
Abstract
Recent studies aim to prevent kimchi spoilage and enhance the sensory and nutritional qualities using lactic acid bacteria, particularly Leuconostoc species, as kimchi starters. However, the factors enabling the successful adaptation and predominance of Leuconostoc species remain unclear. This study investigates the factors that contribute to the successful adaptation of Leuconostoc starter strains WiKim32, WiKim33, WiKim0121 and CBA3628 during kimchi fermentation using a comprehensive multi-omics approach. Our findings reveal that ATP-dependent molecular chaperones, which respond to cold and acidic kimchi environments, play crucial roles in successfully adapting Leuconostoc starter strains. Moreover, genes involved in carbohydrate metabolic pathways enhance ATP production, thereby supporting chaperone activity and bacterial growth. This study highlights the practical use of Leuconostoc starter strains WiKim32, WiKim33 and WiKim0121 and identifies essential factors for their successful adaptation and predominance during kimchi fermentation.
Similar content being viewed by others


Introduction
Kimchi, a popular traditional Korean food, is consumed worldwide for its health benefits, unique flavours, and tastes1,2,3,4. These benefits can be primarily attributed to spontaneous kimchi fermentation facilitated by lactic acid bacteria (LAB) originating from kimchi ingredients, including kimchi cabbage, radish, garlic, red pepper powder, ginger, and salted seafood5,6. However, spontaneous fermentation can lead to microbial contamination and inconsistent quality, prompting the development of kimchi starter cultures to ensure safety and consistency7. Researchers and commercial food companies have developed various kimchi starter cultures to prevent spoilage by undesirable microorganisms and enhance the nutritional and sensory properties of kimchi8.
Studies have identified numerous LAB, including Leuconostoc (Leu.) mesenteroides, Leuconostoc (Leu.) citreum, Leuconostoc gelidum, Latilactobacillus sakei, and Lactiplantibacillus plantarum, as predominant microbes in the kimchi fermentation environment9,10,11,12. Consequently, the use of LAB-based starters has garnered significant attention13,14,15,16,17,18. Weissella (Wei.) cibaria and Weissella (Wei.) koreensis are also prevalent in kimchi; however, they are rarely used as starter strains as they are not classified as 'Generally recognised as safe' (GRAS) by the Food and Drug Administration (FDA) (https://www.fda.gov/food/generally-recognized-safe-gras/microorganisms-microbial-derived-ingredients-used-food-partial-list) or included in the 'Qualified Presumption of Safety' (QPS) list by the European Food Safety Authority (EFSA) (https://www.efsa.europa.eu/en/efsajournal/pub/7747)19. Unlike homofermentative Lactobacillus species, heterofermentative Leuconostoc species produce lactate, acetate, ethanol, carbon dioxide, and mannitol, which enhance both the taste and nutritional properties of kimchi20,21,22,23.
Among Leuconostoc species, Leu. mesenteroides is predominant during the early to middle stages of kimchi fermentation24,25,26. Specific strains, such as Leu. mesenteroides strain B1 (KACC 16355)27, LK9328, and ATCC 8293T29, have been developed as starter strains to accelerate the fermentation, enhance metabolite production, and prevent spoilage. These strains also improve sensory qualities by increasing sourness and reducing bitterness. Notably, while several studies have highlighted the predominance of Leu. mesenteroides during spontaneous fermentation24,25,26 and its positive effects on sensory quality27,30 and prevention of spoilage28 when used as a starter, research is yet to identify what factors enable the predominance of Leu. mesenteroides in starters during kimchi fermentation.
This study aimed to investigate factors contributing to Leuconostoc predominance by inoculating kimchi samples with Leuconostoc starter strains and conducting multi-omics analyses, including metataxonomics, metagenomics, metatranscriptomics, and metabolomics4. Our goal is to assess bacterial diversity, metabolite production, and significant genes influencing the adaptation and predominance of starter strains during fermentation. This is the first study to elucidate the crucial factors contributing to the differential dominance of inoculated starter strains in a kimchi fermentation environment.
Results
Changes in pH, LAB colony-forming units (CFUs), and metabolite concentrations
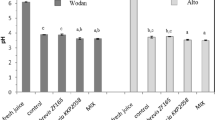
During kimchi fermentation, significant changes in pH and CFUs of LAB were observed (Fig. 1a, b). From days 3 to 33, pH decreased in all samples (W32, W33, W0121, C3628, and CTR—the samples inoculated with starter strains Leu. mesenteroides WiKim32, Leu. mesenteroides WiKim33, Leu. mesenteroides WiKim0121, Leuconostoc sp. CBA3628, and saline solution instead of starter strain, respectively) owing to organic acids produced by LAB31. A slight increase in pH was observed from days 0 to 3 in most samples, except in W33. This phenomenon could be attributed to osmotic pressure caused by salt, which drew water from cabbage and diluted acidic compounds. Initial CFUs were similar across samples; however, after day 11, LAB counts in the starter samples increased over 70 times compared to those in the CTR, indicating that inoculated starter cultures either multiplied or stimulated the growth of other LABs. Higher LAB amounts in the starter samples caused greater acidification from day 0 to 22, resulting in a faster decrease in pH than in CTR.

Changes in a pH levels, b colony-forming units of lactic acid bacteria, and the concentrations of c fructose, d glucose, e acetate, f lactate, g mannitol, and h ethanol in the CTR, W32, W33, W0121, and C3628 samples during kimchi fermentation. The kimchi samples inoculated with strains WiKim32, WiKim33, WiKim0121, and CBA3628, and saline solution were labelled as 'W32', 'W33', 'W0121', 'C3628', and 'CTR', respectively. Data were presented as mean ± standard deviation. Experiments were performed in triplicate.
Concentrations of main kimchi metabolites during fermentation were measured (Fig. 1c–h). Fructose content rapidly decreased in starter samples, whereas it decreased slowly in CTR (Fig. 1c). Acetate, lactate, mannitol, and ethanol levels increased in all samples (Fig. 1e–h); acetate and lactate lowered the pH, while mannitol enhanced the taste and nutritional value of kimchi. Starter samples showed a faster increase in acetate and mannitol levels compared with CTR. The non-metric multidimensional scaling (NMDS) plots revealed dynamic changes in metabolite profiles, with CTR showing slower fermentation than starter samples (Supplementary Fig. 1). By the end stage, metabolite profiles were similar across all samples. Although the profiles of pH, CFUs of LAB, and fermentation speed varied depending on whether the sample was inoculated with a starter strain, all samples followed the typical kimchi fermentation pattern2,5.
Changes in bacterial community succession and alpha diversity
The study investigated the impact of starter strains on bacterial succession during kimchi fermentation (Fig. 2). At the amplicon sequence variant (ASV) level, ASV001 was abundant in the WiKim samples (W32, W33, and W0121), but low in C3628 and CTR (Fig. 2a). ASV002 was only observed in C3628, while ASV003 was prevalent in C3628 and CTR. Phylogenetic tree analysis identified ASV001, ASV002, and ASV003 as Leu. mesenteroides, Leuconostoc sp. CBA3628, and Leu. citreum, respectively (Fig. 2b). The high abundance of ASV001 in WiKim samples represents the inoculated Leu. mesenteroides starter strains, whereas ASV001 in C3628 and CTR originated from kimchi ingredients. ASV002 represented the inoculated CBA3628 strain, showing low relative abundance. ASV003 (Leu. citreum) was prevalent in the bacterial community profile of CTR between days 0 and 6. The WiKim starters adapted and predominated better than the spontaneously derived Leu. citreum, indicating their practicality as kimchi starter cultures, unlike the less effective CBA3628 strain.

a Bacterial community profiles at amplicon sequence variant (ASV) level in the CTR, W32, W33, W0121, and C3628 samples during kimchi fermentation. ‘F’ indicates family level. b A phylogenetic tree of Leuconostoc species, including Leuconostoc ASV001, Leuconostoc ASV002, Leuconostoc ASV003, and four starter strains (WiKim32, WiKim33, WiKim0121, and CBA3628). The tree was inferred using the neighbour-joining method and bootstrap values were calculated from 1000 replicates. Weissella cibaria CBA3612 and Weissella koreensis KACC 15510 were used as the outgroup. Bar, 0.01 substitution per nucleotide position. The kimchi samples inoculated with strains WiKim32, WiKim33, WiKim0121, and CBA3628, and saline solution were labelled as 'W32', 'W33', 'W0121', 'C3628', and 'CTR', respectively.
Additionally, ASV008 and ASV009, identified as Wei. cibaria and Wei. koreensis, respectively (Supplementary Fig. 2), were markedly present in the CTR and C3628 samples. Microbial alpha diversity analysis32 showed that WiKim samples initially exhibited higher diversity indices than C3628 and CTR (Supplementary Table 1). CTR and C3628 samples, dominated by ASV003 and ASV008, exhibited relatively stable diversity indices throughout the fermentation process. In contrast, inoculated WiKim strains appeared to be in competition with ASV003 and ASV008, resulting in higher diversity and lower evenness during the early fermentation. However, ASV001 (starter strains in WiKim samples) became predominant during the later stages of fermentation, leading to a stabilising trend in diversity indices during the late fermentation period. This is explained by the effect of inoculated WiKim strains (WiKim32, WiKim33, and WiKim0121) instead of the dominance of spontaneously derived LAB, including ASV003, ASV008, and ASV009 (Fig. 2a). Strain CBA3628 could not significantly alter natural microbial succession, showing a diversity similar to that seen in CTR.
Genetic differences between starter strains
Comparative genomic analyses between starter strains were conducted to identify the factors influencing the differences in adaptation and predominance in kimchi fermentation environments. Despite high 16S rRNA gene sequence similarity, Leuconostoc sp. CBA3628 showed distinct genomic differences with lower average nucleotide identity (ANI) and in silico DNA–DNA hybridisation (DDH) values compared with other Leu. mesenteroides strains (Supplementary Fig. 3a–c).
The general genomic features of starter strains indicated that Leuconostoc sp. CBA3628 had fewer total genes and coding sequences (CDSs) than WiKim strains (Supplementary Table 2). Functional features of the four starter strains were compared using clusters of orthologous groups (COG), carbohydrate-active enzymes (CAZymes), and Kyoto Encyclopaedia of Genes and Genomes (KEGG) analyses. COG analysis showed that strain CBA3628 had the lowest functional gene counts in key categories for fermentative metabolism (C, E, G, J, and K; Fig. 3a)33, making its fermentative capacity weaker. CAZyme comparison revealed that strain CBA3628 contained fewer genes encoding glycoside hydrolases (GH) and glycosyl transferases (GT); this reduced its carbohydrate metabolism efficiency (Fig. 3b).

a Clusters of orthologous groups (COG) functional categories display the number of genes in each starter strain, classified as follows: C, energy production and conversion; D, cell cycle control, cell division, chromosome partitioning; E, amino acid transport and metabolism; F, nucleotide transport and metabolism; G, carbohydrate transport and metabolism; H, coenzyme transport and metabolism; I, lipid transport and metabolism; J, translation, ribosomal structure and biogenesis; K, transcription; L, replication, recombination and repair; M, cell wall/membrane/envelope biogenesis; N, cell motility; O, post-translational modification, protein turnover, and chaperones; P, inorganic ion transport and metabolism; Q, secondary metabolites biosynthesis, transport, and catabolism; S, function unknown; T, signal transduction mechanisms; U, intracellular trafficking, secretion, and vesicular transport; and V, defence mechanisms. b The number of CAZyme genes in each starter strain is classified as follows: GH glycoside hydrolases, GT glycosyl transferases, AA auxiliary activities, CE carbohydrate esterases, PL polysaccharide lyases, and CBM carbohydrate-binding molecules.
Metabolic pathway analysis displayed using iPath based on KEGG Orthology (KO) numbers showed that all strains shared genes for phosphoketolase and incomplete glycolysis pathways (Fig. 4), which are key pathways in Leu. mesenteroides33. WiKim strains also harboured genes related to energy metabolism, including galactose and pyruvate metabolism, which CBA3628 lacked (red lines in Fig. 4). The absence of these genes might result in lower ATP production in Leuconostoc sp. CBA3628 than WiKim strains.

Metabolic pathways of Leuconostoc mesenteroides WiKim32, Leuconostoc mesenteroides WiKim33, Leuconostoc mesenteroides WiKim0121, and Leuconostoc sp. CBA3628 were generated using the iPath v2 module based on Kyoto Encyclopaedia of Genes and Genomes (KEGG) Orthology (KO) numbers. The pathways are displayed in different colours depending on the presence and/or absence of genes in each starter strain, as depicted in the table inside the figure.
Expression levels of genes involved in carbohydrate metabolism
Based on the genetic differences between WiKim strains and Leuconostoc sp. CBA3628, we constructed a pathway map focusing on energy metabolism (Fig. 5a). As shown in red, yellow, purple, and green pathways in the map, strain CBA3628 could not use various carbon sources, such as raffinose, lactose, maltose, xylose, and arabinose, owing to the absence of related genes, unlike WiKim strains. The expression levels of these genes were investigated and visualised using a heatmap (Fig. 5b). Compared with strain CBA3628, no additional genes related to energy metabolism (including Arabic and KO numbers) were identified in the WiKim strains: pyruvate oxidase (14, K00158); alpha-galactosidase (30, K07407); beta-galactosidase (36, K01190); galactokinase (37, K00849); maltose phosphorylase (38, K00691); UDP-glucose-hexose-1-phosphate uridylyltransferase (39, K00965); xylose isomerase (51, K01805); xylulokinase (52, K00854); l-ribulose-5-phosphate 4-epimerase (53, K03077); l-arabinose isomerase (54, K01804); l-ribulokinase (55, K00853); ascorbate PTS system EIIA component (56, K02821); ascorbate PTS system EIIB component (57, K02822); l-ascorbate 6-phosphate lactonase (58, K03476); 3-dehydro-l-gulonate-6-phosphate decarboxylase (59, K03078); and l-ribulose-5-phosphate 3-epimerase (60, K03079). These genes are involved in ATP production from raffinose, galactose, lactose, maltose, pyruvate, ribose, xylose, arabinose, and ascorbate. The differential expression levels of these genes may result in distinct ATP yields, contributing to the growth and predominance of the WiKim strains during fermentation12. Indeed, these carbon sources did exist in the kimchi samples, although present in small amounts, and were consumed during the fermentation (Supplementary Fig. 4).

a Reconstructed carbohydrate metabolic pathways of Leuconostoc mesenteroides, Leuconostoc citreum, Weissella cibaria, and Weissella koreensis. The pathways are represented by different colours depending on the presence (P) and/or absence (A) of genes in each strain, as depicted in the table inside the figure. b Expression levels of genes in Leuconostoc mesenteroides, Leuconostoc citreum, Weissella cibaria, and Weissella koreensis in response to carbohydrate availability. Arabic numbers and KO numbers are positioned close to the lines in a reconstructed pathways and to b the heatmap, showing the expression levels of each gene across the strains. The kimchi samples inoculated with strains WiKim32, WiKim33, WiKim0121, and CBA3628, and saline solution were labelled as 'W32', 'W33', 'W0121', 'C3628', and 'CTR', respectively.
Additionally, some of these genes—such as pyruvate oxidase, alpha-galactosidase, beta-galactosidase, galactokinase, maltose phosphorylase, UDP-glucose-hexose-1-phosphate uridylyltransferase, xylose isomerase, xylulokinase, l-ribulose-5-phosphate 4-epimerase, L-arabinose isomerase, l-ribulokinase, ascorbate PTS system EIIA or EIIB component, l-ascorbate 6-phosphate lactonase, 3-dehydro-l-gulonate-6-phosphate decarboxylase, and l-ribulose-5-phosphate 3-epimerase—were also expressed in LAB derived from raw ingredients, including Leu. citreum, Wei. cibaria, and/or Wei. koreensis, which compete with strain CBA3628 for carbon sources during fermentation (Fig. 5b). This suggests that strain CBA3628 is unable to utilise various carbon sources for ATP production as efficiently as the WiKim strains and the LAB from raw ingredients, leading to the non-dominance of strain CBA3628.
Selection of putative genes associated with predominance of starter strains
Gene expression from each starter strain was categorised based on KEGG BRITE hierarchical classifications to identify putative genes affecting the adaptation and predominance of starter strains in the kimchi fermentation environment. The 1238 genes annotated within the WiKim32, WiKim33, WiKim0121, CBA3628, and ATCC 8293T strains across 33 categories were analysed to calculate the average transcripts per million (TPM) values. Among them, eight categories—oxidative phosphorylation, messenger RNA biogenesis, ribosomes, translation factors, chaperones and folding catalysts, membrane trafficking, mitochondrial biogenesis, and exosomes—exceeded the average TPM values of 2000 (Supplementary Fig. 5). In these categories, 83 genes had expression levels above 2000 TPM, and subsequent network analysis revealed 12 functional genes—encoding pyruvate kinase (K00873); glucose-6-phosphate isomerase (GPI; K01810); F-type H+-transporting ATPase subunit delta (K02113); molecular chaperones DnaJ (K03686), GprE (K03687), DnaK (K04043), ClpL (K04086), and Hsp20 (K13993); translation initiation factor IF-2 (K02520), small subunit ribosomal protein S2 (K02967) and S9 (K02996); and electron transfer flavoprotein alpha subunit (K03522)—closely correlated with bacterial adaptation and predominance (Fig. 6a). Most of them (8 of 12) encode stress-response proteins, which may help maintain protein homoeostasis under the cold and acidic environments of kimchi; therefore, we selected these genes as putative genes associated with the predominance of starter strains.

The average transcripts per million (TPM) values of 1238 genes in 33 categories based on KEGG BRITE hierarchical classifications were calculated, and 83 genes that exhibited transcript levels above 2000 TPM were selected. Spearman correlation coefficients were calculated between the relative abundance of Leuconostoc ASV001 and the TPM values mapped to Leuconostoc mesenteroides ATCC 8293T, Leuconostoc mesenteroides WiKim32, Leuconostoc mesenteroides WiKim33, and Leuconostoc mesenteroides WiKim0121 in the kimchi samples CTR, W32, W33, and W0121 samples, respectively, and between the relative abundance of Leuconostoc ASV002 and the TPM values mapped to Leuconostoc sp. CBA3628 in the C3628 sample. Then, a a network analysis was performed. The colours of nodes represent the following: red, bacterial relative abundances; yellow-green, KO number within oxidative phosphorylation category; grey, KO number within ribosome category; white-yellow, KO number within translation factors category; pink, KO number within chaperones and folding catalysts category; green, KO number within membrane trafficking category; blue, KO number within mitochondrial biogenesis category; apricot, KO number within exosome category; and sky-blue, KO number within the categories more than two. The edges represent Spearman correlations, with thicker edges indicating a stronger positive correlation and thinner edges displaying a weaker correlation or a negative correlation, as per the correlation scale ranging from −1.0 to 1.0. The highly ‘strong’ positive correlation of over 0.6 is visualised with red edges. b The TPM values of genes encoding F-type ATPase and chaperone proteins from five strains in each kimchi sample during fermentation were visualised using the heatmap. Gene expression levels are indicated by colour intensity, with red representing higher TPM values (0 to 50,000 or greater). The pH values at each time point are provided above the heatmap, with the intensity of colour (yellow–black) indicating the pH, as shown on the scale bar (4.06‒5.14). The boxes of gene names are filled with colours corresponding to each category described in the network analysis. The kimchi samples inoculated with strains WiKim32, WiKim33, WiKim0121, and CBA3628, and saline solution were labelled as 'W32', 'W33', 'W0121', 'C3628', and 'CTR', respectively.
The impact of the genes encoding stress-response proteins on the adaptation and predominance of WiKim strains compared to CBA3628 during kimchi fermentation was evaluated (Fig. 6b). We first assessed the expression of genes encoding F-type ATPase and Chaperones DnaJ, GrpE, and DnaK, which were correlated at day 6 of fermentation (Fig. 6a). F-type ATPase is likely a stress-response protein that maintains bacterial intracellular pH homoeostasis under acidic conditions34,35. Chaperones DnaJ, GrpE, and DnaK coordinate a crucial protein folding and repair cycle, ensuring cellular resilience and resistance under stress conditions like cold environments36,37,38,39,40,41. The expression levels of F-type ATPase complex genes were similar in all samples; however, the expression pattern of chaperone genes (dnaJ, grpE, and dnaK) was significantly different only in WiKim kimchi samples. WiKim strains showed higher expression of these genes encoding chaperones at day 6 than strains CBA3628 and ATCC 8293T, indicating greater resistance to low temperatures in early fermentation (Fig. 6b).
Higher expression of clpL in WiKim strains than in strains CBA3628 and ATCC 8293T during late fermentation indicates their significant impact on consistent predominance. The findings suggest that acidic and cold-stress-response proteins significantly contribute to bacterial adaptation and predominance during kimchi fermentation. In early fermentation, the F-type ATPase complex and chaperones DnaJ, DnaK, and GrpE play a crucial role, with cold-stress resistance by chaperones being more critical for initial adaptation. Chaperone ClpL plays a pivotal role both in cold- and acidic-stress resistance during late fermentation, impacting consistent predominance as pH decreases.
These stress-response proteins require ATP42,43, emphasising the importance of ATP production by starter strains in the kimchi fermentation environment, as mentioned earlier. In addition, the genes encoding pyruvate kinase and GPI, which showed a close correlation with bacterial relative abundance (Fig. 6a), are also key metabolic genes involved in ATP production. These points are further elaborated in the discussion section.
Dominance of Wei. koreensis in a kimchi environment without starter strains
Weissella ASV009 (Wei. koreensis) was predominant in CTR from day 11 to 33 (Fig. 2a). As transcript levels of Wei. koreensis in carbohydrate fermentative pathways were unremarkable, the arginine deiminase (ADI) pathway in Wei. koreensis was explored44. During kimchi fermentation, TPM values of genes involved in the ADI pathway were highest in CTR (Fig. 7a), corresponding to the high relative abundances of ASV009 (Fig. 2a). Simultaneously, arginine levels decreased, and ornithine levels increased at day 11 in CTR (Fig. 7b, c). Weissella ASV008 (Wei. cibaria) did not affect arginine and ornithine concentrations. Wei. koreensis can produce one molecule of ATP while metabolising arginine to produce ornithine, which can be a metabolic energy source advantageous for survival. In addition, NH3 is produced during this process, which can contribute to buffering the lowered pH around the cell.

a Putative schematic diagram of arginine deiminase pathway of Weissella cibaria and Weissella koreensis with their TPM values observed in each pathway and visualised by the heatmap during kimchi fermentation. Changes in the concentrations of b arginine and c ornithine in the CTR, W32, W33, W0121, and C3628 kimchi samples. The kimchi samples inoculated with strains WiKim32, WiKim33, WiKim0121, and CBA3628, and saline were labelled as 'W32', 'W33', 'W0121', 'C3628', and 'CTR', respectively. Data were presented as mean ± standard deviation. Experiments were performed in triplicate.
Discussion
In this study, we aimed to identify the key factors enabling the dominance of Leuconostoc strains in the kimchi fermentation environment. While it is well-established that inoculated Leuconostoc starter cultures dominate during kimchi fermentation16, this study uniquely reveals the molecular and metabolic mechanisms underlying the successful predominance of Leu. mesenteroides WiKim32, WiKim33, and WiKim0121, compared with Leuconostoc sp. CBA3628, which failed to establish dominance. Comparison of the genomes of WiKim and CBA3628 strains revealed that CBA3628 harboured fewer genes involved in crucial metabolic pathways, especially carbohydrate metabolism (Figs. 3, 4), which likely contributed to its reduced adaptability and lower dominance in the kimchi environment (Fig. 2a)33. The transcript-level expression of these genes, present in WiKim strains but absent in strain CBA3628, demonstrated that WiKim strains can use a higher number of carbon sources such as raffinose, lactose, maltose, xylose, and arabinose than strain CBA3628, likely contributing to their higher ATP production (Fig. 5).
The network analysis further highlighted the importance of genes encoding pyruvate kinase and GPI, both of which are involved in ATP production (Fig. 6a). Considering that fructose is the main carbon source for starter strains in this study (Fig. 1c), GPI plays a critical role in ATP production as a key component of fructose metabolism. Pyruvate kinase is a crucial rate-limiting enzyme in glycolysis45. As rate-limiting enzymes determine the overall speed of a metabolic pathway, the expression levels of pyruvate kinase directly affect both the quantities and rates of ATP production in the starter strains. Based on the transcript-level expression and network analysis results, ATP production by the starter cultures is essential for their adaptation and predominance during kimchi fermentation. Large amounts of ATP not only promote bacterial growth but are also utilised by stress-response proteins to ensure the survival and consistent predominance of these bacteria in the fermentation environment, as further discussed below.
Considering that kimchi fermentation is typically conducted at low temperatures (0–10 °C) with pH decreasing to around 4, stress-response proteins might be crucial for adapting to kimchi’s cold and acidic environments. Based on network analysis (Fig. 6a), ATPase and molecular chaperones play crucial roles in helping bacteria adapt to various environmental stresses46,47,48. ATPase, typically associated with oxidative phosphorylation in aerobic conditions49,50, is unlikely to function in this capacity in Leuconostoc owing to the anaerobic nature of the kimchi environment. Instead, it likely serves as a proton pump, maintaining bacterial intracellular pH homoeostasis in acidic environments34,35. Analysis of genes encoding the F-type ATPase complex revealed that the overall expression levels decreased from day 6 to 33 in WiKim32, WiKim33, WiKim0121, and CBA3628, whereas ATCC 8293T showed an increase from day 6 to 11, followed by a decrease (Fig. 6b). These patterns can be explained by pH changes in each kimchi sample during fermentation. Prior research indicates that bacterial ATPase activity is pH-dependent, with certain Bifidobacterium strains showing increased ATPase expression at pH 4 than in more acidic (pH 3) or more alkaline (pH 5) conditions51. Similarly, the TPM values of the genes encoding ATPase complex from strains WiKim32, WiKim0121, and CBA3628 on day 6, and those from strain ATCC 8293T on day 11 were similar, reflecting similar pH conditions (Fig. 6b). This suggests that the variations in ATPase activity among strains could be attributed to different pH values of each kimchi sample.
Chaperones, including DnaJ, DnaK, GrpE, and ClpL, play vital roles in helping bacteria adapt to cold and acidic conditions37,40,41,52,53 typical of the kimchi environment. This study highlights that the expression levels of these chaperone genes were significantly higher in WiKim strains than in strain CBA3628 during kimchi fermentation (Fig. 6b). This enhanced expression might be advantageous in maintaining protein homoeostasis and cellular functions under stress conditions, thereby promoting the adaptation and predominance of starter strains in kimchi environments. Particularly, DnaJ, DnaK, and GrpE may contribute to the cold resistance of WiKim strains for their initial adaptation in an early kimchi environment, while ClpL plays a crucial role in their predominance by mediating both cold and acidic stress responses as pH decreases. These stress-response proteins require ATP42,43,54, indicating that ATP production is important for bacterial adaptation and predominance not only for their growth but also for their resistance in the kimchi fermentation environment.
Additionally, the dominance of Wei. koreensis was confirmed in the CTR kimchi environment (Fig. 2a). The arginine deamination pathway of Wei. koreensis uses arginine to produce ornithine, ATP, and NH3 together (Fig. 7)44. In a highly acidic environment such as kimchi fermentation, NH3 can help protect cells from acid stress55. Wei. koreensis secures metabolic energy and overcomes stressful conditions in a competitive fermentation environment, using strategies similar to those used by the WiKim strains, although via different genes, to gain an advantage in competition with other microorganisms. This could potentially be the reason Weissella ASV009 (Wei. koreensis) became dominant in the late kimchi fermentation environment, where there was no particularly dominant starter strain.
This study provides valuable insights into the molecular and metabolic mechanisms underpinning the adaptation and predominance of Leuconostoc starter strains in kimchi fermentation. The results of this study emphasise that the ability to cope with stress conditions, such as low temperature and pH, and efficient carbohydrate metabolism are key factors determining the growth and dominance of kimchi LAB in fermentation environments. The importance of ATP production in maintaining stress-response activities in kimchi starter cultures further underscores the complex interplay between metabolism and adaptation in fermentation. These insights could inform the development of more robust and effective starter cultures for kimchi production, potentially contributing to quicker acidification, reducing safety risks, and minimising flavour defects caused by spoilage microorganisms. Starter cultures may also improve the sensory and nutritional properties of the final product, but this remains to be investigated.
Despite these insights, certain limitations remain. While our findings identify key contributors that confer competitive advantages to Leu. mesenteroides WiKim32, WiKim33, and WiKim0121 as kimchi starter strains, it remains unclear whether these key metabolic pathways are strain-specific or conserved at the species level among Leuconostoc spp. and Weissella spp. Future studies should investigate whether these pathways and stress-response mechanisms are widespread among Leu. mesenteroides strains or unique to strains WiKim32, WiKim33, and WiKim0121, as well as how they compare to those in Leu. citreum, Wei. cibaria, and Wei. koreensis. Understanding the species- and strain-specific contributions of these factors could further refine microbial selection criteria for optimal starter cultures, ultimately enhancing the stability and functional properties of kimchi fermentation.
Methods
Preparation of starter strains and kimchi samples
Four strains, Leu. mesenteroides WiKim32, Leu. mesenteroides WiKim33, Leu. mesenteroides WiKim0121, and Leuconostoc sp. CBA3628, which were previously isolated at the World Institute of Kimchi in Gwangju, Korea, were used as kimchi starter cultures in this study. The strains WiKim32, WiKim33, and WiKim0121 are well-studied for their use as kimchi starters and have been deposited as patent strains with the numbers KR101836365B1, KR101660847B1, and KR102488052B1, respectively, owing to their ability to dominate during kimchi fermentation and to enhance sensory and/or nutritional properties. These strains were cultured in de Man–Rogosa–Sharpe (MRS; BD, USA) medium at 30 °C before inoculation into the kimchi samples.
Kimchi ingredients, including kimchi cabbages, red pepper powders, garlic, ginger, green onions, and radishes, were purchased from a commercial market in Gwangju. The preparation followed a previously established method with slight modifications56. Specifically, sugar and starch were not added to kimchi samples, and the fermentation temperature was lowered to 5 °C. Briefly, kimchi cabbages were soaked in 10% (w/v) solar salt solution for 10 h, washed three times with tap water, and drained. Garlic, ginger, and radishes were ground using a homogeniser (Hanil, Seoul, Korea) and mixed with salted kimchi cabbages. The final kimchi mixture consisted of 70% salted kimchi cabbages, 4.5% red pepper powders, 3.75% ground garlic, 1.2% ground ginger, 3.75% green onions, 4.5% ground radishes, and 12.3% distilled water.
Each kimchi sample was inoculated with one of the four Leuconostoc strains at 107 CFUs per gram of kimchi. The control sample was inoculated with 0.9% (w/v) saline instead of the starter strain. The samples inoculated with strains WiKim32, WiKim33, WiKim0121, and CBA3628, and saline solution instead of starter strain were labelled as 'W32', 'W33', 'W0121', 'C3628', and 'CTR', respectively. The experiments were conducted in triplicate, resulting in a total of fifteen kimchi samples. All samples were pre-fermented at room temperature for 6 h to allow the starter strains to adapt well in the kimchi samples before being fermented at 5 °C for 33 days. In this study, sampling time at day 0 corresponded to the beginning of fermentation at 5 °C.
Sample collection
Approximately 8 mL of kimchi soup samples were collected at days 0, 3, 6, 11, 22, and 33 during fermentation and filtered through sterile stomacher filter bags (Nasco, WI, USA) to remove large kimchi particles. A portion of the filtered kimchi soup was used to determine pH and LAB CFUs using a pH metre (Thermo Scientific, MA, USA) and a 3MTM PetrifilmTM Lactic Acid Bacteria Count Plate (3M™, MN, USA), respectively. Of the remaining filtrate, 4 mL was subject to centrifugation at 8000×g for 10 min at 4 °C. The pellets were used for metataxonomic and metatranscriptomic analyses, whereas the supernatants were utilised for metabolomic analysis.
Bacterial community succession observation
Total genomic DNA (gDNA) was extracted from the pellets using the DNeasy PowerSoil Kit (Qiagen, Hilden, Germany), following the manufacturer’s instructions. The extracted gDNA was used for amplifying the V3 to V4 regions of the 16S rRNA gene through polymerase chain reaction (PCR) using the following primer sets: 341F and 805R57,58. Subsequently, PCR products were purified using the QIAquick PCR Purification Kit (Qiagen, Hilden, Germany). Library preparation was performed using the Nextera XT DNA Library Preparation Kit v2 with i5 and i7 primers (Illumina) according to the manufacturer’s instructions, with modifications in the PCR amplification step (San Diego, CA, USA) to attach barcodes. After a second PCR, products were purified and pooled in one tube for adjusting to equal concentrations. The pooled amplicons were sequenced by Macrogen (Seoul, Korea) on a MiSeq™ platform (Illumina, CA, USA).
Following the trimming of barcode and adaptor sequences from FASTQ reads, the trimmed reads were demultiplexed through the bcl2fastq2 conversion software (version 2.20.0; Illumina, CA, USA). The Illumina paired-end sequencing reads were assembled and filtered for quality scores less than q20 using VSEARCH. Subsequent analyses of the reads were performed with the Qiime2 version 2023.0259. The DADA2 plugin was used for quality filtering, denoising, and clustering of the imported paired reads60, exclusion of chimeric sequences, and singleton ASVs for further analyses. Taxonomic classification was performed with the q2-feature classifier plugin, employing the classify-sklearn method61 and the pre-trained SILVA version 138.1 database62 with 99% identity. Sequences from non-target sources, including cyanobacteria, mitochondria, and chloroplasts, were removed in silico. Sample libraries were normalised by rarefying to the lowest sequencing depth. Bacterial community composition and alpha diversity were analysed using the q2-diversity plugin in the Qiime2. The sequences of the V3–V4 region of the 16S rRNA genes of the strains and those of ASVs from the metataxonomic results were aligned and used to construct phylogenetic trees using the neighbour-joining (NJ) algorithm in the MEGA 11 software package63.
Metabolites measurement
Proton nuclear magnetic resonance (1H-NMR) spectroscopy was used to determine metabolites generated during kimchi fermentation, following a previously established method with slight modifications64. First, 0.5 mL of each kimchi sample supernatant was mixed with 0.5 mL of a solution containing 99.9% deuterium oxide (D2O; Sigma-Aldrich, MO, USA) and 2.9 mM 3-(trimethylsilyl) propionic-2,2,3,3-d4 acid (TSP; Sigma-Aldrich, MO, USA) (pH adjusted to 7.0). Samples were then transferred into 5-mm NMR tubes, and 1H-NMR spectra were obtained using the Varian Inova 600-MHz 1H-NMR spectrometer (Varian, CA, USA) with the standard PRESAT pulse sequence at 25 °C. Spectral data, collected over 32 k data points across a spectral width of 9,615 Hz, were subjected to manual phase adjustment and baseline correction using the Chenomx Processor NMR Suite Programme version 8.3 (Chenomx, Edmonton, Canada). Spectral intensities were segmented into 0.04-ppm bins spanning 0.5‒10 ppm and normalised to the TSP signal intensity at 0 ppm for comparison. For metabolite identification and quantification in kimchi samples, 1H-NMR spectra were analysed using the Chenomx Profiler NMR Suite Programme version 9.0 (Chenomx, Edmonton, Canada), with TSP serving as an internal standard against the reference library 9 for 600-MHz compounds65.
The relationships between bacterial taxa and all metabolites across fermentation periods were visualised using NMDS based on the Bray–Curtis dissimilarity index, employing metaMDS and envfit functions from the vegan package within the R package version 4.3.266. In the NMDS plot, the points differentiated by colour represent the metabolite profiles of kimchi samples during fermentation, with the proximity of points indicating their similarity. The distances between the points of each kimchi sample reflect the dynamic changes in metabolite profiles during fermentation.
Comparative genome analyses
For whole-genome sequence analysis of four Leuconostoc starter strains, gDNA was extracted using the Wizard Genomic DNA Purification Kit (Promega, WI, USA), following the manufacturer’s instructions. As described previously55, the genomes extracted from four starter strains were sequenced by Macrogen using a combination of Illumina HiSeq 2500 sequencing and PacBio RS single-molecule real-time sequencing based on a 20-kb library. The quality of the genomes, i.e. their completeness and contamination rates, was verified using the CheckM software version 1.0.467. Whole-genome sequences of Leu. mesenteroides WiKim32, Leu. mesenteroides WiKim33, Leu. mesenteroides WiKim0121, and Leuconostoc sp. CBA3628, have been deposited in GenBank with the accession numbers CP037750–4, CP021491–4, CP098784–9, and CP042404–7, respectively. These sequences were automatically annotated through the NCBI prokaryotic genome annotation pipeline (PGAP)68.
For comparative genomic analyses, sequences of 16S rRNA gene and whole genomes from other Leuconostoc strains were retrieved from GenBank. Genome qualities were verified using the CheckM software. 16S rRNA gene sequence similarity, ANI values, and in silico DDH values of 23 Leuconostoc strains were calculated using BLASTn (NCBI; MD, USA), the Orthologous ANI Tool (OAT; https://www.ezbiocloud.net/tools/orthoani)69, and the Genome-to-Genome Distance Calculator version 3.0 (GGDC 3.0; https://ggdc.dsmz.de/ggdc.php)70, respectively, according to previously described parameters33.
In order to compare the genomic features of the starter strains—WiKim32, WiKim33, WiKim0121, and CBA3628—COG, CAZyme, and KEGG analyses were performed using corresponding amino acid sequences. For clustering functional genes of four starter strains, the corresponding amino acid sequences were analysed using the Evolutionary Genealogy of Genes: Non-supervised Orthologous Groups (eggNOG) mapper version 2.0, hosted on the public database eggNOG version 5.071. Each starter strain’s gene numbers were assigned to COG categories, and their functional characteristics were compared. The CAZyme gene profiles of strains were analysed using DIAMOND and HMMER tools, referencing the pre-annotated CAZyme sequence database in dbCAN372.
Functional annotation of predicted proteins in four starter strains was performed using the Blast KEGG Orthology And Links Annotation (BlastKOALA)73, and the metabolic pathways of these strains were displayed using the iPath version 3.0, based on KO. Pathway visualisation was achieved by varying line thickness and colour depending on the presence and/or absence of genes in each strain.
Transcriptional expression analysis
Total RNA was extracted from the kimchi pellets using Trizol reagent (Invitrogen, MA, USA). Subsequently, the clear aqueous phase was collected and precipitated with ethanol. RNA quality was assessed using an Agilent 2100 bioanalyzer with an RNA 6000 Nano Chip (Agilent Technologies, Amstelveen, The Netherlands). Total RNA extraction was followed by the removal of ribosomal RNA (rRNA) and transfer RNA (tRNA) to isolate messenger RNA (mRNA) using the NEBNext® Ultra™ II Directional RNA Library Prep Kit (NEB, USA). The isolated mRNAs were used for cDNA synthesis and shearing following the manufacturer’s instructions. Indexing was performed using the Illumina indexes 1–12. The enrichment step was carried out using PCR. After pooling the products into one tube, high-throughput sequencing was performed as paired-end 100 sequencing using NovaSeq 6000 (Illumina, CA, USA) at Ebiogen (Seoul, Korea).
The raw sequencing reads were trimmed, and those shorter than 30 nucleotides were removed using Sickle software74. The Burrows–Wheeler Aligner (BWA) software75 was used to map the putative mRNA reads from each starter kimchi sample with the whole-genome sequences of the respective strains WiKim32, WiKim33, WiKim0121, and CBA3628. For the control sample, the reads were annotated to the complete genomes of type strain Leu. mesenteroides subsp. mesenteroides ATCC 8293T. Following the added starter strains, Leu. citreum, Wei. cibaria, and Wei. koreensis were identified as the prevalent lactic acid bacteria during the kimchi fermentation process, and their reads were mapped to the whole-genome sequences of Leu. citreum CBA3621 (GenBank accession no. CP042410–11), Wei. cibaria CBA3612 (CP041193–6), and Wei. koreensis KACC 15510 (CP002899–900), respectively. From the BWA-aligned reads, TPM values were calculated to quantify relative gene expression against the strains in the respective kimchi samples. In this study, TPM values from days 0 and 3 could not be obtained owing to insufficient mRNA sequencing reads. The metatranscriptomic analyses of kimchi fermentation were conducted from early (day 6) to late stages (day 33).
The carbohydrate fermentative pathways were reconstructed and metatranscriptome reads were mapped to the whole-genome sequences of Leu. mesenteroides ATCC 8293T (in the CTR), Leu. mesenteroides WiKim32 (in W32), Leu. mesenteroides WiKim33 (in W33), Leu. mesenteroides WiKim0121 (in W0121), Leuconostoc sp. CBA3628 (in C3628), and Leu. citreum CBA3621, Wei. cibaria CBA3612, and Wei. koreensis KACC 15510 (in all samples) with their transcriptional expression levels in each pathway. The pathways were illustrated with lines of different colours to represent the presence and/or absence of genes in each strain, and their TPM values were visualised using a heatmap. Arabic numbers and KO numbers are positioned close to the lines in the pathways and the heatmap, showing the expression levels of each gene across the strains.
Putative genes associated with the dominance of starter strains
Owing to the vast number of genes in each starter strain, a threshold was initially established to select putative genes potentially related to the adaptation of starter strains and their predominance during kimchi fermentation. The genes from each strain were categorized based on KEGG BRITE hierarchical classifications, and then gene expression levels were quantified. Within each functional category, the average expression level was calculated by summing the TPM values of the assigned genes from each starter strain and dividing by the total number of genes within that category. Gene expression levels are typically categorized as ‘low’ (below 100 TPM), ‘intermediate’ (between 100 and 1000 TPM), and ‘high’ (above 1000 TPM)76. In this study, the threshold was set to 2000 TPM to identify expressions significantly exceeding the ‘high’ threshold. The categories surpassing the established average TPM values were first selected, and then individual genes above 2000 TPM within the selected categories were further analysed for network diagram construction.
Spearman correlation coefficients were calculated using the Prism software version 9.5.1 (GraphPad Software, CA, USA) to assess the relationships between the relative abundances of four starter strains and their respective TPM values at each fermentation period. Network analysis was then performed using the Cytoscape version 3.10.177 based on these correlation results and visualised by varying the colour of nodes and the width of edges to represent relative abundances and putative genes of Leuconostoc starter strains. The red edges indicate Spearman correlation coefficients above 0.6, suggesting a ‘strong’ positive correlation78. The potential effects of the genes encoding stress-response proteins F-type ATPase and molecular chaperones on the adaptation and predominance of the starter strains were visualised using a heatmap based on the Spearman correlation coefficients.
Fermentative metabolic pathways were reconstructed based on annotated KEGG pathways and associated KO numbers to analyse the carbohydrate metabolism of four starter strains during kimchi fermentation. Leu. citreum, Wei. cibaria, and Wei. koreensis were also included for a comprehensive comparison with the metabolic profiles of the starter strains. A heatmap based on TPM values was used to illustrate expression levels across different fermentation periods. Arabic numbers and KO numbers corresponding to the respective genes involved in each pathway were annotated alongside the heatmap.
Data availability
The kimchi metatranscriptome sequencing and the 16S rRNA gene sequencing data are publicly available in the NCBI Short Read Archive (SRA). Accession no. of metatranscriptome sequencing data are SRR28544770–28544789, and those of 16S rRNA gene sequencing data are SRR22317905, SRR22317916–22317939, SRR22317950 and SRR22317956–22317959.
Code availability
Not applicable.
References
Cha, J. et al. Does kimchi deserve the status of a probiotic food?. Crit. Rev. Food Sci. Nutr. 64, 6512–6525 (2024).
Jung, M. Y. et al. Role of jeotgal, a Korean traditional fermented fish sauce, in microbial dynamics and metabolite profiles during kimchi fermentation. Food Chem. 265, 135–143 (2018).
Kim, H. J. et al. A review of the health benefits of kimchi functional compounds and metabolites. Microbiol. Biotechnol. Lett. 51, 353–373 (2023).
Lee, S. H., Whon, T. W., Roh, S. W. & Jeon, C. O. Unraveling microbial fermentation features in kimchi: from classical to meta-omics approaches. Appl. Microbiol. Biotechnol. 104, 7731–7744 (2020).
Lee, S. H., Jung, J. Y. & Jeon, C. O. Source tracking and succession of kimchi lactic acid bacteria during fermentation. J. Food Sci. 80, M1871–M1877 (2015).
Song, H. S. et al. Microbial niches in raw ingredients determine microbial community assembly during kimchi fermentation. Food Chem. 318, 126481 (2020).
Cheigh, H. S. & Park, K. Y. Biochemical, microbiological, and nutritional aspects of kimchi (Korean fermented vegetable products). Crit. Rev. Food Sci. Nutr. 34, 175–203 (1994).
Lee, M.-E. et al. Starter cultures for kimchi fermentation. J. Microbiol. Biotechnol. 25, 559–568 (2015).
Jeong, S. H., Jung, J. Y., Lee, S. H., Jin, H. M. & Jeon, C. O. Microbial succession and metabolite changes during fermentation of dongchimi, traditional Korean watery kimchi. Int. J. Food Microbiol. 164, 46–53 (2013).
Jeong, S. H., Lee, S. H., Jung, J. Y., Choi, E. J. & Jeon, C. O. Microbial succession and metabolite changes during long-term storage of kimchi. J. Food Sci. 78, M763–M769 (2013).
Jung, J. Y. et al. Metatranscriptomic analysis of lactic acid bacterial gene expression during kimchi fermentation. Int. J. Food Microbiol. 163, 171–179 (2013).
Jung, J. Y., Lee, S. H. & Jeon, C. O. Kimchi microflora: history, current status, and perspectives for industrial kimchi production. Appl. Microbiol. Biotechnol. 98, 2385–2393 (2014).
Bong, Y.-J., Jeong, J.-K. & Park, K.-Y. Fermentation properties and increased health functionality of kimchi by kimchi lactic acid bacteria starters. J. Korean Soc. Food Sci. Nutr. 42, 1717–1726 (2013).
Choi, I.-K. et al. Novel Leuconostoc citreum starter culture system for the fermentation of kimchi, a fermented cabbage product. Antonie Van. Leeuwenhoek 84, 247–253 (2003).
Jin, H.-S., Kim, J.-B., Yun, Y.-J. & Lee, K.-J. Selection of kimchi starters based on the microbial composition of kimchi and their effects. J. Korean Soc. Food Sci. Nutr. 37, 671–675 (2008).
Lee, J.-J. et al. Effects of combining two lactic acid bacteria as a starter culture on model kimchi fermentation. Food Res. Int. 136, 109591 (2020).
Lee, K. & Lee, Y. Effect of Lactobacillus plantarum as a starter on the food quality and microbiota of kimchi. Food Sci. Biotechnol. 19, 641–646 (2010).
Park, S.-E. et al. Changes of microbial community and metabolite in kimchi inoculated with different microbial community starters. Food Chem. 274, 558–565 (2019).
Fessard, A. & Remize, F. Why are Weissella spp. not used as commercial starter cultures for food fermentation?. Fermentation 3, 38 (2017).
Cho, J. et al. Microbial population dynamics of kimchi, a fermented cabbage product. FEMS Microbiol. Lett. 257, 262–267 (2006).
Grobben, G. J. et al. Spontaneous formation of a mannitol-producing variant of Leuconostoc pseudomesenteroides grown in the presence of fructose. Appl. Environ. Microbiol. 67, 2867–2870 (2001).
Jung, J. Y. et al. Metagenomic analysis of kimchi, a traditional Korean fermented food. Appl. Environ. Microbiol. 77, 2264–2274 (2011).
Moon, S. H., Kim, C. R. & Chang, H. C. Heterofermentative lactic acid bacteria as a starter culture to control kimchi fermentation. LWT 88, 181–188 (2018).
Hong, Y., Li, J., Qin, P., Lee, S.-Y. & Kim, H.-Y. Predominant lactic acid bacteria in mukeunji, a long-term-aged kimchi, for different aging periods. Food Sci. Biotechnol. 24, 545–550 (2015).
Jeong, S. H. et al. Effects of red pepper powder on microbial communities and metabolites during kimchi fermentation. Int. J. Food Microbiol. 160, 252–259 (2013).
Kim, T.-W. et al. Identification and distribution of predominant tactic acid bacteria in Kimchi, a Korean traditional fermented food. J. Microbiol. Biotechnol. 12, 635–642 (2002).
Jung, J. Y. et al. Effects of Leuconostoc mesenteroides starter cultures on microbial communities and metabolites during kimchi fermentation. Int. J. Food Microbiol. 153, 378–387 (2012).
Park, J.-M. et al. Effect of a Leuconostoc mesenteroides strain as a starter culture isolated from the kimchi. Food Sci. Biotechnol. 22, 1729–1733 (2013).
Li, L., Yan, Y., Ding, W., Gong, J. & Xiao, G. Improvement in the quality of kimchi by fermentation with Leuconostoc mesenteroides ATCC 8293 as starter culture. Microbiol. Biotechnol. Lett. 48, 533–538 (2020).
Lee, K. W. et al. S. Y. Isolation and characterization of kimchi starters Leuconostoc mesenteroides PBio03 and Leuconostoc mesenteroides PBio104 for manufacture of commercial kimchi. J. Microbiol. Biotechnol. 30, 1060–1066 (2020).
Shim, S.-M. et al. Profiling of fermentative metabolites in kimchi: volatile and non-volatile organic acids. J. Korean Soc. Appl. Biol. Chem. 55, 463–469 (2012).
Thukral, A. K. A review on measurement of Alpha diversity in biology. Agric. Res. J. 54, 1–10 (2017).
Chun, B. H., Kim, K. H., Jeon, H. H., Lee, S. H. & Jeon, C. O. Pan-genomic and transcriptomic analyses of Leuconostoc mesenteroides provide insights into its genomic and metabolic features and roles in kimchi fermentation. Sci. Rep. 7, 11504 (2017).
Baker-Austin, C. & Dopson, M. Life in acid: pH homeostasis in acidophiles. Trends Microbiol. 15, 165–171 (2007).
Guan, N. & Liu, L. Microbial response to acid stress: mechanisms and applications. Appl. Microbiol. Biotechnol. 104, 51–65 (2020).
Liberek, K., Marszalek, J., Ang, D., Georgopoulos, C. & Zylicz, M. Escherichia coli DnaJ and GrpE heat shock proteins jointly stimulate ATPase activity of DnaK. Proc. Natl Acad. Sci. USA 88, 2874–2878 (1991).
Maillot, N. J., Honoré, F. A., Byrne, D., Méjean, V. & Genest, O. Cold adaptation in the environmental bacterium Shewanella oneidensis is controlled by a J-domain co-chaperone protein network. Commun. Biol. 2, 323 (2019).
Schröder, H., Langer, T., Hartl, F. U. & Bukau, B. DnaK, DnaJ and GrpE form a cellular chaperone machinery capable of repairing heat-induced protein damage. EMBO J. 12, 4137–4144 (1993).
Sugimoto, S., Saruwatari, K., Higashi, C. & Sonomoto, K. The proper ratio of GrpE to DnaK is important for protein quality control by the DnaK–DnaJ–GrpE chaperone system and for cell division. Microbiology 154, 1876–1885 (2008).
Sung, M.-S., Im, H.-N. & Lee, K.-H. Molecular cloning and chaperone activity of DnaK from cold-adapted bacteria, KOPRI22215. Bull. Korean Chem. Soc. 32, 1925–1930 (2011).
Yoshimune, K., Galkin, A., Kulakova, L., Yoshimura, T. & Esaki, N. Cold-active DnaK of an Antarctic psychrotroph Shewanella sp. Ac10 supporting the growth of dnaK-null mutant of Escherichia coli at cold temperatures. Extremophiles 9, 145–150 (2005).
Imamoglu, R., Balchin, D., Hayer-Hartl, M. & Hartl, F. U. Bacterial Hsp70 resolves misfolded states and accelerates productive folding of a multi-domain protein. Nat. Commun. 11, 365 (2020).
Park, S.-S. et al. ClpL is a chaperone without auxiliary factors. FEBS J. 282, 1349–1568 (2015).
Mun, S. Y. & Chang, H. C. Characterization of Weissella koreensis SK isolated from kimchi fermented at low temperature (around 0 °C) based on complete genome sequence and corresponding phenotype. Microorganisms 8, 1147 (2020).
Jeremy, M. B., John, L. T. & Lubert, S. Biochemistry 5th edn (W. H. Freeman, 2002).
Ellis, R. J. & van der Vies, S. M. Molecular chaperones. Annu. Rev. Biochem. 60, 321–347 (1991).
Sugimoto, S., Abdullah-Al-Mahin & Sonomoto, K. Molecular chaperones in lactic acid bacteria: physiological consequences and biochemical properties. J. Biosci. Bioeng. 106, 324–336 (2008).
Voth, W. & Jakob, U. Stress-activated chaperones: a first line of defense. Trends Biochem. Sci. 42, 899–913 (2017).
Senior, A. E. ATP synthesis by oxidative phosphorylation. Physiol. Rev. 68, 177–231 (1988).
Voet, D., & Voet, J. G. Biochemistry: Biomolecules, Mechanisms of Enzyme Action, and Metabolism Vol. 3 (John Wiley & Sons, 2004).
Matsumoto, M., Ohishi, H. & Benno, Y. H+-ATPase activity in Bifidobacterium with special reference to acid tolerance. Int. J. Food Microbiol. 93, 109–113 (2004).
Varcamonti, M. et al. Expression of the heat shock gene clpL of Streptococcus thermophilus is induced by both heat and cold shock. Microb. Cell Factories 5, 6 (2006).
Wall, T. et al. The early response to acid shock in Lactobacillus reuteri involves the ClpL chaperone and a putative cell wall-altering esterase. Appl. Environ. Microbiol. 73, 3924–3935 (2007).
Sun, Y. in Regulation of Ca2+-ATPases, V-ATPases and F-ATPases (eds Dhalla, N. S. & Chakraborti, S.) Ch. 22 (Springer International Publishing, 2016).
Jeong, S. E. et al. Genomic and metatranscriptomic analyses of Weissella koreensis reveal its metabolic and fermentative features during kimchi fermentation. Food Microbiol. 76, 1–10 (2018).
Chang, J. Y. & Chang, H. C. Improvements in the quality and shelf life of kimchi by fermentation with the induced bacteriocin-producing strain, Leuconostoc citreum GJ7 as a starter. J. Food Sci. 75, M103–M110 (2010).
Fadrosh, D. W. et al. An improved dual-indexing approach for multiplexed 16S rRNA gene sequencing on the Illumina MiSeq platform. Microbiome 2, 6 (2014).
Shin, J. et al. Analysis of the mouse gut microbiome using full-length 16S rRNA amplicon sequencing. Sci. Rep. 6, 29681 (2016).
Bolyen, E. van et al. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat. Biotechnol. 37, 852–857 (2019).
Callahan, B. J. et al. DADA2: high-resolution sample inference from Illumina amplicon data. Nat. Methods 13, 581–583 (2016).
Fabian, P. et al. Scikit-learn: machine learning in Python. J. Mach. Learn. Res. 12, 2825–2830 (2011).
Quast, C. et al. The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res. 41, D590–D596 (2013).
Tamura, K., Stecher, G. & Kumar, S. MEGA11: molecular evolutionary genetics analysis version 11. Mol. Biol. Evol. 38, 3022–3027 (2021).
Jung, M.-J. et al. Viral community predicts the geographical origin of fermented vegetable foods more precisely than bacterial community. Food Microbiol. 76, 319–327 (2018).
Lee, E.-J. et al. Quality assessment of ginseng by 1H NMR metabolite fingerprinting and profiling analysis. J. Agric. Food Chem. 57, 7513–7522 (2009).
Oksanen, J. Multivariate analysis of ecological communities in R: vegan tutorial. R. package version 1, 1–43 (2011).
Parks, D. H., Imelfort, M., Skennerton, C. T., Hugenholtz, P. & Tyson, G. W. CheckM: assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res. 25, 1043–1055 (2015).
Tatusova, T. et al. NCBI prokaryotic genome annotation pipeline. Nucleic Acids Res. 44, 6614–6624 (2016).
Lee, I., Kim, Y. O., Park, S.-C. & Chun, J. OrthoANI: an improved algorithm and software for calculating average nucleotide identity. Int. J. Syst. Evolut. Microbiol. 66, 1100–1103 (2016).
Meier-Kolthoff, J. P., Auch, A. F., Klenk, H.-P. & Göker, M. Genome sequence-based species delimitation with confidence intervals and improved distance functions. BMC Bioinformatics 14, 60 (2013).
Huerta-Cepas, J. et al. eggNOG 5.0: a hierarchical, functionally and phylogenetically annotated orthology resource based on 5090 organisms and 2502 viruses. Nucleic Acids Res. 47, D309–D314 (2019).
Zheng, J. et al. dbCAN3: automated carbohydrate-active enzyme and substrate annotation. Nucleic Acids Res. 51, W115–W121 (2023).
Kanehisa, M., Sato, Y. & Morishima, K. BlastKOALA and GhostKOALA: KEGG tools for functional characterization of genome and metagenome sequences. J. Mol. Biol. 428, 726–731 (2016).
Joshi, N. A. & Fass, J. N. Sickle: a sliding-window, adaptive, quality-based trimming tool for FastQ files (Version 1.33). (2011).
Li, H. & Durbin, R. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 25, 1754–1760 (2009).
Wang, Y., Jiménez, D. J., Zhang, Z. & van Elsas, J. D. Functioning of a tripartite lignocellulolytic microbial consortium cultivated under two shaking conditions: a metatranscriptomic study. Biotechnol. Biofuels Bioprod. 16, 54 (2023).
Shannon, P. et al. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 13, 2498–2504 (2003).
Statstutor. Spearmans’ correlation. http://www.statstutor.ac.uk/resources/uploaded/spearmans.pdf (2020).
Acknowledgements
This research was supported by the World Institute of Kimchi (grant number: KE2501-1-1) and the National Research Foundation of Korea (NRF) (grant number: 2021R1C1C1013859), which was funded by the Ministry of Science and ICT, and the Korea Institute of Planning and Evaluation for Technology in Food, Agriculture, Forestry, and Fisheries (IPET) through the Agricultural Microbiome R&D Programme supported by the Ministry of Agriculture, Food and Rural Affairs (MAFRA) (grant number: RS-2024-00397218), Korea. The funder played no role in the study design, data collection, analysis and interpretation of data or the writing of this manuscript.
Author information
Authors and Affiliations
Contributions
J.L. wrote the original draft, and J.L., M.-J.J., B.H.R., H.-J.C., T.W.W. and S.H.L. reviewed and edited the manuscript. J.L., M.J.L., T.W.W. and S.H.L. conducted formal analysis and investigation. J.L., Y.B.K., S.W.R., T.W.W. and S.H.L. prepared figures and visualisations. C.O.J. and S.H.L. conceptualised and designed the methodology of this study. S.H.L. supervised the project and managed its administration.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Lee, J., Lee, M.J., Jung, MJ. et al. Unravelling the key factors for the dominance of Leuconostoc starters during kimchi fermentation. npj Sci Food 9, 61 (2025). https://doi.org/10.1038/s41538-025-00415-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41538-025-00415-w
