
Abstract
Degeneration of substantia nigra pars compacta dopaminergic neurons is the “hallmark” of Parkinson’s disease (PD) and is responsible for motor signs. Other neurotransmitter systems are responsible for non-motor symptoms that may precede by decades the clinical onset of motor symptoms. The pathophysiology is complex and neurodegeneration involves excitotoxicity mechanisms and neuroinflammation. L-DOPA is the “gold” symptomatic therapy but does not halt the progression of the disease. Therefore, neuroprotective strategies are highly demanded. Metabotropic glutamate (mGlu) receptors have emerged as potential pharmacological targets because they modulate glutamatergic, GABAergic, and dopaminergic neurotransmissions, and have been implicated in mechanisms of neurodegeneration and neuroinflammation. Thus, mGlu receptors represent valuable targets for the development of new disease-modifying and symptomatic therapies for PD. This review highlights the role of individual mGlu receptor subtypes in the pathophysiology of motor and non-motor symptoms of PD and in mechanisms that contribute to the progression of the disease.
Similar content being viewed by others


Searching for broad-spectrum drugs targeting mechanisms that underlie motor and non-motor signs, neuroinflammation and neurotoxicity in Parkinson’s disease
L-DOPA combined with peripheral inhibitors of L-amino acid aromatic decarboxylase (LAAD) is the “gold” standard in the treatment of Parkinson’s disease (PD), but, in the long-term, L-DOPA treatment is complicated by the onset of motor and non-motor fluctuations and involuntary movements (L-DOPA-induced dyskinesias or LIDs). The efficacy of other drugs, e.g., dopamine receptor agonists, MAOB inhibitors and anticholinergic agents, is suboptimal, and there are no drugs that slow the progression of PD acting as disease-modifying agents1. Phase 2 studies with monoclonal antibodies directed against aggregates of α-synuclein (e.g., cimpanemab and prasinezumab) showed no significant changes in the Movement Disorder Society-sponsored Revision of the Unified Parkinson’s Disease Rating Scale (MDS-UPDRS) score, although prasinezumab caused a near-to-significant improvement in motor symptoms (MDS-UPDRS III), and clinical studies with prasinezumab are still ongoing (PADOVA trial NCT04777331). Other drugs, including monoclonal antibodies, small molecule inhibitors/activators, antisense oligonucleotides, and siRNAs, are under development as disease-modifying agents in PD2,3. A poor brain penetration and high costs are potential pitfalls in the clinical development of these new therapeutic agents. Thus, there are several unmet needs in the treatment of PD, and the development of drugs targeting mechanisms that lie at the core of the disorder is urgently needed. New broad-spectrum agents should be effective on motor and non-motor symptoms, and target some of the mechanisms underlying the progression of PD.
PD is a multifactorial disorder characterized by a progressive degeneration of dopaminergic neurons in the substantia nigra pars compacta (SNpc) and neuronal degeneration in other brainstem nuclei, such as the locus coeruleus (LC), the raphe nuclei, and the dorsal nucleus of vagus nerve. Dopamine denervation in the dorsal striatum causes a hyperactivity of the indirect pathway, which is physiologically inhibited by D2 dopamine receptors, and a reduced activity of the direct pathway, which is physiologically activated by D1 receptors. The resulting abnormality in the basal ganglia motor circuit causes the classical motor signs in PD. Formation of a long-lasting enhancement of excitatory synaptic transmission at the synapse between cortico-striatal glutamatergic fibers and D1-expressing neurons of the direct pathway is a key mechanism in the pathogenesis of LIDs. Thus, new drugs developed for an optimal and long-lasting control of motor symptoms in PD should target regulatory mechanisms that restrains the hyperactivity of the indirect pathway and/or support the activity of the direct pathway without enhancing the occurrence of LIDs if given in combination with L-DOPA.
It is currently believed that neuronal death in PD is caused by the formation of pathological aggregates of α-synuclein (found in Lewy bodies), which can spread to neighbor neurons through a mechanism of seeding and template4,5. However, Lewy bodies are not found in monogenic PD caused by mutations of parkin6, a protein playing a key role in the mitochondrial quality control (MQC)7. Aggregates of α-synuclein may inactivate parkin, leading to mitochondrial dysfunction8. The MQC impairment (i.e., the impairment of mitophagy, mitochondrial fusion, fission, and biogenesis, and formation of mitochondrial vesicles) may have devastating consequences for SNpc neurons, which are not protected against oxidative stress. Damaged mitochondria generate reactive oxygen species (ROS) through a defective respiratory chain, and, in addition, the opening of mitochondrial pores may trigger different types of cell death, such as apoptosis, necrosis, ferroptosis and pyroptosis. Interestingly, the exit of formylated mitochondrial proteins, cardiolipin, and mitochondrial DNA and RNA may trigger innate immunity in SNpc and other vulnerable neurons, contributing to the cascade of events causing neuroinflammation since the early phase of neurodegeneration9,10.
Microglia is a key player in neuroinflammation associated with PD and other neurodegenerative disorders11. A sustained low-grade neuroinflammation mediated by cells of innate immunity, such as activated microglia and macrophages, is increasingly recognized as a driver of PD progression12.
Activated microglia produces pro-inflammatory cytokines, chemokines and complement proteins, contributing to the overall formation of ROS in PD11,12. Microglia-derived interferon-γ may damage SNpc neurons, as shown by the use of an in vitro model in which mixed microglia/midbrain neurons were challenged with rotenone13. In macaques treated with the parkinsonian toxin, 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP), microglial cells are polarized toward dopaminergic neurons with engulfing gliaptic contacts, even years after the neurotoxic insult14. Interestingly, there is a reciprocal cause-to-effect relationship between α-synuclein aggregates and microglia activation. Aggregated α-synuclein can amplify microglia activation induced by pro-inflammatory cytokines, and can also activate the NLRP-3 inflammasome in microglia10,15,16,17,18. On the other hand, microglia-derived neuroinflammation may enhance tyrosine nitrosylation of α-synuclein, with ensuing protein aggregation19. Nitrosylated α-synuclein is detected in cervical lymph nodes from MPTP-lesioned mice and can by-pass immunological tolerance, thereby inducing an adaptive immune response20. In patients affected by PD, T cells with specific MHC alleles can induce α-synuclein-related autoimmunity21, with the Y39 epitope being close to α-synuclein mutations causing monogenic PD22. Aggregation of α-synuclein resulting from Tyr-nitrosylation or other mechanisms causes a loss of immune tolerance, leading to CD4+ and CD8+ T cell infiltrates23,24,25. T cell infiltration is also observed in the mouse striatum and SNpc after acute injection of MPTP26. Thus, neuroinflammation is a potential target for novel disease-modifying drugs in PD.
Excitotoxicity resulting from overactivation of N-methyl-D-aspartate (NMDA) receptors and other types of glutamate receptors is another established component of neurodegeneration in PD27,28,29. In addition, a glutamatergic hyperactivity contributes to the pathophysiology of motor and non-motor symptoms in PD and to the induction and expression of maladaptive synaptic plasticity underlying LIDs30,31. Hence, drugs that modulate glutamatergic neurotransmission cater the potential to act as both symptomatic and disease-modifying agents in PD.
Drugs targeting ionotropic glutamate receptors (e.g., AMPA, NMDA or kainate receptors) have limited value as novel broad-spectrum drugs in PD for their strong impact on excitatory synaptic transmission in the CNS. In contrast, metabotropic glutamate (mGlu) receptors modulate synaptic transmission and display pleiotropic functions in neurons, astrocytes and microglia.
mGlu receptors form a family of eight subtypes (mGlu1 to -8), which are found, with the exception of mGlu6, in brain regions involved in the pathophysiology of motor and non-motor signs of PD32,33.
Here, we discuss the role of individual mGlu receptor subtypes in the pathophysiology of motor and non-motor symptoms of PD, and their involvement in mechanisms of neuroinflammation and excitotoxicity, highlighting their potential value as therapeutic drug targets in PD. A list of mGlu receptor ligands used in preclinical models of Parkinsonism and in clinical trials is shown in Fig. 1 (see also Tables 1 and 2).

Mavoglurant, dipraglurant, and foliglurax have been clinically developed for the treatment of PD; basimglurant has been clinically developed for the treatment of major depressive disorder (see text). NAM negative allosteric modulator, PAM positive allosteric modulator. Chemical names are detailed in the text.
Expression and function of mGlu receptors in the basal ganglia motor circuit
mGlu receptor subtypes are divided into 3 groups on the basis of their amino acid sequence, pharmacological profile, and intracellular signaling mechanisms. Group I mGlu receptor subtypes (mGlu1 and mGlu5) are coupled to Gq/11 proteins and their activation leads to phospholipase-Cβ-mediated hydrolysis of phosphatidylinositol-4,5-bisphosphate, with ensuing formation of inositol-1,4-5-trisphosphate (InsP3) and diacylglycerol (DAG). InsP3 releases Ca2+ from intracellular stores, whereas DAG activates protein kinase C. Both subtypes are predominantly found in the extrasynaptic regions of dendritic spines, where mGlu5 receptors are linked to NMDA receptors and InsP3 receptors through a chain of scaffolding and anchoring proteins, such as PSD-95, Shank, and Homer proteins33,34. There is a reciprocal functional interaction between mGlu5 and NMDA receptors. Activation of mGlu5 receptors enhances NMDA receptor function by relieving the Mg2+ blockade of the NMDA-gated ion channel through a phosphorylation mechanism35,36,37,38,39,40, whereas activation of NMDA receptors amplifies mGlu5 receptor activity by restraining mGlu5 receptor desensitization41. mGlu5 receptors are highly expressed by reactive astrocytes, where their activation generates intracellular Ca2+ oscillations42, and enhances the release of pro-inflammatory cytokines43.
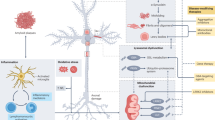
mGlu1 and mGlu5 receptors are expressed by SNpc dopaminergic neurons, medium spiny GABAergic neurons, cholinergic interneurons, globus pallidus (GP) GABAergic neurons, and subthalamic nucleus (STN) glutamatergic neurons44. mGlu1 receptors are expressed at high levels in the SNpc and cerebellum45. Stimulation of mGlu1 receptors restrains the activity of nigrostriatal neurons, by activating a Ca2+-dependent K+ conductance46. In striatal projection neurons, mGlu5 receptors are associated with A2A adenosine receptors, and the combined activation of mGlu5, NMDA and A2A receptors counteracts the inhibitory action of D2 receptors on the indirect pathway (Fig. 2).

In dendritic spines of striatal medium spiny neurons (MSN) of the indirect pathway, mGlu5, NMDA, and A2A receptors counteract the inhibitory activity of D2 receptors. Glial (EAAT1/2) and neuronal (EAAT3) glutamate transporters are also shown. Formation of the heteromeric complex between mGlu5 and D1 receptors in MSN of the direct pathway in response to dopaminergic denervation is shown in box. Within the heteromeric complex, D1 receptors share with mGlu5 receptors the ability to stimulate a PLCβ/ERK pathway underlying L-DOPA-induced dyskinesias (LIDs)86. For simplicity mGlu5 receptors are illustrated as monomers.
Group II mGlu receptor subtypes (mGlu2 and mGlu3) are coupled to Gi/o proteins, and their activation inhibits adenylyl cyclase activity and negatively modulates voltage-sensitive Ca2+ channels. Both receptors are found in the preterminal region of axon terminals, where they inhibit neurotransmitter release33 (Fig. 3). Presynaptic mGlu2/3 receptors are not easily accessible to synaptic glutamate, and might be activated by the glutamate released from astrocytes via the glutamate:cysteine antiporter47. In the basal ganglia motor circuit, presynaptic mGlu2/3 receptors inhibit glutamate release from cortico-striatal fibers, acetylcholine release from striatal cholinergic interneurons, glutamate release at the synapse between excitatory neurons of the STN and the substantia nigra pars reticulata (SNpr)44,48,49,50. The convergence of these effects underlies the symptomatic activity of mGlu2/3 receptor agonists in preclinical models of Parkinsonism44.

Presynaptic mGlu3 receptors are localized in the preterminal region of the axon terminal, and share with mGlu2 receptors the ability to inhibit glutamate release. This mechanism converges with the anti-inflammatory effect in microglia and the induction of TGF-β and GDNF in astrocytes to support the potential disease-modifying effect of mGlu3 receptor agonists or PAMs in PD. Postsynaptic mGlu3 receptors functionally interact with mGlu5 receptors, a mechanism implicated in the potential pro-cognitive effects of mGlu3 receptor agonists or PAMs. For simplicity, mGlu3 receptors are illustrated as monomers.
mGlu3 receptors are also found in post-synaptic densities, and they functionally interact with mGlu5 receptors by boosting mGlu5 receptor signaling (Fig. 3). This mechanism might concur to excitotoxic neuronal damage and generation of LIDs51,52,53. However, the overall effect of mGlu3 receptor activation is neuroprotective because mGlu3 receptors expressed in astrocytes stimulate the production of neurotrophic factors, such as transforming growth factor-β (TGF-β)54 and glial-derived neurotrophic factor (GDNF)55 (Fig. 3), and activation of mGlu3 receptors drives microglial cells towards an anti-inflammatory phenotype56,57 (Fig. 3). mGlu3 receptors display the highest affinity for glutamate among all ionotropic and mGlu receptors, and might respond to ambient glutamate concentrations in the nanomolar range58.
Group III mGlu receptor subtypes (mGlu4, mGlu6, mGlu7, and mGlu8) are all coupled to Gi/o proteins and their activation inhibits cAMP formation. mGlu4, mGlu7, and mGlu8 receptors are found in nerve terminals, and their activation inhibits neurotransmitter release (Fig. 4). mGlu4, mGlu7 and mGlu8 receptors show a widespread expression in the basal ganglia motor circuit. These receptors are presynaptically localized in cortico-striatal glutamatergic terminals, and in GABAergic terminals afferent to the internal globus pallidus (iGP), external globus pallidus (eGP) and SNpr, inhibiting both glutamate and GABA release33,34. Activation of mGlu7 receptors inhibits GABA release at the striatal:SNpr synapse, thereby restraining the activity of the direct pathway of the basal ganglia motor circuit59. However, this regulatory mechanism may only be engaged if extracellular glutamate concentrations are very high because mGlu7 receptors display a very low affinity for glutamate58,60.

Group III mGlu receptors (mGlu4, mGlu7, and mGlu8 receptors) are found in the active zone of presynaptic terminals, where their activation inhibits neurotransmitter release.
mGlu4 receptors are expressed presynaptically in the striato-pallidal synapse of the indirect pathway, and their activation inhibits GABA release61,62,63,64,65,66 (Fig. 5A). In the neostriatum, almost 70% of mGlu4 receptor-immunoreactive cortico-striatal terminals target projection neurons of the indirect pathway67 (Fig. 5B). The combined inhibition of GABA release in the striato-pallidal synapse and glutamate release in the cortico-striatal synapse restrains the activity of the indirect pathway, accounting for the antiparkinsonian effect of mGlu4 receptor activation64,66,68,69 (see below).

Through this mechanism, mGlu4 receptor agonists or PAMs may improve motor symptoms in PD and restrain excitotoxic degeneration of nigral neurons (A). Inhibition of glutamate release by heterodimeric mGlu2/mGlu4 receptors in cortico-striatal fibers (B). Whether mGlu2/mGlu4 heteromers are localized in the pre-terminal region of the axon terminal or in the active zone of neurotransmitter release is unknown.
Targeting mGlu5 receptors to restrain neuroinflammation and neurotoxicity in PD
mGlu5 receptors are physically and functionally linked to NMDA receptors, and play a key role in mechanisms of activity-dependent synaptic plasticity underlying learning and memory processes70,71. However, an excessive activation of mGlu5 receptors may cause excitotoxic neuronal death41,72, although the precise role of mGlu5 receptors in mechanisms of neurodegeneration/neuroprotection is context-dependent73. We have shown that endogenous activation of mGlu5 receptors contributes to nigro-striatal degeneration in mice challenged with MPTP74. Similar findings were found in primates, where systemic treatment with the mGlu5 receptor negative allosteric modulator (NAM), 3-[(2-methyl-1,3-thiazol-4-yl) ethynyl] pyridine (MTEP), protects dopaminergic neurons of the SNpc, noradrenergic neurons of the locus coeruleus, and the A5/A7 nuclei against MPTP toxicity75.
This finding is interesting because depletion of noradrenergic neurons has been implicated in the pathophysiology of non-motor symptoms in PD (see below).
The simplest explanation for these findings is that mGlu5 receptor blockade reduces the excitatory overdrive in the SNpc, and perhaps noradrenergic nuclei, in PD31,76. However, mGlu5 receptors are also found in reactive astrocytes and activated microglia, and, in both cell types, receptor expression was found to be upregulated in a neonatal rat model of excitotoxicity characterized by extensive neuroinflammation43,77. In astrocytes, activation of mGlu5 receptors downregulates the expression of the glial glutamate transporter, GLAST and GLT-178 (now named excitatory amino acid transporter, EAAT1 and EAAT2, respectively) (Fig. 2), and inhibition of this mechanism might restrain neuronal damage by reducing synaptic glutamate levels. In contrast, microglial mGlu5 receptors appear to reduce neuroinflammation and neurotoxicity (Fig. 2). Accordingly, selective activation of mGlu5 receptors with the orthosteric agonist, (RS)-2-chloro-5-hydroxyphenylglycine (CHPG), significantly reduced lipopolysaccharide (LPS)-induced microglial activation and neurotoxicity79. In addition, the anti-cancer drug triptolide restrains microglial activation by upregulating mGlu5 receptors in the LPS model of Parkinsonism80. Thus, mGlu5 receptor NAMs might enhance microglia-induced neuroinflammation in PD. Of note, there are models of Parkinsonism in which mGlu5 receptor blockade fails to cause neuroprotection. For example, systemic treatment with the mGlu5 receptor NAM, 2-methyl-6-(phenylethynyl)pyridine (MPEP), in rats challenged with 6-hydroxy-dopamine (6-OH-DA) reduced akinesia, without affecting neuronal survival or neuroinflammation81.
Targeting mGlu5 receptors in the treatment of LIDs
LIDs are abnormal involuntary movements, which may occur at the time of maximal plasma concentrations of L-DOPA (peak-dose dyskinesias), or, less frequently, during the ascending and decreasing phases of plasmatic L-DOPA levels (diphasic dyskinesias)82. The pathophysiology of LIDs is complex, and likely reflects the formation of a maladaptive form of synaptic plasticity leading to a hyperactivity of striatal projection neurons of the direct pathway expressing both D1 and mGlu5 receptors83,84. The seminal work by Paolo Calabresi and his associates has shown that induction of LTP at the synapse between cortico-striatal fibers and striatal projection neurons of the direct pathway is mediated by D1, NMDA, mGlu5 and mGlu1 receptors85. An increased glutamatergic tone in the striatum and SNpr has been shown in rodent models of parkinsonian patients with LIDs86.
The NMDA receptor antagonist amantadine shows efficacy in reducing LIDs in experimental animals and PD patients, and is the only drug approved for the treatment of LIDs. However, mGlu5 receptor blockade is an interesting option because, as opposed to NMDA receptors, mGlu5 receptors modulate, and not mediate, excitatory synaptic transmission34. Interestingly, D1 and mGlu5 receptors form functional heterodimers, which are upregulated in the dopamine denervated striatum. This results into a mechanism of signaling bias, in which D1 receptors stimulate phosphoinositide (PI) hydrolysis instead of enhancing cAMP formation. In turn, this leads to an overactivation of the extracellular signal-related kinase (ERK) pathway with ensuing formation of LIDs87 (Fig. 2). Cortical mGlu5 receptors may also be involved in the pathophysiology of LIDs, as suggested by a positron emission tomography (PET) study showing changes in the density of mGlu5 receptors in the motor and somatosensory cortex of “parkinsonian” rats treated with L-DOPA88. The use of selective mGlu5 receptor NAMs supports the key role for mGlu5 receptors in the generation of LIDs. For example, in a rat model of LIDs, treatment with MTEP reduces striatal mRNA levels of both prodynorphin and proenkephalin, which are biochemical markers of projection neurons of the direct and indirect pathway, respectively89. Treatment with MTEP also reduced LIDs and the associated increase in the activity of the direct pathway, assessed by measuring extracellular GABA levels in the SNpr90. mGlu5 receptor expression was found to be increased in the striatum of MPTP-lesioned monkeys developing dyskinesias in response to L-DOPA, as well as in the putamen, iGp and eGP of PD patients with LIDs91. mGlu5 receptor NAMs reduced LIDs in MPTP-lesioned monkeys without affecting the therapeutic efficacy of L-DOPA82,83,84,85,86,87,88,89,90,91,92,93,94,95,96,97,98.
One of the leading hypotheses on the pathogenesis of LIDs states that under conditions of extensive dopaminergic denervation L-DOPA is taken up by striatal serotonergic fibers, where it is converted to dopamine by LAAD-mediated decarboxylation99. Hence, agonists of autoreceptors that inhibit the activity of serotonergic neurons (i.e., 5-HT1A/B/D serotonin receptor subtypes) hold promise for the treatment of LIDs100,101.
The association between 5-HT1A/B serotonin receptor PAMs and mGlu5 receptor NAMs might represent a new valuable approach in the treatment of LIDs102.
Clinical studies with mGlu5 receptor NAMs in PD patients with LIDs
Preclinical studies paved the way to the clinical development of two mGlu5 receptor NAMs, mavoglurant (AFQ-056) and dipraglurant, for the treatment of LIDs. Data of 6 randomized clinical trials with mavoglurant vs. placebo have been published103,104,105,106. All studies enrolled patients with PD and moderate/severe LIDs (items 32 and 33 of UPDRS-IV), and the use of antipsychotic or antidyskinetic drugs was not allowed in the 15 days preceding randomization. However, the duration of treatment ranged from 2 to 13 weeks and the dose of mavoglurant from 20 to 300 mg daily in the different studies. Efficacy measures were (i) patient diaries reporting information on the duration of “off-time” “on-time”, “on-time without dyskinesia”, “on-time with non-troublesome dyskinesia”, and “on-time with troublesome dyskinesia”; (ii) the modified abnormal involuntary movement scale (mAIMS); (iii) UPDRS part 3 and 4 (items 32 and 33); and, (iv) the Lang-Fahn activities of daily living dyskinesia scale (LFALDS). An accurate metanalysis taking into account all the possible bias that might have influence the data was recently published107. In the pooled data, treatment with mavoglurant caused no changes in the off-time, on-time, and on-time with troublesome dyskinesia; in one study106 mavoglurant was superior to placebo in enhancing the duration of on-time without dyskinesia. Pooled data showed no effect of mavoglurant on the LFADS scale and UPDRS-IV (items 32 and 33). However, mavoglurant caused a small but significant improvement in LIDs evaluated with the mAIMS107. Mavoglurant was in general well tolerated, with dizziness, euphoria, fatigue, hallucination, insomnia, and nausea being the most frequent adverse effects. Only changes in dizziness (RR = 4.19) were statistically significant107. The metanalysis provided class-one evidence that mavoglurant did not improve LIDs107. The clinical development of mavoglurant for the treatment of LIDs was terminated and the drug has been repositioned for the treatment of cocaine use disorder108,109.
A phase 2a, double-blind, dose escalation study with dipraglurant was more promising, with the drug reducing peak dose dyskinesias on day 1 and 14 of treatment with an acceptable profile of safety and tolerability110 (Table 1). An ongoing study is evaluating the safety and efficacy of a 12-week treatment with dipraglurant in PD patients with dyskinesia (NCT04857359).
mGlu5 receptor ligands in other dystonic/dyskinetic disorders
Perl et al. 111 examined the effect of the mGlu5 receptor NAM, fenobam, and the mGlu5 receptor positive allosteric modulator (PAM), 3-cyano-N-(1,3-diphenyl-1H-pyrazol-5-yl)benzamide (CDPPB), in mutant dtsz hamsters, which develop age-dependent dystonia. Neither drug had any effect on the severity of dystonia, but CDPPB induced axial dyskinesias in mutant hamsters111. Thus, at least in this model, mGlu5 receptors appear to be linked to the pathophysiology of dyskinesias, rather than dystonias. Interestingly, genetic deletion of mGlu5 receptors worsens some biochemical and behavioral alterations in BACHD mice, which model Huntington’s disease (HD)112, whereas pharmacological activation of mGlu5 receptors with the PAMs, CDPPB or [6,7-dihydro-2-(phenox-ymethyl)oxazolo[5,4-c]pyridin-5(4H)-yl](fluorophenyl)methanone (VU0409551), improved the pathological phenotype of BACHD mice113,114. In contrast, a 12-week treatment with the mGlu5 receptor NAM, 2-chloro-4-((2,5-dimethyl-1-(4-(trifluoromethoxy)phenyl)-1H-imidazol-4-yl)ethynyl)pyridine (CTEP), prevented disease progression in the zQ175 mouse model of HD115. In a randomized, double-blind, placebo controlled, clinical study, a 32-day treatment with the mGlu5 receptor NAM, mavoglurant, failed to reduce choreatic movements in patients affected by HD116. These contrasting findings do not encourage the development of mGlu5 receptor ligands for the treatment of HD.
The mGlu1 receptor: an unexplored potential drug target in PD
The mGlu1a receptor, the main splice variant of mGlu1 receptors45, is expressed in the SNpc and SNpr of rodents and primates117, and the mGlu1 receptor transcript is reduced in the rat SNpc after 6-OH-DA-induced nigrostriatal lesions118. Longitudinal PET studies in transgenic mice carrying the A53T mutation of human α-synuclein gene showed a transient increase in striatal mGlu1 receptors preceding the decline in motor activity followed by a large decrease in receptor expression. Changes in mGlu1, but not mGlu5, receptor expression showed a high correlation with motor symptoms and the expression of the high affinity dopamine transporter (a marker of integrity of striatal dopaminergic fibers)119. Although this evidence suggests that mGlu1 receptors shape the vulnerability of nigro-striatal dopaminergic neurons, pharmacological blockade of mGlu1 receptors failed to protect the nigro-striatal system against MPTP toxicity in mice74. Interestingly, neuregulin 1 (NRG1), a trophic factor that regulates neurodevelopment and synaptic plasticity, selectively increases mGlu1 receptor-activity in mesencephalic dopaminergic neurons by inducing the synthesis and membrane trafficking of mGlu1 receptors through the activation of the ErbB4-mediated phosphatidylinositol-3-kinase-Akt-mammalian target of rapamycin (PI-3-K-Akt-mTOR) pathway120. This finding links mGlu1 receptors to the biological function of NRG1, known to have a protective role in mouse models of Parkinsonism121. Finally, it has been reported that dopamine depletion downregulates mGlu1 receptors in striatal cholinergic interneurons at synapses where dopamine is co-released with glutamate. The loss of mGlu1 receptors in cholinergic interneurons might contribute to motor impairments in early-stage of Parkinsonism in mice122. Taken together, these data suggest that mGlu1 receptors might be targeted at least by symptomatic drugs in the treatment of PD, although the precise role of mGlu1 receptors in the regulation of synaptic transmission and plasticity in the basal ganglia motor circuit remains to be elucidated. This warrants further studies with potent and selective mGlu1 receptor PAMs or NAMs such as compounds N-(4-(trifluoromethyl)oxazol-2-yl)-9H-xanthene-9-carboxamide (Ro 07-11401), 3-chloro-N-[3-chloro-4-(4-chloro-1,3-dihydro-1,3-dioxo-2H-isoindol-2-yl)phenyl]-2-pyridinecarboxamide (VU0483605), and N-(3-chloro-4-(4-chloro-1,3-dioxoisoindolin-2-yl)phenyl)-3-methylpicolinamide (VU0483737), and 2-cyclopropyl-5-[1-(2-fluoro-3-pyridinyl)-5-methyl-1H-1,2,3-triazol-4-yl]-2,3-dihydro-1H-isoindol-1-one (CFMTI) and (3,4-dihydro-2H-pyrano[2,3-b]quinolin-7-yl)(cis-4-methoxycyclohexyl)-methanone (JNJ16259685), respectively123.
Activation of mGlu3, but not mGlu2, receptors restrains neuroinflammation and neurotoxicity in PD
Early studies of mGlu2 and mGlu3 receptors on mechanisms of neurodegeneration/neuroprotection in the basal ganglia motor circuit have been confounded by the use of orthosteric agonists, such as (1R,4R,5S,6R)-4-amino-2-oxabicyclo[3.1.0]hexane-4,6-dicarboxylic acid (LY379268), that does not differentiate between the two receptor subtypes60. For example, LY379268 has been used for the study of long-term depression (LTD) of excitatory synaptic transmission at the synapse between the STN and SNpr. The use of mGlu2 and mGlu3 receptor knockout mice allowed to establish that this form of synaptic plasticity was mediated by mGlu2 receptors124. Treatment with LY379268 was neuroprotective in the 6-OH-DA and MPTP models of Parkinsonism125,126, and also reduced akinesia in reserpine-treated rats125. What the two receptors have in common is their ability to inhibit neurotransmitter release from nerve terminals. However, mGlu2 and mGlu3 receptors diverge in the regulation of glial-mediated mechanisms that shape neuroinflammation and toxicity. Only mGlu3 receptors are expressed by astrocytes, and their activation enhances the production of TGF-β and GDNF (Fig. 3). Through this mechanism, activation of mGlu3 receptors in cultured astrocytes protects neurons against excitotoxic death32,54. In contrast, selective activation of mGlu2 receptors was shown to be harmful to neurons in in vitro models and in mice challenged with MPTP127,128. GDNF is a potent neurotrophic factor for survival and axonal growth of mesencephalic dopaminergic neurons, and has powerful neuroprotective and neurorestorative properties because it promotes recovery of the lesioned nigro-striatal system and improves motor functions in both rodents and non-human primates129,130,131. The neuroprotective action of GDNF is likely mediated by TGF-β, suggesting that activation of mGlu3 receptors in astrocytes promotes a powerful mechanism of neuroprotection mediated by two synergic neurotrophic factors55,132. The potential clinical relevance of this mechanism is supported by the development of strategies for the CNS delivery of GDNF in the treatment of PD. A phase 1 study showed that surgical delivery of adeno-associated virus (AAV2) encoding GDNF into the putamen of subjects with advanced PD was safe and well tolerated (NCT01621581). A phase 2 study is underway to evaluate the efficacy, safety and biodistribution of AAV2-GDNF in patients with PD (NCT06285643).
mGlu3 receptors are also expressed by microglia, and their activation drives microglia differentiation towards an anti-inflammatory phenotype (Fig. 3). Perinatal brain injury induced by gestational low-protein diet (LPD) combined with interleukin-1β injection (IL-1β) in rats caused a substantial downregulation of mGlu3 receptors in microglia resulting into neuroinflammation. Genetic deletion or pharmacological blockade of mGlu3 receptors in microglia mimicked the proinflammatory phenotype induced by LPD/IL-1β56. Microglial mGlu3 receptors may also protect against neuroinflammation associated with PD, as suggested by the evidence that mice lacking mGlu3 receptors were more sensitive to MPTP-induced nigrostriatal damage, and showed an increased microglial activation in the SNpc and a reduced expression of genes encoding anti-inflammatory cytokines in the striatum57. We found that two haplotypes of GRM3 (the gene encoding the mGlu3 receptor) were associated with PD, and that individual gene variants correlated with motor and non-motor signs of PD. In addition, patients carrying one of the PD-associated haplotypes showed an impaired cortical plasticity evaluated by magnetic transcranial stimulation57. This strongly encourages the development of mGlu3 receptor-selective PAMs as disease-modifying agents in PD. These drugs are expected to restrain neuroinflammation, and halt the progression of nigrostriatal degeneration in experimental models of Parkinsonism.
Targeting group III mGlu receptors for the treatment of motor symptoms in PD
Preclinical studies with mGlu4 receptor ligands in models of Parkinsonism
A large body of evidence indicates that activation of mGlu4 receptors provides a valuable strategy to improve motor symptoms in PD by restraining the activity of the indirect pathway of the basal ganglia motor circuit (see above).
Systemic treatment with N-phenyl-7-(hydroxyimino)cyclopropa[b]chromen-1a-carboxamide (PHCCC), N-(3-chlorophenyl)picolinamide (VU0364770) or 5-methyl-N-(4-methylpyrimidin-2-yl)-4-(1H-pyrazol-4-yl)thiazol-2-amine (ADX88178), which are all selective mGlu4 receptor PAMs, was found to reverse reserpine-induced akinesia65, haloperidol-induced catalepsy, and forelimb asymmetry in 6-OH-DA lesioned rats133,134. The mGlu4 receptor PAM, ((1S,2R)-N1-(3,4-dichlorophenyl)cyclohexane-1,2-dicarboxamide) (Lu AF21934), inhibited cortico-striatal synaptic transmission, reduced haloperidol-induced catalepsy, and lowered the threshold of L-DOPA in improving akinesia133,135. Interestingly, mGlu4 receptors can form heteromeric complexes with mGlu2 receptors expressed in the cortico-striatal pathway, whereas homodimeric mGlu4 receptors predominate at the striato-pallidal synapses136,137,138,139 (Fig. 5A,B). Striato-pallidal mGlu4 receptors should be targeted in the treatment of PD because a robust antiparkinsonian effect was reported with compound N-(3-chloro-4-fluorophenyl)-1H-pyrazolo[4,3-b]pyridin-3-amine (VU0418506), which selectively amplifies the activity of mGlu4 homodimers136. Orthosteric group III mGlu receptor agonists, such as (2S)-2-amino-4-{hydroxy[hydroxy(4-hydroxy-3-methoxy-5-nitrophenyl)methyl] phosphoryl} butanoic acid (LSP1-2111), have also been proposed as potentially useful agents in the treatment of PD. LSP1-2111 displays a preferential affinity for mGlu4 receptors, it was found to restrain striato-pallidal transmission and its systemic administration reversed haloperidol-induced catalepsy63. The interest in mGlu4 receptor activation is growing more and more, and a novel radiolabeled ligand, [11C]PXT012253, has been developed for the study of mGlu4 receptors in the monkey brain. The use of this PET tracer in human studies is warranted140.
The ability of mGlu4 receptors to negatively modulate glutamate release from cortico-striatal fibers134 suggests a potential use of mGlu4 receptor PAMs or orthosteric agonists in the treatment of LIDs (see above). However, neither the mGlu4 receptor PAM, VU0364770, nor the mGlu4 receptor orthosteric agonist, LSP1-2111, showed antidyskinetic activity in rats lesioned with 6-OH-DA and treated with L-DOPA, and only VU0364770 enhanced the therapeutic efficacy of L-DOPA141. Perhaps the combined use of a selective mGlu4 receptor PAM may have an indirect beneficial effect on LIDs allowing to reduce the daily dose of L-DOPA in PD patients.
The mGlu4 receptor PAM, foliglurax, failed to improve motor symptoms in PD patients
The highly potent and selective mGlu4 receptor PAM, PXT002331/foliglurax, showed a high brain exposure after oral administration142, and improved motor symptoms and LIDs in macaques challenged with MPTP143. Foliglurax is fully active at mGlu4 homodimers, but fails to enhance responses at mGlu2/mGlu4 and mGlu3/mGlu4 heterodimers144. So far, foliglurax has been the only mGlu4 receptor ligand clinically developed for the treatment of PD. In a phase 2a double-blind, randomized study, 153 patients with advanced PD (disease duration >3 years) treated with a stable dose of L-DOPA were randomly assigned to receive either 10 or 30 mg foliglurax or placebo twice a day for 28 days. The primary endpoint was the change in off-time assessed by the Hauser diaries. Secondary endpoints were changes in the UPDRS and the on-time without troublesome dyskinesia. Foliglurax treatment did not cause significant changes in the off-time, and secondary endpoints were also not met, and the development of foliglurax for the treatment of PD was terminated145,146. However, in the phase 2a study, the least-squares differences in daily awake time were greater with foliglurax vs. placebo, and 44.7% of patients treated with 30 mg foliglurax had a decrease in daily awake off-time >1 h vs. 30.4% with placebo146. Perhaps the termination of the foliglurax program has been premature.
Targeting mGlu4 receptors to restrain neurotoxicity and neuroinflammation
mGlu4 receptor activation reduces GABA release in striato-pallidal synapses, an effect that may restrain the overactivation of STN neurons, and, therefore, excitotoxic neuronal death in the SNpc (see above). Using the MPTP model in mice, we could demonstrate a neuroprotective activity of the mGlu4 receptor PAM, PHCCC147. Interestingly, PHCCC could also induce neuroprotection when locally infused in the eGP147. Neuroprotection could also be observed in response to systemic treatment with 3 mg/kg of foliglurax in mice challenged with MPTP148. In 6-OH-DA-lesioned mice, pharmacological activation of mGlu4 receptors could also induce neuroprotection associated with an anti-inflammatory response149,150.
mGlu4 receptor activation may have a profound impact on mechanisms of innate and adaptive immunity underlying neuroinflammation in PD. In mice developing experimental autoimmune encephalomyelitis (EAE, a model of multiple sclerosis), genetic deletion of mGlu4 receptors enhanced EAE severity and associated neuroinflammation in the spinal cord, whereas treatment with PHCCC in wild-type mice was protective151. Neuroprotection was also observed in EAE mice treated with the mGlu4 receptor PAM, ADX88178152. It has been demonstrated that activation of mGlu4 receptors in antigen-presenting cells drives T cell differentiation into T regulatory cells underlying immune tolerance151. Interestingly, application of ADX88178 to cultured microglial cells could also attenuate microglial activation and inflammation in response to LPS153. Thus, drugs that selectively activate mGlu4 receptors cater the potential to behave as disease-modifying agents in PD by reducing neuroinflammation and excitotoxic neuronal death.
Study of mGlu7 and mGlu8 receptors in preclinical models of Parkinsonism
The mGlu7 receptor PAM, N,N’-bis(diphenylmethyl)-1,2-ethanediamine dihydrochloride (AMN082), showed anti-Parkinsonian activity in reversing reserpine- and haloperidol-induced akinesia154,155,156. However, in some studies, AMN082 produced electrophysiological and behavioral effects in mice lacking mGlu7 receptors, suggesting that the drug may have off-target effects157. The mGlu8 receptor orthosteric agonist, (S)-3,4-dicarboxyphenylglycine (DCPG), failed to reduce catalepsy and akinesia induced by an acute treatment with haloperidol or reserpine, but became highly effective in animals repeatedly challenged with haloperidol or reserpine154,158. These findings suggest that adaptive changes occurring in the basal ganglia motor system in response to dopamine depletion or dopamine receptor blockade shape the behavioral response to mGlu8 receptor activation. In addition, mGlu8 receptors display high affinity for glutamate58, which can compete with DCPG in behavioral studies.
In conclusion, the study of mGlu7 and mGlu8 receptors in the pathophysiology and treatment of PD requires further investigation with the aid of more selective and appropriate pharmacological tools.
mGlu receptors as a potential target in the treatment of non-motor symptoms of PD
PD is associated with a broad-spectrum of non-motor symptoms (NMS). Some NMS, such as hyposmia, rapid eye movement sleep behavior disorder, excessive daytime sleeping, depression and constipation may precede motor symptoms by several years. Other NMS, such as global cognitive impairment, alterations in executive function and working memory, autonomic dysfunction and sleep disorders are more frequent in late stages of PD159. Hallucinations, dysphagia, and cognitive impairment develop as a result of the complex interactions between progressive neuronal degeneration and dopaminergic replacement therapy. Sensory symptoms, like pain and hyposmia, are more common in early-onset PD, whereas depression and anxiety are seen in all stages of the disease and are linked to the OFF phase in patients with motor fluctuations. Autonomic dysfunction including bladder, bowel and sexual dysfunction, and cardiovascular dysautonomia, usually precedes motor symptoms, but become more relevant with disease progression160. Targeting mGlu receptors may offer a new potential strategy for the management of NMS in PD. Cognitive impairment in PD has been linked to functional abnormalities in glutamatergic transmission and plasticity161, and might reflect alterations in the signal-to-noise ratio in learning and memory processes. This may help explain the paradoxical improvement of cognition observed with drugs acting as fast NMDA receptor channel blockers, such as memantine and amantadine. Memantine is approved worldwide for the treatment of Alzheimer’s disease, and improves attention and episodic memory in disorders characterized by dementia and Parkinsonian features, such as Lewy body dementia and Parkinson’s Disease Dementia162,163. Treatment with amantadine could enhance the speed of visual cognitive processing in non-Caucasian PD patients164. mGlu5 receptors are physically and functionally linked to NMDA receptors and cooperate with NMDA receptors in the regulation of activity-dependent synaptic plasticity165. Hence, we can predict that mGlu5 receptors behave similarly to NMDA receptor antagonists in improving cognition in chronic neurodegenerative disorders. Pharmacological blockade or genetic deletion of mGlu5 receptors was found to improve cognitive function in mouse models of Alzheimer’s disease166,167. However, this effect might be an indirect consequence of neuroprotection because oligomeric forms of the β-amyloid peptide (Aβ1-42) cause neurotoxicity by interacting with mGlu5 receptors and the cellular prion protein in neuronal membranes165,166,167. In rats locally injected with MPTP in the SNpc, treatment with the mGlu5 NAM, MPEP, improved cognition in the T maze and in an object recognition test, and was also protective against nigrostriatal damage, neuroinflammation, and loss of CA1 hippocampal neurons168. Studies in which a pro-cognitive effect is dissociated from neuroprotection are needed to disclose the activity of mGlu5 receptor antagonists on cognitive function in models of Parkinsonism.
The mGlu3 receptor is another potential drug target to improve cognition in PD. Postsynaptic mGlu3 receptors interact with mGlu5 receptors by boosting mGlu5 receptor-mediated PI hydrolysis51. This interaction is involved in the induction of LTD in the prefrontal cortex (PFC)51,169, a form of activity-dependent synaptic plasticity that underlies PFC-dependent cognitive functions169,170,171. Genetic variants of GRM3 are associated with lower performance in PFC-dependent cognitive tasks171,172,173.
Interestingly, activation of mGlu3 receptors with the endogenous agonist, N-acetylaspartylglutamate (NAAG), enhances delay in cell firing in the dorsolateral PFC during a working memory task, improving cognitive function174. In virally-suppressed HIV-infected individuals with persistent cognitive impairment, higher NAAG levels in the frontal white matter were associated with better attention and working memory, and higher NAAG levels in the left basal ganglia were related to a better verbal fluency175. Interestingly, we found an association between an intronic polymorphic variant of GRM3 (rs1989796, C>T) and attention and memory in patients with PD57. Taken together, these findings encourage the development of selective mGlu3 receptor agonists or PAMs for the treatment of cognitive dysfunction in PD.
Chronic pain is another NMS, which has a high impact on the quality of life, occurs in >80% of PD patients, and is difficult to treat. The King’s PD Pain Scale recognizes 7 pain domains: musculoskeletal, chronic, fluctuation-related, nocturnal, orofacial, discoloration (edema/swelling) and radicular pain, but the exact pathophysiological mechanisms underlying each of these domains are unknown176,177. Glutamate has a key role in pain transmission and in the induction and maintenance of nociceptive sensitization underlying chronic pain171,178. Multiple mGlu receptor subtypes are involved in the pathophysiology of chronic pain and are potential drug targets for pain management. mGlu5 receptor NAMs are candidate drugs in the treatment of pain associated with PD, because these agents have consistently shown analgesic activity in models of chronic inflammatory and neuropathic pain179,180. mGlu1 receptor antagonists also cause analgesia in models of inflammatory and neuropathic pain, but their use was associated with motor impairment and cognitive adverse effects181,182,183. mGlu5 receptor antagonists are more effective than mGlu1 receptor antagonists in reducing anxiety, which is frequently associated with chronic pain184.
Drugs that activate mGlu2/3185,186, mGlu454,187,188 or mGlu7189 receptors have shown analgesic activity in models of chronic inflammatory and/or neuropathic pain. Interestingly, L-acetylcarnitine (LAC), a drug that epigenetically enhances mGlu2 receptor expression, causes long-lasting analgesia in rodents190,191, and is used for the treatment of diabetic neuropathy and other painful peripheral neuropathies. This drug has an excellent safety profile and might be repositioned for the treatment of pain associated with PD.
The prevalence of depression exceeds 35% in PD, is associated with freezing of gait, apathy, and fatigue, and frequently occurs in patients with β-glucocerebrosidase mutations192,193. Depression in PD reflects a dysfunction of central monoaminergic systems194, and, in some patients, can be efficiently managed with a dopaminergic replacement therapy. Selective Serotonin Reuptake Inhibitors (SSRIs) or Serotonin Noradrenaline Reuptake Inhibitors (SNRIs) are drugs of choice in the treatment of depression in PD, but their use may worsen motor symptoms195 and enhance apathy196. Glutamate receptors have emerged as novel targets for the treatment of depression, and three NMDA receptor antagonists are either approved (S-ketamine, dextromethorphane) or under development (S-methadone) for the treatment of depression197. mGlu5 receptor blockade was shown to improve depressive symptoms in animal models198,199,200, and changes in brain density of mGlu5 receptors have been consistently detected by PET imaging or biochemical analysis of post-mortem tissues in individual affected by major depressive disorders200,201,202,203,204,205. The mGlu5 receptor NAM, basimglurant, has been clinically developed for the treatment of major depressive disorder. In a phase 2b, double-blind, randomized clinical trial, a 6-week add-on treatment with basimglurant failed to meet the primary endpoint (change on the score from baseline in the Montgomery-Asberg Depression Rating Scale) in patients with major depressive disorder. However, basimglurant treatment caused significant changes in patient-rated, depression-related secondary endpoints206, suggesting that the use of mGlu5 receptor NAMs in depression may be promising and warrants further investigation.
mGlu2 and mGlu3 receptors are also considered as candidate drug targets in the treatment of depression, but data with mGlu2/3 receptor ligands are not uniform. mGlu2/3 receptor antagonists show fast and sustained antidepressant-like effects similar to those of ketamine in rodents52, but systemic injection of the mGlu2/3 receptor agonist, LY379268, facilitates the induction of neuroadaptive mechanisms in response to antidepressants207,208,209. In addition, the selective mGlu2 receptor PAM, N-(4-((2-(trifluoromethyl)-3-hydroxy-4-(isobutyryl)phenoxy)methyl)benzyl)-1-methyl-1H-imidazole-4 carboxamide (THIIC), showed anxiolytic/antidepressant efficacy in different assays, indicating that selective activation of mGlu2 receptors may have therapeutic potential by normalizing excessive glutamatergic neurotransmission210. LAC, which induces the expression of mGlu2 receptors in the CNS, has consistently shown antidepressant-like activity in animal models of stress-related disorders, and plasma LAC levels are reduced in individual affected by major depressive disorder211,212,213. Perhaps modulation of mGlu2 receptor expression by LAC may provide a new strategy for the combined management of pain and depression in PD.
Conclusion
Many decades have passed since mGlu receptor identification, but much information is still missing. Modulation of neurotransmission and synaptic plasticity by mGlu receptors is still a potential target for the development of neuroprotective, anti-inflammatory and symptomatic drugs in PD, because these receptors are highly expressed in the basal ganglia motor circuit, and have been implicated in the pathophysiology of motor symptoms, NMS and LIDs (Fig. 6). The mGlu5 receptor NAMs, mavoglurant and dipraglurant, and the mGlu4 receptor PAM, foliglurax, have been clinically developed with disappointing results104,105,110,145,146 in spite of promising preclinical studies. A potential explanation may reside in the design of clinical studies, the differential affinity and efficacy of allosteric modulators for mGlu receptor homodimers and heterodimers214, or potential off-target effects of mGlu receptor ligands. We believe that mGlu5 receptor NAMs or mGlu4 receptor PAMs still cater the potential to behave as broad-spectrum antiparkinsonian agents, combining symptomatic and disease-modifying effects, taking into consideration that studies of neuroprotection in PD await the identification of early biomarkers of the disease, which allow treatments in the presymptomatic stage. The so called Silent Allosteric Modulators (SAMs) of mGlu5 receptors, which inhibit pathological mGlu5 receptors signaling sparing the physiological actions of mGlu5 receptors (e.g., their role in learning and memory processes)215,216 may be even better candidate drugs for PD. The study of mGlu3 receptors in PD is limited by the lack of potent and selective PAMs. These drugs might combine a robust neuroprotective and anti-inflammatory effect with a potential pro-cognitive effect, and their development is supported by the association of GRM3 gene variants and motor and non-motor symptoms in PD patients.

Putative candidate drugs for broad-spectrum treatments of Parkinson’s disease.
Data availability
No datasets were generated or analysed during the current study.
References
Vijiaratnam, N., Simuni, T., Bandmann, O., Morris, H. R. & Foltynie, T. Progress towards therapies for disease modification in Parkinson’s disease. Lancet Neurol. 20, 559–572 (2021).
Stocchi, F., Bravi, D., Emmi, A. & Antonini, A. Parkinson disease therapy: current strategies and future research priorities. Nat. Rev. Neurol. 20, 695–707 (2024).
Longardner, K. et al. Interoception in Parkinson’s disease: a narrative review and framework for translational research. Auton. Neurosci. 259, 103258 (2025).
Li, J. Y. et al. Lewy bodies in grafted neurons in subjects with Parkinson’s disease suggest host-to-graft disease propagation. Nat. Med. 14, 501–503 (2008).
Kordower, J. H., Chu, Y., Hauser, R. A., Freeman, T. B. & Olanow, C. W. Lewy body-like pathology in long-term embryonic nigral transplants in Parkinson’s disease. Nat. Med. 14, 504–506 (2008).
Madsen, D. A., Schmidt, S. I., Blaabjerg, M. & Meyer, M. Interaction between parkin and α-synuclein in PARK2-mediated Parkinson’s disease. Cells 10, 283 (2012).
Narendra, D. P. & Youle, R. J. The role of PINK1-Parkin in mitochondrial quality control. Nat. Cell. Biol. 26, 1639–165 (2024).
Vozdek, R. et al. Fluorescent reporter of Caenorhabditis elegans Parkin: Regulators of its abundance and role in autophagy-lysosomal dynamics. Open Res. Eur. 2, 23 (2023).
De Virgilio, A. et al. Parkinson’s disease: Autoimmunity and neuroinflammation. Autoimmun. Rev. 15, 1005–1011 (2016).
Nasrolahi, A. et al. Immune system and new avenues in Parkinson’s disease research and treatment. Rev. Neurosci. 30, 709–727 (2019).
Samant, R. R., Standaert, D. G. & Harms, A. S. The emerging role of disease-associated microglia in Parkinson’s disease. Front. Cell. Neurosci. 18, 1476461 (2024).
Zeng, K. W., Zhao, M. B., Ma, Z. Z., Jiang, Y. & Tu, P. F. Protosappanin A inhibits oxidative and nitrative stress via interfering the interaction of transmembrane protein CD14 with Toll-like receptor-4 in lipopolysaccharide-induced BV-2 microglia. Int. Immunopharmacol. 14, 558–569 (2012).
Mount, M. P. et al. Involvement of interferon-gamma in microglial-mediated loss of dopaminergic neurons. J. Neurosci. 27, 3328–3337 (2007).
Barcia, C. et al. Persistent phagocytic characteristics of microglia in the substantia nigra of long-term Parkinsonian macaques. J. Neuroimmunol. 261, 60–66 (2013).
Isik, S., Yeman Kiyak, B., Akbayir, R., Seyhali, R. & Arpaci, T. Microglia mediated neuroinflammation in Parkinson’s Disease. Cells 12, 1012 (2023).
Codolo, G. et al. Triggering of inflammasome by aggregated α-synuclein, an inflammatory response in synucleinopathies. PLoS ONE 8, e55375 (2013).
Wang, S., Yuan, Y. H., Chen, N. H. & Wang, H. B. The mechanisms of NLRP3 inflammasome/pyroptosis activation and their role in Parkinson’s disease. Int. Immunopharmacol. 67, 458–464 (2019).
Huang, Q. et al. The interplay between α-Synuclein and NLRP3 inflammasome in Parkinson’s disease. Biomed. Pharmacother. 168, 115735 (2023).
Zhang, W. et al. Aggregated alpha-synuclein activates microglia: a process leading to disease progression in Parkinson’s disease. FASEB J. 19, 533–542 (2005).
Benner, E. J. et al. Nitrated alpha-synuclein immunity accelerates degeneration of nigral dopaminergic neurons. PLoS ONE 3, e1376 (2008).
Sulzer, D. et al. T cells from patients with Parkinson’s disease recognize α-synuclein peptides. Nature 546, 656–661 (2017).
Dikiy, I. et al. Semisynthetic and in vitro phosphorylation of alpha-synuclein at Y39 promotes functional partly helical membrane-bound states resembling those induced by PD mutations. ACS Chem. Biol. 11, 2428–2437 (2016).
Bandres-Ciga, S. & Cookson, M. R. Alpha-synuclein triggers T-cell response. Is Parkinson’s disease an autoimmune disorder?. Mov. Disord. 32, 1327 (2017).
Fiszer, U., Mix, E., Fredrikson, S., Kostulas, V. & Link, H. Parkinson’s disease and immunological abnormalities: increase of HLA-DR expression on monocytes in cerebrospinal fluid and of CD45RO+ T cells in peripheral blood. Acta Neurol. Scand. 90, 160–166 (1994).
Seo, J. et al. Chronic infiltration of T lymphocytes into the brain in a non-human primate model of Parkinson’s disease. Neuroscience 431, 73–85 (2020).
Chandra, G., Roy, A., Rangasamy, S. B. & Pahan, K. Induction of adaptive immunity leads to nigrostriatal disease progression in MPTP mouse model of Parkinson’s disease. J. Immunol. 198, 4312–4326 (2017).
Caudle, W. M. & Zhang, J. Glutamate, excitotoxicity, and programmed cell death in Parkinson disease. Exp. Neurol. 220, 230–233 (2009).
Guatteo, E., Berretta, N., Monda, V., Ledonne, A. & Mercuri, N. B. Pathophysiological features of nigral dopaminergic neurons in animal models of Parkinson’s Disease. Int. J. Mol. Sci. 23, 4508 (2022).
Verma, M., Lizama, B. N. & Chu, C. T. Excitotoxicity, calcium and mitochondria: a triad in synaptic neurodegeneration. Transl. Neurodegener. 11, 3 (2022).
Calabresi, P., Pisani, A., Mercuri, N. B. & Bernardi, G. The cortico-striatal projection: from synaptic plasticity to dysfunctions of the basal ganglia. Trends Neurosci. 19, 19–24 (1996).
Campanelli, F., Natale, G., Marino, G., Ghiglieri, V. & Calabresi, P. Striatal glutamatergic hyperactivity in Parkinson’s disease. Neurobiol. Dis. 168, 105697 (2022).
Bruno, V. et al. The impact of metabotropic glutamate receptors into active neurodegenerative processes: A “dark side” in the development of new symptomatic treatments for neurologic and psychiatric disorders. Neuropharmacology 115, 180–192 (2017).
Gregory, K. J. & Goudet, C. International Union of Basic and Clinical Pharmacology. CXI. Pharmacology, signaling, and physiology of metabotropic glutamate receptors. Pharmacol. Rev. 73, 521–569 (2021).
Nicoletti, F. et al. Metabotropic glutamate receptors: from the workbench to the bedside. Neuropharmacology 60, 1017–1041 (2011).
Doherty, A. J., Palmer, M. J., Henley, J. M., Collingridge, G. L. & Jane, D. E. RS)-2-chloro-5-hydroxyphenylglycine (CHPG) activates mGlu5, but no mGlu1, receptors expressed in CHO cells and potentiates NMDA responses in the hippocampus. Neuropharmacology 36, 265–267 (1997)..
Ugolini, A., Corsi, M. & Bordi, F. Potentiation of NMDA and AMPA responses by the specific mGluR5 agonist CHPG in spinal cord motoneurons. Neuropharmacology 38, 1569–1576 (1999).
Awad, H., Hubert, G. W., Smith, Y., Levey, A. I. & Conn, P. J. Activation of metabotropic glutamate receptor 5 has direct excitatory effects and potentiates NMDA receptor currents in neurons of the subthalamic nucleus. J. Neurosci. 20, 7871–7879 (2000).
Attucci, S., Carlà, V., Mannaioni, G. & Moroni, F. Activation of type 5 metabotropic glutamate receptors enhances NMDA responses in mice cortical wedges. Br. J. Pharmacol. 132, 799–806 (2001).
Mannaioni, G., Marino, M. J., Valenti, O., Traynelis, S. F. & Conn, P. J. Metabotropic glutamate receptors 1 and 5 differentially regulate CA1 pyramidal cell function. J. Neurosci. 21, 5925–5934 (2001).
Pisani, A. et al. Metabotropic glutamate receptor 5 mediates the potentiation of N-methyl-D-aspartate responses in medium spiny striatal neurons. Neuroscience 106, 579–587 (2001).
Alagarsamy, S. et al. Activation of N-methyl-D-aspartate receptors reverses desensitization of metabotropic glutamate receptor, mGluR5, in native and recombinant systems. Ann. N. Y. Acad. Sci. 868, 526–530 (1999).
Pasti, L., Volterra, A., Pozzan, T. & Carmignoto, G. Intracellular calcium oscillations in astrocytes: a highly plastic, bidirectional form of communication between neurons and astrocytes in situ. J. Neurosci. 17, 7817–7830 (1997).
Budgett, R. F., Bakker, G., Sergeev, E., Bennett, K. A. & Bradley, S. J. Targeting the type 5 metabotropic glutamate receptor: a potential therapeutic strategy for neurodegenerative diseases?. Front. Pharmacol. 13, 893422 (2022).
Conn, P. J., Battaglia, G., Marino, M. J. & Nicoletti, F. Metabotropic glutamate receptors in the basal ganglia motor circuit. Nat. Rev. Neurosci. 6, 787–798 (2005).
Ferraguti, F., Crepaldi, L. & Nicoletti, F. Metabotropic glutamate 1 receptor: current concepts and perspectives. Pharmacol. Rev. 60, 536–581 (2008).
Fiorillo, C. D. & Williams, J. T. Glutamate mediates an inhibitory postsynaptic potential in dopamine neurons. Nature 394, 78–82 (1998).
Baker, D. A., Xi, Z. X., Shen, H., Swanson, C. J. & Kalivas, P. W. The origin and neuronal function of in vivo nonsynaptic glutamate. J. Neurosci. 22, 9134–9141 (2002).
Battaglia, G., Monn, J. A. & Schoepp, D. D. In vivo inhibition of veratridine-evoked release of striatal excitatory amino acids by the group II metabotropic glutamate receptor agonist LY354740 in rats. Neurosci. Lett. 229, 161–164 (1997).
Bradley, S. R. et al. Activation of group II metabotropic glutamate receptors inhibits synaptic excitation of the substantia nigra pars reticulata. J. Neurosci. 20, 3085–3094 (2000).
Rouse, S. T. et al. Distribution and roles of metabotropic glutamate receptors in the basal ganglia motor circuit: implications for treatment of Parkinson’s disease and related disorders. Pharmacol. Ther. 88, 427–435 (2000).
Di Menna, L. et al. Functional partnership between mGlu3 and mGlu5 metabotropic glutamate receptors in the central nervous system. Neuropharmacology 128, 301–313 (2018).
Dogra, S. & Conn, P. J. Targeting metabotropic glutamate receptors for the treatment of depression and other stress-related disorders. Neuropharmacology 196, 10868 (2021).
Joffe, M. E. et al. Mechanisms underlying prelimbic prefrontal cortex mGlu3/mGlu5-dependent plasticity and reversal learning deficits following acute stress. Neuropharmacology 144, 19–28 (2019).
Bruno, V. et al. Neuroprotection by glial metabotropic glutamate receptors is mediated by transforming growth factor-beta. J. Neurosci. 18, 9594–9600 (1998).
Battaglia, G. et al. Activation of mGlu3 receptors stimulates the production of GDNF in striatal neurons. PLoS ONE 4, e6591 (2009).
Zinni, M. et al. mGlu3 receptor regulates microglial cell reactivity in neonatal rats. J. Neuroinflammation 18, 13 (2021).
Di Menna, L. et al. Preclinical and clinical study on type 3 metabotropic glutamate receptors in Parkinson’s disease. Nat. Park. Dis. 11, 9 (2025).
Reiner, A. & Levitz, J. Glutamatergic signaling in the central nervous system: ionotropic and metabotropic receptors in concert. Neuron 98, 1080–1098 (2018).
Kosinski, C. M. et al. Localization of metabotropic glutamate receptor 7 mRNA and mGluR7a protein in the rat basal ganglia. J. Comp. Neurol. 415, 266–284 (1999).
Schoepp, D. D., Jane, D. E. & Monn, J. A. Pharmacological agents acting at subtypes of metabotropic glutamate receptors. Neuropharmacology 38, 1431–1476 (1999).
Bradley, S. R. et al. Immunohistochemical localization of subtype 4a metabotropic glutamate receptors in the rat and mouse basal ganglia. J. Comp. Neurol. 407, 33–46 (1999).
Bogenpohl, J., Galvan, A., Hu, X., Wichmann, T. & Smith, Y. Metabotropic glutamate receptor 4 in the basal ganglia of parkinsonian monkeys: ultrastructural localization and electrophysiological effects of activation in the striatopallidal complex. Neuropharmacology 66, 242–252 (2013).
Beurrier, C. et al. Electrophysiological and behavioral evidence that modulation of metabotropic glutamate receptor 4 with a new agonist reverses experimental Parkinsonism. FASEB J. 23, 3619–3628 (2009).
Amalric, M. et al. Group III and subtype 4 metabotropic glutamate receptor agonists: discovery and pathophysiological applications in Parkinson’s disease. Neuropharmacology 66, 53–64 (2013).
Marino, M. J. et al. Allosteric modulation of group III metabotropic glutamate receptor 4: a potential approach to Parkinson’s disease treatment. Proc. Natl Acad. Sci. USA 100, 13668–13673 (2003).
Valenti, O. et al. Group III metabotropic glutamate receptor-mediated modulation of the striatopallidal synapse. J. Neurosci. 23, 7218–7226 (2003).
Iskhakova, L. & Smith, Y. mGluR4-containing cortico-striatal terminals: synaptic interactions with direct and indirect pathway neurons in mice. Brain Struct. Funct. 221, 4589–4599 (2016).
Bradley, S. R., Standaert, D. G., Levey, A. I. & Conn, P. J. Distribution of group III mGluRs in rat basal ganglia with subtype-specific antibodies. Ann. N. Y. Acad. Sci. 868, 531–534 (1999).
Gubellini, P., Melon, C., Dale, E., Doller, D. & Kerkerian-Le Goff, L. Distinct effects of mGlu4 receptor positive allosteric modulators at cortico-striatal vs. striatopallidal synapses may differentially contribute to their antiparkinsonian action. Neuropharmacology 85, 166–177 (2014).
Ayala, J. E. et al. mGluR5 positive allosteric modulators facilitate both hippocampal LTP and LTD and enhance spatial learning. Neuropsychopharmacology 34, 2057–2071 (2009).
Jia, Z. et al. Selective abolition of the NMDA component of long-term potentiation in mice lacking mGluR5. Learn. Mem. 5, 331–343 (1998).
Tu, J. C. et al. Coupling of mGluR/Homer and PSD-95 complexes by the Shank family of postsynaptic density proteins. Neuron 23, 583–592 (1999).
Nicoletti, F. et al. Group-I metabotropic glutamate receptors: hypotheses to explain their dual role in neurotoxicity and neuroprotection. Neuropharmacology 38, 1477–1484 (1999).
Battaglia, G. et al. Endogenous activation of mGlu5 metabotropic glutamate receptors contributes to the development of nigro-striatal damage induced by 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine in mice. J. Neurosci. 24, 828–835 (2004).
Masilamoni, G. J. et al. Metabotropic glutamate receptor 5 antagonist protects dopaminergic and noradrenergic neurons from degeneration in MPTP-treated monkeys. Brain 134, 2057–2073 (2011).
Roshan, M. H., Tambo, A. & Pace, N. P. Potential role of caffeine in the treatment of Parkinson’s disease. Open Neurol. J. 10, 42–58 (2016).
Drouin-Ouellet, J. et al. Neuroinflammation is associated with changes in glial mGluR5 expression and the development of neonatal excitotoxic lesions. Glia 59, 188–199 (2011).
Aronica, E. et al. Expression and functional role of mGluR3 and mGluR5 in human astrocytes and glioma cells: opposite regulation of glutamate transporter proteins. Eur. J. Neurosci. 17, 2106–2118 (2003).
Byrnes, K. R. et al. Metabotropic glutamate receptor 5 activation inhibits microglial associated inflammation and neurotoxicity. Glia 57, 550–560 (2009).
Huang, Y. Y. et al. Triptolide up-regulates metabotropic glutamate receptor 5 to inhibit microglia activation in the lipopolysaccharide-induced model of Parkinson’s disease. Brain Behav. Immun. 71, 93–107 (2018).
Ambrosi, G. et al. Effects of early and delayed treatment with an mGluR5 antagonist on motor impairment, nigrostriatal damage and neuroinflammation in a rodent model of Parkinson’s disease. Brain Res. Bull. 82, 29–38 (2010).
Cenci, M. A., Ohlin, K. E. & Odin, P. Current options and future possibilities for the treatment of dyskinesia and motor fluctuations in Parkinson’s disease. CNS Neurol. Disord. Drug Targets 10, 670–684 (2011).
Guigoni, C., Doudnikoff, E., Li, Q., Bloch, B. & Bezard, E. Altered D(1) dopamine receptor trafficking in parkinsonian and dyskinetic non-human primates. Neurobiol. Dis. 26, 452–463 (2007).
Aubert, I. et al. Increased D1 dopamine receptor signaling in levodopa-induced dyskinesia. Ann. Neurol. 57, 17–26 (2005).
Gubellini, P., Pisani, A., Centonze, D., Bernardi, G. & Calabresi, P. Metabotropic glutamate receptors and striatal synaptic plasticity: implications for neurological diseases. Prog. Neurobiol. 74, 271–300 (2004).
Sgambato-Faure, V. & Cenci, M. A. Glutamatergic mechanisms in the dyskinesias induced by pharmacological dopamine replacement and deep brain stimulation for the treatment of Parkinson’s disease. Prog. Neurobiol. 96, 69–86 (2012).
Sebastianutto, I. et al. D1-mGlu5 heteromers mediate noncanonical dopamine signaling in Parkinson’s disease. J. Clin. Invest. 130, 1168–1184 (2020).
Crabbé, M. et al. Altered mGluR5 binding potential and glutamine concentration in the 6-OH-DA rat model of acute Parkinson’s disease and levodopa-induced dyskinesia. Neurobiol. Aging 61, 82–92 (2018).
Lin, J. Y., Liu, Z. G., Xie, C. L., Song, L. & Yan, A. J. Antidyskinetic treatment with MTEP affects multiple molecular pathways in the Parkinsonian striatum. Parkinsons Dis. 2017, 5798734 (2017).
Mela, F. et al. Antagonism of metabotropic glutamate receptor type 5 attenuates l-DOPA-induced dyskinesia and its molecular and neurochemical correlates in a rat model of Parkinson’s disease. J. Neurochem. 101, 483–497 (2007).
Ouattara, B. et al. Metabotropic glutamate receptor type 5 in levodopa-induced motor complications. Neurobiol. Aging 32, 1286–1295 (2011).
Morin, N., Grégoire, L., Gomez-Mancilla, B., Gasparini, F. & Di Paolo, T. Effect of the metabotropic glutamate receptor type 5 antagonists MPEP and MTEP in Parkinsonian monkeys. Neuropharmacology 58, 981–986 (2010).
Morin, N. et al. MPEP, an mGlu5 receptor antagonist, reduces the development of L-DOPA-induced motor complications in de novo Parkinsonian monkeys: biochemical correlates. Neuropharmacology 66, 355–364 (2013).
Pourmirbabaei, S., Dolatshahi, M. & Rahmani, F. Pathophysiological clues to therapeutic applications of glutamate mGlu5 receptor antagonists in levodopa-induced dyskinesia. Eur. J. Pharmacol. 855, 149–159 (2019).
Rylander, D. et al. A mGluR5 antagonist under clinical development improves L-DOPA-induced dyskinesia in Parkinsonian rats and monkeys. Neurobiol. Dis. 39, 352–361 (2010).
Ko, W. K. et al. Combined fenobam and amantadine treatment promotes robust antidyskinetic effects in the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP)-lesioned primate model of Parkinson’s disease. Mov. Disord. 29, 772–779 (2014).
Epping-Jordan, M. P. et al. Effect of the metabotropic glutamate receptor type 5 negative allosteric modulator dipraglurant on motor and non-motor symptoms of Parkinson’s disease. Cells 12, 1004 (2023).
Bezard, E. et al. The mGluR5 negative allosteric modulator dipraglurant reduces dyskinesia in the MPTP macaque model. Mov. Disord. 29, 1074–1079 (2014).
Calabresi, P., Di Filippo, M., Ghiglieri, V., Tambasco, N. & Picconi, B. Levodopa-induced dyskinesias in patients with Parkinson’s disease: filling the bench-to-bedside gap. Lancet Neurol. 9, 1106–1117 (2010).
Dupre, K. B., Eskow, K. L., Negron, G. & Bishop, C. The differential effects of 5-HT(1A) receptor stimulation on dopamine receptor-mediated abnormal involuntary movements and rotations in the primed hemiparkinsonian rat. Brain Res. 1158, 135–143 (2007).
Thomsen, M. et al. Synergistic effect of serotonin 1A and serotonin 1B/D receptor agonists in the treatment of L-DOPA-induced dyskinesia in 6-hydroxydopamine-lesioned rats. Exp. Neurol. 358, 114209 (2022).
Iderberg, H., Rylander, D., Bimpisidis, Z. & Cenci, M. A. Modulating mGluR5 and 5-HT1A/1B receptors to treat l-DOPA-induced dyskinesia: effects of combined treatment and possible mechanisms of action. Exp. Neurol. 250, 116–124 (2013).
Berg, D. et al. AFQ056 treatment of levodopa-induced dyskinesias: results of 2 randomized controlled trials. Mov. Disord. 26, 1243–1250 (2011).
Stocchi, F. et al. AFQ056 in Parkinson patients with levodopa-induced dyskinesia: 13-week, randomized, dose-finding study. Mov. Disord. 28, 1838–1846 (2013).
Trenkwalder, C. et al. Mavoglurant in Parkinson’s patients with l-Dopa-induced dyskinesias: two randomized phase 2 studies. Mov. Disord. 31, 1054–1058 (2016).
Kumar, R. et al. Mavoglurant (AFQ056) in combination with increased levodopa dosages in Parkinson’s disease patients. Int. J. Neurosci. 126, 20–24 (2016).
Negida, A. et al. Mavoglurant (AFQ056) for the treatment of levodopa-induced dyskinesia in patients with Parkinson’s disease: a meta-analysis. Neurol. Sci. 42, 3135–3143 (2021).
Vendruscolo, L. F. et al. The mGlu5 receptor negative allosteric modulator mavoglurant reduces escalated cocaine self-administration in male and female rats. Psychopharmacology241, 2303–2313 (2024).
Gomez-Mancilla, B. et al. Mavoglurant reduces cocaine use in patients with cocaine use disorder in a phase 2 clinical trial. Sci. Transl. Med. 17, eadi4505 (2025).
Tison, F. et al. A Phase 2A trial of the novel mGluR5-negative allosteric modulator dipraglurant for Levodopa-induced dyskinesia in Parkinson’s disease. Mov. Disord. 31, 1373–1380 (2016).
Perl, S., Richter, F., Gericke, B. & Richter, A. Expression of metabotropic glutamate 5 receptors in the striatum and cortex and effects of modulators on the severity of dystonia in the phenotypic dtsz model. Eur. J. Pharmacol. 859, 172527 (2019).
de Souza, J. M. et al. mGluR5 ablation leads to age-related synaptic plasticity impairments and does not improve Huntington’s disease phenotype. Sci. Rep. 12, 8982 (2022).
Doria, J. G. et al. The mGluR5 positive allosteric modulator, CDPPB, ameliorates pathology and phenotypic signs of a mouse model of Huntington’s disease. Neurobiol. Dis. 73, 163–173 (2015).
Doria, J. G. et al. The mGluR5 positive allosteric modulator VU0409551 improves synaptic plasticity and memory of a mouse model of Huntington’s disease. J. Neurochem. 147, 222–239 (2018).
Abd-Elrahman, K. S. et al. mGluR5 antagonism increases autophagy and prevents disease progression in the zQ175 mouse model of Huntington’s disease. Sci. Signal. 10, eaan6387 (2017).
Reilmann, R. et al. A randomized, placebo-controlled trial of AFQ056 for the treatment of chorea in Huntington’s disease. Mov. Disord. 30, 427–431 (2015).
Hubert, G. W., Paquet, M. & Smith, Y. Differential subcellular localization of mGluR1a and mGluR5 in the rat and monkey Substantia nigra. J. Neurosci. 21, 1838–1847 (2001).
Messenger, M. J., Dawson, L. G. & Duty, S. Changes in metabotropic glutamate receptor 1-8 gene expression in the rodent basal ganglia motor loop following lesion of the nigrostriatal tract. Neuropharmacology 43, 261–271 (2002).
Yamasaki, T. et al. Dynamic changes in striatal mGluR1 but not mGluR5 during pathological progression of Parkinson’s disease in human alpha-synuclein A53T transgenic rats: a multi-PET imaging study. J. Neurosci. 36, 375–384 (2016).
Ledonne, A. et al. Neuregulin 1 signalling modulates mGluR1 function in mesencephalic dopaminergic neurons. Mol. Psychiatry 20, 959–973 (2015).
Carlsson, T. et al. Systemic administration of neuregulin-1β1 protects dopaminergic neurons in a mouse model of Parkinson’s disease. J. Neurochem. 117, 1066–1074 (2011).
Cai, Y., Nielsen, B. E., Boxer, E. E., Aoto, J. & Ford, C. P. Loss of nigral excitation of cholinergic interneurons contributes to Parkinsonian motor impairments. Neuron 109, 1137–1149.e5 (2021).
Dogra, S. & Conn, P. J. Metabotropic glutamate receptors as emerging targets for the treatment of schizophrenia. Mol. Pharmacol. 101, 275–285 (2022).
Johnson, K. A., Niswender, C. M., Conn, P. J. & Xiang, Z. Activation of group II metabotropic glutamate receptors induces long-term depression of excitatory synaptic transmission in the substantia nigra pars reticulata. Neurosci. Lett. 504, 102–106 (2011).
Murray, T. K. et al. Evaluation of the mGluR2/3 agonist LY379268 in rodent models of Parkinson’s disease. Pharmacol. Biochem. Behav. 73, 455–466 (2002).
Battaglia, G. et al. Protective role of group-II metabotropic glutamate receptors against nigro-striatal degeneration induced by 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine in mice. Neuropharmacology 45, 155–166 (2003).
Caraci, F. et al. Targeting group II metabotropic glutamate (mGlu) receptors for the treatment of psychosis associated with Alzheimer’s disease: selective activation of mGlu2 receptors amplifies beta-amyloid toxicity in cultured neurons, whereas dual activation of mGlu2 and mGlu3 receptors is neuroprotective. Mol. Pharmacol. 79, 618–626 (2011).
Corti, C. et al. The use of knock-out mice unravels distinct roles for mGlu2 and mGlu3 metabotropic glutamate receptors in mechanisms of neurodegeneration/neuroprotection. J. Neurosci. 27, 8297–8308 (2007).
Gash, D. M., Zhang, Z. & Gerhardt, G. Neuroprotective and neurorestorative properties of GDNF. Ann. Neurol. 44, S121–S125 (1998).
Tome, D., Fonseca, C. P., Campos, F. L. & Baltazar, G. Role of neurotrophic factors in Parkinson’s disease. Curr. Pharm. Des. 23, 809–838 (2017).
Drinkut, A. et al. Ret is essential to mediate GDNF’s neuroprotective and neuroregenerative effect in a Parkinson disease mouse model. Cell Death Dis. 7, e2359 (2016).
Schober, A. et al. GDNF applied to the MPTP-lesioned nigrostriatal system requires TGF-beta for its neuroprotective action. Neurobiol. Dis. 25, 378–391 (2007).
Le Poul, E. et al. A potent and selective metabotropic glutamate receptor 4 positive allosteric modulator improves movement in rodent models of Parkinson’s disease. J. Pharmacol. Exp. Ther. 343, 167–177 (2012).
Jones, C. K. et al. The metabotropic glutamate receptor 4-positive allosteric modulator VU0364770 produces efficacy alone and in combination with L-DOPA or an adenosine 2A antagonist in preclinical rodent models of Parkinson’s disease. J. Pharmacol. Exp. Ther. 340, 404–421 (2012).
Bennouar, K. E. et al. Synergy between L-DOPA and a novel positive allosteric modulator of metabotropic glutamate receptor 4: implications for Parkinson’s disease treatment and dyskinesia. Neuropharmacology 66, 158–169 (2013).
Niswender, C. M. et al. Development and antiparkinsonian activity of VU0418506, a selective positive allosteric modulator of metabotropic glutamate receptor 4 homomers without activity at mGlu2/4 heteromers. ACS Chem. Neurosci. 7, 1201–1211 (2016).
Yin, S. et al. Selective actions of novel allosteric modulators reveal functional heteromers of metabotropic glutamate receptors in the CNS. J. Neurosci. 34, 79–94 (2014).
Engers, D. W. et al. Discovery, synthesis and preclinical characterization of N-(3-chloro-4-fluorophenyl)-1H-pyrazolo[4,3-b]pyridin-3-amine (VU0418506), a novel positive allosteric modulator of the metabotropic glutamate receptor 4 (mGlu4). ACS Chem. Neurosci. 7, 1192–1200 (2016).
Conn, P. J., Lindsley, C. W., Meiler, J. & Niswender, C. M. Opportunities and challenges in the discovery of allosteric modulators of GPCRs for treating CNS disorders. Nat. Rev. Drug Discov. 13, 692–708 (2014).
Takano, A. et al. Characterization of [11C]PXT012253 as a PET radioligand for mGlu4 allosteric modulators in nonhuman primates. Mol. Imaging Biol. 21, 500–508 (2019).
Iderberg, H. et al. Pharmacological stimulation of metabotropic glutamate receptor type 4 in a rat model of Parkinson’s disease and L-DOPA-induced dyskinesia: comparison between a positive allosteric modulator and an orthosteric agonist. Neuropharmacology 95, 121–129 (2015).
Charvin, D. et al. Discovery, structure-activity relationship, and antiparkinsonian effect of a potent and brain-penetrant chemical series of positive allosteric modulators of metabotropic glutamate receptor 4. J. Med. Chem. 60, 8515–8537 (2017).
Charvin, D. et al. An mGlu4-positive allosteric modulator alleviates Parkinsonism in primates. Mov. Disord. 33, 1619–1631 (2018).
McCullock, T.W., Couch, T. & Kammermeier, P.J. Metabotropic glutamate receptor homo- and heterodimers exhibit distinct responses to orthosteric and allosteric ligands. Mol. Pharmacol. 107, 100063 (2025).
Doller, D., Bespalov, A., Miller, R., Pietraszek, M. & Kalinichev, M. A case study of foliglurax, the first clinical mGluR4 PAM for symptomatic treatment of Parkinson’s disease: translational gaps or a failing industry innovation model?. Expert. Opin. Investig. Drugs 29, 1323–1338 (2020).
Rascol, O., Medori, R., Baayen, C., Such, P. & Meulien, D. A randomized, double-blind, controlled phase II study of foliglurax in Parkinson’s disease. Mov. Disord. 37, 1088–1093 (2022).
Battaglia, G. et al. Pharmacological activation of mGlu4 metabotropic glutamate receptors reduces nigrostriatal degeneration in mice treated with 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine. J. Neurosci. 26, 7222–7229 (2006).
Bourque, M., Morissette, M., Conquet, F., Charvin, D. & Di Paolo, T. Foliglurax, a positive allosteric modulator of the metabotrophic glutamate receptor 4, protects dopaminergic neurons in MPTP-lesioned male mice. Brain Res. 1809, 148349 (2023).
Austin, P. J. et al. Symptomatic and neuroprotective effects following activation of nigral group III metabotropic glutamate receptors in rodent models of Parkinson’s disease. Br. J. Pharmacol. 160, 1741–1753 (2010).
Betts, M. J., O’Neill, M. J. & Duty, S. Allosteric modulation of the group III mGlu4 receptor provides functional neuroprotection in the 6-hydroxydopamine rat model of Parkinson’s disease. Br. J. Pharmacol. 166, 2317–2330 (2012).
Fallarino, F. et al. Metabotropic glutamate receptor-4 modulates adaptive immunity and restrains neuroinflammation. Nat. Med. 16, 897–902 (2010).
Volpi, C. et al. Allosteric modulation of metabotropic glutamate receptor 4 activates IDO1-dependent, immunoregulatory signaling in dendritic cells. Neuropharmacology 102, 59–71 (2016).
Taylor, D. L., Diemel, L. T. & Pocock, J. M. Activation of microglial group III metabotropic glutamate receptors protects neurons against microglial neurotoxicity. J. Neurosci. 23, 2150–2160 (2003).
Broadstock, M., Austin, P. J., Betts, M. J. & Duty, S. Antiparkinsonian potential of targeting group III metabotropic glutamate receptor subtypes in the rodent substantia nigra pars reticulata. Br. J. Pharmacol. 165, 1034–1045 (2012).
Greco, B., Lopez, S., van der Putten, H., Flor, P. J. & Amalric, M. Metabotropic glutamate 7 receptor subtype modulates motor symptoms in rodent models of Parkinson’s disease. J. Pharmacol. Exp. Ther. 332, 1064–1071 (2010).
Konieczny, J. & Lenda, T. Contribution of the mGluR7 receptor to antiparkinsonian-like effects in rats: a behavioral study with the selective agonist AMN082. Pharmacol. Rep. 65, 1194–1203 (2013).
Ahnaou, A., Raeyemaekers, L., Huysmans, H. & Drinkenburg, W. H. I. M. Off-target potential of AMN082 on sleep EEG and related physiological variables: evidence from mGluR7(-/-) mice. Behav. Brain Res. 311, 287–297 (2016).
Johnson, K. A. et al. The metabotropic glutamate receptor 8 agonist (S)-3,4-DCPG reverses motor deficits in prolonged but not acute models of Parkinson’s disease. Neuropharmacology 66, 187–195 (2013).
Kalia, L. V. & Lang, A. E. Parkinson’s disease. Lancet 386, 896–912 (2015).
Schapira, A. H. V., Chaudhuri, K. R. & Jenner, P. Non-motor features of Parkinson disease. Nat. Rev. Neurosci. 18, 435–450 (2017).
Cacabelos, R., Takeda, M. & Winblad, B. The glutamatergic system and neurodegeneration in dementia: preventive strategies in Alzheimer’s disease. Int. J. Geriatr. Psychiatry 14, 3–47 (1999).
Aarsland, D. et al. Memantine in patients with Parkinson’s disease dementia or dementia with Lewy bodies: a double-blind, placebo-controlled, multicentre trial. Lancet Neurol. 8, 613–618 (2009).
Wesnes, K. A., Aarsland, D., Ballard, C. & Londos, E. Memantine improves attention and episodic memory in Parkinson’s disease dementia and dementia with Lewy bodies. Int. J. Geriatr. Psychiatry 30, 46–54 (2015).
Bandini, F., Pierantozzi, M. & Bodis-Wollner, I. The visuo-cognitive and motor effect of amantadine in non-Caucasian patients with Parkinson’s disease. A clinical and electrophysiological study. J. Neural Transm.109, 41–51 (2002).
Abd-Elrahman, K. S. & Ferguson, S. S. G. Noncanonical metabotropic glutamate receptor 5 signaling in Alzheimer’s disease. Annu. Rev. Pharmacol. Toxicol. 62, 235–254 (2022).
Hamilton, A., Esseltine, J. L., DeVries, R. A., Cregan, S. P. & Ferguson, S. S. Metabotropic glutamate receptor 5 knockout reduces cognitive impairment and pathogenesis in a mouse model of Alzheimer’s disease. Mol. Brain 7, 40 (2014).
Hamilton, A. et al. Chronic pharmacological mGluR5 inhibition prevents cognitive impairment and reduces pathogenesis in an Alzheimer disease mouse model. Cell Rep. 15, 1859–1865 (2016).
Hsieh, M. H. et al. Blockade of metabotropic glutamate receptors inhibits cognition and neurodegeneration in an MPTP-induced Parkinson’s disease rat model. Pharmacol. Biochem. Behav. 102, 64–71 (2012).
Walker, A. G. et al. Metabotropic glutamate receptor 3 activation is required for long-term depression in medial prefrontal cortex and fear extinction. Proc. Natl Acad. Sci. USA 112, 1196–1201 (2015).
Kasanetz, F. et al. Prefrontal synaptic markers of cocaine addiction-like behavior in rats. Mol. Psychiatry 18, 729–737 (2013).
Otani, S., Auclair, N., Desce, J. M., Roisin, M. P. & Crépel, F. Dopamine receptors and groups I and II mGluRs cooperate for long-term depression induction in rat prefrontal cortex through converging postsynaptic activation of MAP kinases. J. Neurosci. 19, 9788–9802 (1999).
Egan, M. F. et al. Variation in GRM3 affects cognition, prefrontal glutamate, and risk for schizophrenia. Proc. Natl Acad. Sci. USA 101, 12604–12609 (2004).
Harrison, P. J., Lyon, L., Sartorius, L. J., Burnet, P. W. & Lane, T. A. The group II metabotropic glutamate receptor 3 (mGluR3, mGlu3, GRM3): expression, function and involvement in schizophrenia. J. Psychopharmacol. 22, 308–322 (2008).
Jin, L. E. et al. mGluR2 versus mGluR3 metabotropic glutamate receptors in primate dorsolateral prefrontal cortex: postsynaptic mGluR3 strengthen working memory networks. Cereb. Cortex 28, 974–987 (2018).
Wiseman, R. L. et al. Brain N -acetyl-aspartyl-glutamate is associated with cognitive function in older virally suppressed people with HIV. AIDS 38, 1003–1011 (2024).
Custozzo, A., DiMarzio, M. & Pilitsis, J. G. Addressing Parkinson disease-related pain with deep brain stimulation. World Neurosurg. 135, 381–382 (2020).
Latremoliere, A. & Woolf, C. J. Central sensitization: a generator of pain hypersensitivity by central neural plasticity. J. Pain 10, 895–926 (2009).
Pereira, V. & Goudet, C. Emerging trends in pain modulation by metabotropic glutamate receptors. Front. Mol. Neurosci. 11, 464 (2019).
Montana, M. C. et al. The metabotropic glutamate receptor subtype 5 antagonist fenobam is analgesic and has improved in vivo selectivity compared with the prototypical antagonist 2-methyl-6-(phenylethynyl)-pyridine. J. Pharmacol. Exp. Ther. 330, 834–843 (2009).
Notartomaso, S. et al. Light-induced activation of a specific type-5 metabotropic glutamate receptor antagonist in the ventrobasal thalamus causes analgesia in a mouse model of breakthrough cancer pain. Int. J. Mol. Sci. 23, 8018 (2022).
Sevostianova, N. & Danysz, W. Analgesic effects of mGlu1 and mGlu5 receptor antagonists in the rat formalin test. Neuropharmacology 51, 623–630 (2006).
El-Kouhen, O. et al. Blockade of mGluR1 receptor results in analgesia and disruption of motor and cognitive performances: effects of A-841720, a novel non-competitive mGluR1 receptor antagonist. Br. J. Pharmacol. 149, 761–774 (2006).
Zhu, C. Z. et al. Analgesic activity of metabotropic glutamate receptor 1 antagonists on spontaneous post-operative pain in rats. Eur. J. Pharmacol. 580, 314–321 (2008).
Varty, G. B. et al. The antinociceptive and anxiolytic-like effects of the metabotropic glutamate receptor 5 (mGluR5) antagonists, MPEP and MTEP, and the mGluR1 antagonist, LY456236, in rodents: a comparison of efficacy and side-effect profiles. Psychopharmacology 179, 207–217 (2005).
Sharpe, E. F., Kingston, A. E., Lodge, D., Monn, J. A. & Headley, P. M. Systemic pre-treatment with a group II mGlu agonist, LY379268, reduces hyperalgesia in vivo. Br. J. Pharmacol. 135, 1255–1262 (2002).
Johnson, M. P. et al. Broad spectrum efficacy with LY2969822, an oral prodrug of metabotropic glutamate 2/3 receptor agonist LY2934747, in rodent pain models. Br. J. Pharmacol. 174, 822–835 (2017).
Vilar, B. et al. Alleviating pain hypersensitivity through activation of type 4 metabotropic glutamate receptor. J. Neurosci. 33, 18951–18965 (2013).
Pereira, V., Arias, J. A., Llebaria, A. & Goudet, C. Photopharmacological manipulation of amygdala metabotropic glutamate receptor mGlu4 alleviates neuropathic pain. Pharmacol. Res. 187, 106602 (2023).
Dolan, S., Gunn, M. D., Biddlestone, L. & Nolan, A. M. The selective metabotropic glutamate receptor 7 allosteric agonist AMN082 inhibits inflammatory pain-induced and incision-induced hypersensitivity in rat. Behav. Pharmacol. 20, 596–604 (2009).
Chiechio, S. et al. L-Acetylcarnitine induces analgesia by selectively up-regulating mGlu2 metabotropic glutamate receptors. Mol. Pharmacol. 61, 989–996 (2002).
Notartomaso, S. et al. Analgesia induced by the epigenetic drug, L-acetylcarnitine, outlasts the end of treatment in mouse models of chronic inflammatory and neuropathic pain. Mol. Pain. 13, 1744806917697009 (2017).
Reijnders, J. S., Ehrt, U., Weber, W. E., Aarsland, D. & Leentjens, A. F. A systematic review of prevalence studies of depression in Parkinson’s disease. Mov. Disord. 23, 183–189 (2008).
Cong, S. et al. Prevalence and clinical aspects of depression in Parkinson’s disease: a systematic review and meta-analysis of 129 studies. Neurosci. Biobehav. Rev. 141, 104749 (2022).
Assogna, F. et al. Drug choices and advancements for managing depression in Parkinson’s disease. Curr. Neuropharmacol. 18, 277–287 (2020).
Matić, T., Hendriks, M., Vinke, R. S., Sadikov, A. & Georgiev, D. The effect of serotonin reuptake and serotonin-noradrenaline reuptake inhibitors on motor symptoms in Parkinson’s disease: a PPMI-based matched-subject study. J. Parkinsons Dis. 14, 1642–1651 (2024).
Schade, R. N. et al. Greater apathy associated with selective serotonin reuptake inhibitor use in Parkinson’s disease. J. Geriatr. Psychiatry Neurol. 38, 13–22 (2025).
Marwaha, S. et al. Novel and emerging treatments for major depression. Lancet 401, 141–153 (2023).
Belozertseva, I. V., Kos, T., Popik, P., Danysz, W. & Bespalov, A. Y. Antidepressant-like effects of mGluR1 and mGluR5 antagonists in the rat forced swim and the mouse tail suspension tests. Eur. Neuropsychopharmacol. 17, 172–179 (2007).
Pomierny-Chamioło, L., Poleszak, E., Pilc, A. & Nowak, G. NMDA but not AMPA glutamatergic receptors are involved in the antidepressant-like activity of MTEP during the forced swim test in mice. Pharmacol. Rep. 62, 1186–1190 (2010).
Hughes, Z. A. et al. Negative allosteric modulation of metabotropic glutamate receptor 5 results in broad spectrum activity relevant to treatment resistant depression. Neuropharmacology 66, 202–214 (2013).
Deschwanden, A. et al. Reduced metabotropic glutamate receptor 5 density in major depression determined by [(11)C]ABP688 PET and postmortem study. Am. J. Psychiatry 168, 727–734 (2011).
Matosin, N. et al. Metabotropic glutamate receptor mGluR2/3 and mGluR5 binding in the anterior cingulate cortex in psychotic and nonpsychotic depression, bipolar disorder and schizophrenia: implications for novel mGluR-based therapeutics. J. Psychiatry Neurosci. 39, 407–416 (2014).
DeLorenzo, C. et al. Characterization of brain mGluR5 binding in a pilot study of late-life major depressive disorder using positron emission tomography and [¹¹C]ABP688. Transl. Psychiatry 5, e693 (2015).
Abdallah, C. G. et al. Metabotropic glutamate receptor 5 and glutamate involvement in major depressive disorder: a multimodal imaging study. Biol. Psychiatry Cogn. Neurosci. Neuroimaging 2, 449–456 (2017).
Kim, J. H., Joo, Y. H., Son, Y. D., Kim, H. K. & Kim, J. H. Differences in mGluR5 availability depending on the level of social avoidance in drug-naïve young patients with major depressive disorder. Neuropsychiatr. Dis. Treat. 18, 2041–2053 (2022).
Quiroz, J. A. et al. Efficacy and safety of basimglurant as adjunctive therapy for major depression: a randomized clinical trial. JAMA Psychiatry 73, 675–684 (2016).
Matrisciano, F. et al. Metabotropic glutamate receptors and neuroadaptation to antidepressants: imipramine-induced down-regulation of beta-adrenergic receptors in mice treated with metabotropic glutamate 2/3 receptor ligands. J. Neurochem. 93, 1345–1352 (2005).
Matrisciano, F. et al. Group-II metabotropic glutamate receptor ligands as adjunctive drugs in the treatment of depression: a new strategy to shorten the latency of antidepressant medication?. Mol. Psychiatry 12, 704–706 (2007).
Matrisciano, F. et al. Defective group-II metaboropic glutamate receptors in the hippocampus of spontaneously depressed rats. Neuropharmacology 55, 525–531 (2008).
Fell, M. J. et al. N-(4-((2-(trifluoromethyl)-3-hydroxy-4-(isobutyryl)phenoxy)methyl)benzyl)-1-methyl-1H-imidazole-4-carboxamide (THIIC), a novel metabotropic glutamate 2 potentiator with potential anxiolytic/ antidepressant properties: in vivo profiling suggests a link between behavioral and central nervous system neurochemical changes. J. Pharmacol. Exp. Ther. 336, 165–177 (2011).
Nasca, C. et al. L-acetylcarnitine causes rapid antidepressant effects through the epigenetic induction of mGlu2 receptors. Proc. Natl Acad. Sci. USA 110, 4804–4809 (2013).
Bigio, B. et al. Epigenetics and energetics in ventral hippocampus mediate rapid antidepressant action: Implications for treatment resistance. Proc. Natl Acad. Sci. USA 113, 7906–7911 (2016).
Nasca, C. et al. Acetyl-l-carnitine deficiency in patients with major depressive disorder. Proc. Natl Acad. Sci. USA 115, 8627–8632 (2018).
Belkacemi, K., Rondard, P., Pin, J. P. & Prézeau, L. Heterodimers revolutionize the field of metabotropic glutamate receptors. Neuroscience 25, S0306–452200270–7 (2024).
Haas, L. T. et al. Silent allosteric modulation of mGluR5 maintains glutamate signaling while rescuing Alzheimer’s mouse phenotypes. Cell Rep. 20, 76–88 (2017).
Spurrier, J. et al. Reversal of synapse loss in Alzheimer mouse models by targeting mGluR5 to prevent synaptic tagging by C1Q. Sci. Transl. Med. 14, eabi8593 (2022).
Acknowledgements
The Authors would like to thank all PD patients involved in clinical trials and funding agencies for providing economic support for preclinical and clinical studies. This work was directly supported by the Italian Ministry of Health (funds to IRCCS Neuromed, “Ricerca Corrente”).
Author information
Authors and Affiliations
Contributions
M.A., A.C., M.C.: Conceptualization, Writing-Original Draft. F.N., G.B., V.B.: Conceptualization, Supervision, Writing-Review & Editing.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Alborghetti, M., Ceccherelli, A., Caridi, M. et al. Targeting metabotropic glutamate receptors for symptomatic and disease-modifying treatment in Parkinson’s disease. npj Parkinsons Dis. 11, 290 (2025). https://doi.org/10.1038/s41531-025-01138-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41531-025-01138-1
