).
Schöndorf, D. C. et al. The NAD+ precursor nicotinamide riboside rescues mitochondrial defects and neuronal loss in iPSC and fly models of Parkinson’s disease. Cell Rep. 23, 2976–2988 (2018).
Schwab, A. J. & Ebert, A. D. Neurite aggregation and calcium dysfunction in iPSC-derived sensory neurons with Parkinson’s disease-related LRRK2 G2019S mutation. Stem cell Rep. 5, 1039–1052 (2015).
Seibler, P. et al. Mitochondrial Parkin recruitment is impaired in neurons derived from mutant PINK1 induced pluripotent stem cells. J. Neurosci. 31, 5970–5976 (2011).
Soldner, F. et al. Generation of isogenic pluripotent stem cells differing exclusively at two early onset Parkinson point mutations. Cell 146, 318–331 (2011).
Suzuki, S. et al. Efficient induction of dopaminergic neuron differentiation from induced pluripotent stem cells reveals impaired mitophagy in PARK2 neurons. Biochemical Biophysical Res. Commun. 483, 88–93 (2017).
Busskamp, V. et al. Rapid neurogenesis through transcriptional activation in human stem cells. Mol. Syst. Biol. 10, 760 (2014).
Zhang, Y. et al. Rapid single-step induction of functional neurons from human pluripotent stem cells. Neuron 78, 785–798 (2013).
Liu, X. et al. Direct reprogramming of human fibroblasts into dopaminergic neuron-like cells. Cell Res. 22, 321–332 (2012).
Park, C.-H. et al. Differential actions of the proneural genes encoding Mash1 and neurogenins in Nurr1-induced dopamine neuron differentiation. J. Cell Sci. 119, 2310–2320 (2006).
Xue, Y. et al. Synthetic mRNAs drive highly efficient iPS cell differentiation to dopaminergic neurons. Stem Cells Transl. Med. 8, 112–123 (2019).
Wang, C. et al. Scalable production of iPSC-derived human neurons to identify tau-lowering compounds by high-content screening. Stem Cell Rep. 9, 1221–1233 (2017).
Frattini, E. et al. Lewy pathology formation in patient-derived GBA1 Parkinson’s disease midbrain organoids. Brain p. awae365. (2024).
Kim, M. S. et al. Advanced human iPSC-based preclinical model for Parkinson’s disease with optogenetic alpha-synuclein aggregation. Cell Stem Cell 30, 973–986.e11 (2023).
Morrone Parfitt, G. et al. Disruption of lysosomal proteolysis in astrocytes facilitates midbrain organoid proteostasis failure in an early-onset Parkinson’s disease model. Nat. Commun. 15, 447 (2024).
Muwanigwa, M. N. et al. Alpha-synuclein pathology is associated with astrocyte senescence in a midbrain organoid model of familial Parkinson’s disease. Mol. Cell. Neurosci. 128, 103919 (2024).
Smits, L. M. et al. Modeling Parkinson’s disease in midbrain-like organoids. NPJ Parkinson’s. Dis. 5, 5 (2019).
Wulansari, N. et al. Neurodevelopmental defects and neurodegenerative phenotypes in human brain organoids carrying Parkinson’s disease-linked DNAJC6 mutations. Sci. Adv. 7, eabb1540 (2021).
Barmpa, K. et al. Modeling early phenotypes of Parkinson’s disease by age-induced midbrain-striatum assembloids. Commun. Biol. 7, 1561 (2024).
Kriks, S. et al. Dopamine neurons derived from human ES cells efficiently engraft in animal models of Parkinson’s disease. Nature 480, 547–551 (2011).
Jo, J. et al. Midbrain-like organoids from human pluripotent stem cells contain functional dopaminergic and neuromelanin-producing neurons. Cell Stem Cell 19, 248–257 (2016).
Monzel, A. S. et al. Derivation of human midbrain-specific organoids from neuroepithelial stem cells. Stem cell Rep. 8, 1144–1154 (2017).
Qian, X. et al. Generation of human brain region–specific organoids using a miniaturized spinning bioreactor. Nat. Protoc. 13, 565–580 (2018).
Grenier, K., Kao, J. & Diamandis, P. Three-dimensional modeling of human neurodegeneration: brain organoids coming of age. Mol. Psychiatry 25, 254–274 (2020).
Torrens-Mas, M. et al. Organoids: an emerging tool to study aging signature across human tissues. modeling aging with patient-derived organoids. Int. J. Mol. Sci. 22, 10547 (2021).
Lapasset, L. et al. Rejuvenating senescent and centenarian human cells by reprogramming through the pluripotent state. Genes Dev. 25, 2248–2253 (2011).
Marion, R. M. et al. Telomeres acquire embryonic stem cell characteristics in induced pluripotent stem cells. Cell Stem Cell 4, 141–154 (2009).
Kim, Y. et al. Mitochondrial aging defects emerge in directly reprogrammed human neurons due to their metabolic profile. Cell Rep. 23, 2550–2558 (2018).
Drouin-Ouellet, J. et al. Age-related pathological impairments in directly reprogrammed dopaminergic neurons derived from patients with idiopathic Parkinson’s disease. Stem Cell Rep. 17, 2203–2219 (2022).
Schaffner, S. L. & Kobor, M. S. DNA methylation as a mediator of genetic and environmental influences on Parkinson’s disease susceptibility: Impacts of alpha-Synuclein, physical activity, and pesticide exposure on the epigenome. Front. Genet. 13, 971298 (2022).
Vera, E., Bosco, N. & Studer, L. Generating late-onset human iPSC-based disease models by inducing neuronal age-related phenotypes through telomerase manipulation. Cell Rep. 17, 1184–1192 (2016).
Caiazzo, M. et al. Direct generation of functional dopaminergic neurons from mouse and human fibroblasts. Nature 476, 224–227 (2011).
Rusilowicz-Jones, E. V. et al. Benchmarking a highly selective USP30 inhibitor for enhancement of mitophagy and pexophagy. Life Sci. Alliance 5, e202101287 (2022).
Schwartzentruber, A. et al. Oxidative switch drives mitophagy defects in dopaminergic parkin mutant patient neurons. Sci. Rep. 10, 15485 (2020).
Abyzov, A. et al. Somatic copy number mosaicism in human skin revealed by induced pluripotent stem cells. Nature 492, 438–442 (2012).
Biddy, B. A. et al. Single-cell mapping of lineage and identity in direct reprogramming. Nature 564, 219–224 (2018).
Hersbach, B. A. et al. Probing cell identity hierarchies by fate titration and collision during direct reprogramming. Mol. Syst. Biol. 18, e11129 (2022).
Chanda, S. et al. Generation of induced neuronal cells by the single reprogramming factor ASCL1. Stem Cell Rep. 3, 282–296 (2014).
Bury, A. G. et al. Standard Operating Procedures (SOPs) for Senescence and Mitochondrial Function Assays in Cellular Models of Parkinson’s Disease and Ageing. Zenodo. (2025).
Leng, K. et al. CRISPRi screens in human iPSC-derived astrocytes elucidate regulators of distinct inflammatory reactive states. Nat. Neurosci. 25, 1528–1542 (2022).
Sturm, G. et al. OxPhos defects cause hypermetabolism and reduce lifespan in cells and in patients with mitochondrial diseases. Commun. Biol. 6, 22 (2023).
Tyshkovskiy, A., Zhang, S. & Gladyshev, V. N. Accelerated transcriptional elongation during aging impairs longevity. Cell Res. 33, 817–818 (2023).
Wang, B. et al. The senescence-associated secretory phenotype and its physiological and pathological implications. Nat. Rev. Mol. Cell Biol. 25, 958–978 (2024).
Nagy, A. & K. Turksen, Induced pluripotent stem (iPS) cells: methods and protocols. Vol. 2454. Springer Nature (2022).
Guttikonda, S. R. et al. Fully defined human pluripotent stem cell-derived microglia and tri-culture system model C3 production in Alzheimer’s disease. Nat. Neurosci. 24, 343–354 (2021).
Park, J. et al. A 3D human triculture system modeling neurodegeneration and neuroinflammation in Alzheimer’s disease. Nat. Neurosci. 21, 941–951 (2018).
Leung, C. M. et al. A guide to the organ-on-a-chip. Nat. Rev. Methods Prim. 2, 33 (2022).
Low, L. A. et al. Organs-on-chips: into the next decade. Nat. Rev. Drug Discov. 20, 345–361 (2021).
Ma, C. et al. Organ-on-a-chip: a new paradigm for drug development. Trends Pharmacol. Sci. 42, 119–133 (2021).
Tavakol, D. N., Fleischer, S. & Vunjak-Novakovic, G. Harnessing organs-on-a-chip to model tissue regeneration. Cell Stem Cell 28, 993–1015 (2021).
Vunjak-Novakovic, G., Ronaldson-Bouchard, K. & Radisic, M. Organs-on-a-chip models for biological research. Cell 184, 4597–4611 (2021).
Wu, Q. et al. Organ-on-a-chip: recent breakthroughs and future prospects. Biomed. Eng. online 19, 1–19 (2020).
Zhang, B. et al. Advances in organ-on-a-chip engineering. Nat. Rev. Mater. 3, 257–278 (2018).
Corsini, N. S. & Knoblich, J. A. Human organoids: New strategies and methods for analyzing human development and disease. Cell 185, 2756–2769 (2022).
Hofer, M. & Lutolf, M. P. Engineering organoids. Nat. Rev. Mater. 6, 402–420 (2021).
Kim, J., Koo, B.-K. & Knoblich, J. A. Human organoids: model systems for human biology and medicine. Nat. Rev. Mol. Cell Biol. 21, 571–584 (2020).
Rossi, G., Manfrin, A. & Lutolf, M. P. Progress and potential in organoid research. Nat. Rev. Genet. 19, 671–687 (2018).
Schutgens, F. & Clevers, H. Human organoids: tools for understanding biology and treating diseases. Annu. Rev. Pathol.: Mech. Dis. 15, 211–234 (2020).
McTague, A. et al. Genome editing in iPSC-based neural systems: From disease models to future therapeutic strategies. Front. Genome Edit. 3, 630600 (2021).
Madrid, M. et al. Autologous induced pluripotent stem cell–based cell therapies: Promise, progress, and challenges. Curr. Protoc. 1, e88 (2021).
Schweitzer, J. S. et al. Personalized iPSC-derived dopamine progenitor cells for Parkinson’s disease. N. Engl. J. Med. 382, 1926–1932 (2020).
Schweitzer, J. S., Song, B. & Kim, K.-S. A step closer to autologous cell therapy for Parkinson’s disease. Cell Stem Cell 28, 595–597 (2021).
Oyefeso, F. A. et al. Effects of acute low-moderate dose ionizing radiation to human brain organoids. Plos one 18, e0282958 (2023).
Kreienkamp, R. et al. A cell-intrinsic interferon-like response links replication stress to cellular aging caused by progerin. Cell Rep. 22, 2006–2015 (2018).
Russo, T. et al. The SATB1-MIR22-GBA axis mediates glucocerebroside accumulation inducing a cellular senescence-like phenotype in dopaminergic neurons. Aging Cell 23, e14077 (2024).
Zhang, C. et al. Identifying age-modulating compounds using a novel computational framework for evaluating transcriptional age. Aging Cell 24, e70075 (2025).
Saurat, N. et al. Genome-wide CRISPR screen identifies neddylation as a regulator of neuronal aging and AD neurodegeneration. Cell Stem Cell 31, 1162–1174.e8 (2024).
Fathi, A. et al. Chemically induced senescence in human stem cell-derived neurons promotes phenotypic presentation of neurodegeneration. Aging Cell 21, e13541 (2022).
Leandro, G. S., Sykora, P. & Bohr, V. A. The impact of base excision DNA repair in age-related neurodegenerative diseases. Mutat. Res. Fundam. Mol. Mech. Mutagen. 776, 31–39 (2015).
Maynard, S. et al. DNA damage, DNA repair, aging, and neurodegeneration. Cold Spring Harb. Perspect. Med. 5, a025130 (2015).
Armanville, S. et al. Chemically induced senescence prompts functional changes in human microglia-like cells. J. Immunol. Res. 2025, 3214633 (2025).
Gatto, N. et al. Directly converted astrocytes retain the ageing features of the donor fibroblasts and elucidate the astrocytic contribution to human CNS health and disease. Aging Cell 20, e13281 (2021).
Huh, C. J. et al. Maintenance of age in human neurons generated by microRNA-based neuronal conversion of fibroblasts. elife 5, e18648 (2016).
Mertens, J. et al. Directly reprogrammed human neurons retain aging-associated transcriptomic signatures and reveal age-related nucleocytoplasmic defects. Cell Stem Cell 17, 705–718 (2015).
Pircs, K. et al. Distinct subcellular autophagy impairments in induced neurons from patients with Huntington’s disease. Brain 145, 3035–3057 (2022).
Victor, M. B. et al. Striatal neurons directly converted from Huntington’s disease patient fibroblasts recapitulate age-associated disease phenotypes. Nat. Neurosci. 21, 341–352 (2018).
Capano, L. S. et al. Recapitulation of endogenous 4R tau expression and formation of insoluble tau in directly reprogrammed human neurons. Cell Stem Cell 29, 918–932.e8 (2022).
Allen, S. P. et al. C9orf72 expansion within astrocytes reduces metabolic flexibility in amyotrophic lateral sclerosis. Brain 142, 3771–3790 (2019).
Varghese, N. et al. From young to old: mimicking neuronal aging in directly converted neurons from young donors. Cells 13, 1260 (2024).
Drouin-Ouellet, J. et al. REST suppression mediates neural conversion of adult human fibroblasts via microRNA-dependent and-independent pathways. EMBO Mol. Med. 9, 1117–1131 (2017).
Samoylova, E. & Baklaushev, V. Cell reprogramming preserving epigenetic age: advantages and limitations. Biochemistry 85, 1035–1047 (2020).
Cederquist, G. Y. et al. Specification of positional identity in forebrain organoids. Nat. Biotechnol. 37, 436–444 (2019).
Chanoumidou, K. et al. One-step reprogramming of human fibroblasts into oligodendrocyte-like cells by SOX10, OLIG2, and NKX6. 2. Stem Cell Rep. 16, 771–783 (2021).
Hu, W. et al. Direct conversion of normal and Alzheimer’s disease human fibroblasts into neuronal cells by small molecules. cell stem cell 17, 204–212 (2015).
Sun, Z. et al. Modeling late-onset Alzheimer’s disease neuropathology via direct neuronal reprogramming. Science 385, adl2992 (2024).
López-Otín, C. et al. Hallmarks of aging: An expanding universe. Cell 186, 243–278 (2023).
Rutledge, J., Oh, H. & Wyss-Coray, T. Measuring biological age using omics data. Nat. Rev. Genet. 23, 715–727 (2022).
Poewe, W. et al. Parkinson disease. Nat. Rev. Dis. Prim. 3, 1–21 (2017).
Lu, A. T. et al. Universal DNA methylation age across mammalian tissues. Nat. Aging 3, 1144–1166 (2023).
Acknowledgements
This work was funded by the Michael J Fox Foundation (Grant ID: MJFF-022769, https://www.michaeljfox.org/). We thank all members of PD-AGE.
Author information
Authors and Affiliations
Contributions
All authors took part in the PD-AGE working group 2 workshops. A.B., A.O., C.T. and N.S. wrote the first draft of the manuscript and SOPs. J.D-O. and H.M. gained funding, oversaw the project group, and conceived the working groups and discussion items. E.S. drafted the reports from workshops used as the basis for the manuscript. All authors (A.B., A.O., C.T., N.S., E.S., D.H., J.S., L.S., P.G.M., S.B., T.K., V.K., J.D-O. and H.M.) reviewed the manuscript and suggested changes to the manuscript.
Corresponding author
Ethics declarations
Competing interests
H.M. and E.S. are co-founders and shareholders of Mitotype Precision Labs Limited. H.M. is an inventor on a patent related to bile acids and neurodegenerative diseases. N.S. and L.S. are inventors on a patent filed by MSKCC relating to methods of modulating cellular ageing in neurons. L.S. is also a scientific co-founder and consultant of Bluerock Therapeutics and DaCapo Brainscience. J.C.S. and S.B. are co-founders and shareholders of OrganoTherapeutics SARL. V.I.K. is a Scientific Advisor for Longaevus Technologies. P.G.M. is an inventor on a patent application related to the use of a DNA damage molecular signature in blood for prognostic purposes in Parkinson’s disease. A.G.B., A.O., C.T., D.H., T.K., and J.D.O. declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Bury, A.G., Olejnik, A., Tocco, C. et al. Investigating the ageing-Parkinson’s disease nexus: standardisation of in vitro models and techniques by the PD-AGE network. npj Parkinsons Dis. 11, 289 (2025). https://doi.org/10.1038/s41531-025-01137-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41531-025-01137-2
