
Abstract
Lineage tracing remains an essential approach towards understanding cell fate, tissue formation, and human development. Herein, we review advancements in lineage tracing techniques, the integration of sequencing and imaging technologies, and computational tools for analysis. We report on recent lineage tracing applications, including integrative techniques (MADM-CloneSeq), in situ hybridization (DART-FISH), and modern workflows (BaSISS), which hold an essential role in our aim to unravel lineage hierarchies and, ultimately, human development.
Similar content being viewed by others

Introduction
Lineage tracing cannot be defined in terms of a single technique or approach, but as any experimental design aimed at establishing hierarchical relationships between cells. Modern flagship studies are rigorous and multimodal, validating hypotheses by a multitude of distinct methods. It is not unusual for such studies to incorporate advanced microscopy, state-of-the-art sequencing technology, and multiple biological models. The resulting datasets continue to increase in size and complexity, necessitating sophisticated and integrative approaches to experimental design and analysis. However, when broken into constituent parts, these studies are rooted in the same fundamental concepts that define all lineage-tracing studies. In brief, this review introduces lineage tracing, examines key historical developments, highlights recent advancements in imaging-based lineage-tracing techniques (new and old), examines up-to-date analytical and modelling-based tools, and provides an outlook on an integrative future of lineage tracing.
Studies applying lineage tracing may investigate cell/tissue morphology, differentiation, clonal expansion, and gene inheritance/expression. The resolution and methodological approach will define the limits of an analysis, in either precision or generalisability. For example, the low specificity of a particular label may prevent discrimination between particular cell types. Alternatively, excessive labelling can lead to close proximity of clonal populations in situ, limiting a study’s ability to perform clonal analysis. Experimental constraints such as these often restrict a study to making inferences about a cell ‘population’, which may not represent a uniform population of cells, but a group of cells sharing a particular trait (a shared phenotype, gene-of-interest, etc.). For example, the transcription factor Sox9 identifies osteochondral stem and progenitor cells, remaining present until after commitment to the chondrocyte lineage1—as such, to discuss ‘Sox9+ cells’ would not refer to a single-cell type, but a spectrum of cell types with a shared marker. This is not to imply that higher specificity is unilaterally superior; population-level analyses assist in generalising observations made in single cells (or cell types) or forming associations between discriminant cell types.
Although rooted in developmental biology, lineage tracing has been implemented to answer biological questions for a wide range of topics. Lineage-tracing data can provide insight into cellular origins, proliferation, and differentiation. Depending on experimental design, lineage tracing can answer questions about regenerative models, cancer development, and disease progression. In regeneration, lineage tracing has established cell origin and fate in models spanning embryonic development to adult tissues. Tracing cancer cell lineages has determined mutations critical to cancer progression and lineage-specificity for therapeutics. In disease models, these same techniques are used to measure changes in cell potential and stem cell exhaustion.
Over the past twenty years, non-imaging tools (e.g., scRNAseq) have become a vital tool in formulating and validating lineage-tracing hypotheses2,3. As sequencing technologies improve in their power, accessibility, and cost, what were once expensive and exceptional approaches are increasingly becoming a requisite tool in modern lineage tracing work. Despite this, traditional imaging-based approaches remain central to lineage-tracing studies. When investigating less-studied cell lineages (e.g., skeletal or epithelial stem cells, cutting-edge technologies are frequently not applicable, either due to unavailability or an insufficient understanding of the underlying lineage hierarchy. Therefore, it is important as researchers to have a comprehensive understanding of not only the technological cutting-edge, but the foundational techniques on which our field is built and are largely still applicable and often necessary. Since its conception, lineage tracing has remained of central importance in biology, and imaging has persisted as the fundamental tool in performing lineage tracing both qualitatively and quantitatively. Herein, we highlight recent advancements and implementations of imaging-based lineage-tracing techniques (new and old), provide context in contrast to previous iterations, and offer readers a concise summary of how these techniques have and/or may be applied.
A brief historical perspective
Lineage tracing has remained of central importance in biology since the late 1800s, when Charles Whitman reported the direct observation of germ layer differentiation in leeches4. In 1887, direct observation was the sole method for interpreting cell lineage, and data collection was ultimately dependent on the visual observations of an experimenter in real time. As a result, experimental models would have been limited to those that afforded observable changes via light microscopy (Box 1)4,5. Cell arrangement, differentiation events, and counts would have been visually monitored and recorded manually.
Labelling allows experimenters to distinguish and monitor cells by means other than continuous observation, and has greatly increased in sophistication since its conception. Non-specific labels were originally applied to lineage tracing by Eric Vogt, who fate mapped an amphibian blastula in 1929 using Nile Blue6. A more modern implementation of non-specific labelling is nucleoside analogues (BrdU, EdU, etc.), in which a modified nucleoside is incorporated into cellular DNA and is subsequently labelled with fluorescent dye7,8. Nucleoside analogues are applied to identify proliferating cell populations, which will incorporate the dye more quickly than slow-cycling populations and are still frequently used today, with the natural disadvantage of label dilution proportional to cell proliferation.
The late 20th century was marked by the exponential development of gene editing technologies, which were particularly useful in refining imaging methodologies for lineage analysis. By manipulating the expression of reporter genes, labelling technologies circumvented many of the limitations posed by earlier approaches. Starting in the 1980s, the first transgenic approaches involving enzymatic reporters were produced9, a key example of which is the E. coli-derived β-galactosidase (which can convert substrate X-gal into a dark blue precipitate)10. At this time, the expanding literature on transgenic modalities allowed for gene incorporation either by direct injection, viral transduction, or plasmid transfection by 199011,12. The Cre-loxP recombinase system was also introduced during this period, being implemented in mammalian cells in 198813, and in mice in vivo in 199414. The year 1994 also saw the introduction of green fluorescent protein (GFP) as an endogenous reporter, which marked a massive shift for lineage tracing, giving cells the potential to express reporters without the need for an external stimulus15,16. Taken together, these tools laid the technological foundation for lineage tracing in the modern era (Fig. 1).

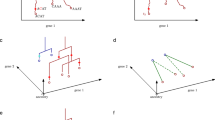
Key technological and methodological advances have driven large shifts in progress since the 1800s (a). Results returned from a PubMed search of the term “lineage tracing” (b). Timeline highlighting key discoveries that have laid the foundation for modern lineage tracing (c). Created using BioRender.
Image-based lineage-tracing techniques
Site-specific recombinases
Central to imaging-based lineage-tracing research are site-specific recombinase (SSR) systems, of which Cre-loxP remains one of the most fundamental and commonly used. The widespread availability, versatility, and ease of use all uphold the Cre-loxP system as the gold standard in lineage-tracing studies. These systems can be used to knock-in/knock-out alleles and influence gene expression with a great degree of cell and temporal specificity. In the context of lineage tracing, Cre-loxP systems are commonly applied in clonal analysis studies, during which Cre recombinase often excises a STOP codon between two adjacent loxP binding sites, activating a fluorescent reporter gene. The specificity of this activation is dependent on Cre, whose expression can be driven by cell-type-specific promoters or ubiquitously expressed. These approaches traditionally consist of a single fluorescent reporter tied to a single gene of interest, the concepts of which have been reviewed at length in the literature17. However, the Cre-loxP system continues to be applied to develop advanced lineage-tracing techniques, including dual SSR systems, multicolour models, and more advanced methods.
Analyses using single fluorescent reporters can provide population data but are largely limited in their ability to resolve cell populations at the single-cell level. This largely stems from the difficulty (and often impossibility) of distinguishing clonal groups within a homogenously labelled population. Despite this, sparse labelling approaches can be used to circumvent this. For example, in sparse labelling, the activating agent in inducible models (e.g., Tamoxifen in CreERT2 models) can be titrated such that recombination is limited to a limited number of cells within the population (thus allowing for spatial separation)18. This has the added effect of increased specificity, as cells with greater promoter expression will be preferentially labelled19. Unfortunately, the natural limitation to sparse labelling approaches is the natural increase in sampling required (meaning increased biological replicates, analyses, and lower reproducibility).
Dual recombinase systems
The Cre-loxP system can also be implemented in combination with analogous technologies to create dual recombinase systems. Dre-rox is a common heterospecific and efficient alternative to Cre-loxP in which Dre recombinase is specific for rox sites20,21. Dual recombinase systems take advantage of the site specificity of recombinases and offer multiple experimental design strategies beneficial to lineage tracing (i.e., expression occurs following recombination of (i) either Cre or Dre, (ii) both Cre and Dre, (iii) Cre in the absence of Dre)22. Several dual systems have been implemented, Cre-loxP/Dre-rox being one of the most common, and possess numerous applications in lineage tracing. For example, a Cre/Dre dual system was recently used to determine the origin of regenerative cells in remodelled bone, distinguishing otherwise homogenous periosteal tissue into distinct layers and evaluating layer contributions in fracture regeneration23. Cre/Dre was also applied to investigate the cellular origins of alveolar epithelial stem cells post-injury and distinguish contributions made by multiple epithelial cell populations simultaneously24. Dual recombinases were also applied to develop novel genetic techniques for evaluating senescence during liver fibrosis, offering an approach to discriminating between senescent cell populations expressing analogous senescent markers25.
Multicolour lineage-tracing approaches
A major advance in imaging-based lineage tracing was the introduction of multicolour reporter cassettes. The first of these was ‘Brainbow’, capable of expressing up to four different fluorescent proteins and driven by stochastic Cre-loxP-mediated excision and/or inversion26. In this design, multiple pairs of loxP sites are arranged within the cassette, facilitating mutually exclusive recombination events. Recombination will ultimately result in a reordering/removal of fluorescent protein sequences and—given only the first fluorophore in sequence will be transcribed—expression of a different fluorescent protein. This technology has since been adapted to several experimental models and inspired numerous analogous technologies27,28. One of the most popular adaptations is the R26R-Confetti reporter, given its widespread applicability to existing Cre models. Lineage-tracing studies now incorporate confetti reporters as a means to perform clonal analysis at the single-cell level in a broad range of tissues, including hematopoetic29, epithelial30, kidney31, and skeletal32 cells. Multicolour models are also being applied in live-imaging studies, for example, confetti reporters have recently been used in intravital imaging to trace macrophage origin and proliferation in mammary glands in real time33. The protocol used in this study was subsequently published by the authors and carries promising applications in future organogenesis studies34.
Mosaic analysis with a repressible cell marker (MARCM) is a tracing technique that identifies lineage tree branches based on mitotic recombination. Originally developed in Drosophila, MARCM uses the FLP-FRT system (analogous to Cre-loxP) to induce recombination between homologous chromosomes during mitosis, ultimately resulting in a single daughter cell inheriting a reporter35. Pre-recombination, a ubiquitously expressed transcriptional repressor prevents reporter expression; following mitotic recombination, the repressor and reporter genes are partitioned into corresponding daughter cells by chromosomal segregation. This technology was improved to incorporate two repressor/reporter pairs (twin-spot MARCM) and analogously developed for the Cre-loxP system as mosaic analysis with double markers (MADM)36,37. Both systems offer unparalleled specificity in fate mapping by labelling branches of a lineage tree in a mitosis-dependent manner. Recombination events exclusively occur during mitosis, and daughter cells are asymmetrically labelled, meaning lineage branches are labelled at their origin point and sibling cells can be independently traced. A major limitation of mitotic recombination systems was their availability for each chromosome, but as of 2021, a genome-wide library for MADM is available, with knock-in cassettes for all 19 autosomes38. MADM has now facilitated highly specific lineage tracing of neural stem and progenitor cells during cortical development39,40 and seed populations responsible for breast and ovarian cancer (these and other applications are summarised in Table 1)41,42. MADM-based clonal analysis has recently been paired with single-cell sequencing techniques (MADM-CloneSeq) to perform lineage tracing of the murine midbrain at the single-cell level43. In this multimodal technique, imaging is used to identify clones (via MADM), after which RNA is isolated from individual cells-of-interest for sequencing. Techniques such as MADM-CloneSeq bridge the gap between imaging and sequencing technologies—an approach built on foundational technologies that will likely prove versatile in a broad range of models. In parallel to Cre and other SSR-based approaches, advances in the field of image-based spatial transcriptomics have expanded our ability to spatiotemporally visualise transcriptional targets in situ.
Spatial transcriptomics and its applications to lineage tracing
In situ hybridisation
In situ hybridisation (ISH) encompasses a broad range of techniques based on detecting nucleic acid (NA) sequences (DNA or RNA) of interest through complementary binding of labelled single-stranded sequences. Once bound, probes may then be detected in situ during imaging. Originally developed in the late 1960s, early approaches to ISH involved autoradiography of tritium (3H)-incorporated RNA sequences44. The advent of fluorescent probes together with technological advancements in microscopy led to the development of fluorescent in situ hybridisation (FISH). The early 1980s saw two major advancements to ISH, in which (1) detection of biotin-labelled probes by immunofluorescence45 and (2) direct fluorescent labelling of RNA probes46 were used to detect NA sequences. The decades that followed saw the development of single-molecule FISH (smFISH), enabling the detection of individual copies of mRNA47,48. smFISH was then improved on once again to develop sequential FISH (seqFISH), a multiplexing approach capable of detecting hundreds of genes (at a single mRNA level) via barcoding over sequential rounds of hybridisation and imaging49,50,51. These developments culminated in transforming a once cumbersome technique into a broad spectrum of methodologies with applications spanning basic science to clinical diagnostics.
In 2017, seqFISH was applied in combination with CRISPR/Cas9-based mutagenesis to develop a method named Memory by Engineered Mutagenesis with Optical in Situ Readout (MEMOIR)52. This groundbreaking technique enables in situ recording of lineage information via permanent modifications to a set of cellular barcoded ‘scratchpad’ recording elements. Scratchpads are irreversibly collapsed by Cas9/gRNA as cells proliferate in a stochastic order, enabling cells to record their own lineage information. Scratchpads are then analysed in situ via seqFISH to determine the now-modified scratchpad state for each cell, in parallel with spatial and gene expression data, providing a multimodal illustration of single-cell behaviour. For interested readers, the concepts of cellular recording on which MEMOIR is based have recently been reviewed in detail53.
MEMOIR has since undergone several methodological improvements to improve memory capacity and accuracy. An integrase-editable version (intMEMOIR) has been developed, which improves on the scratchpad, introducing a three-state memory element with an initial and two end barcode states (in contrast to the scratchpad’s single end state), which greatly increases storage and the accuracy of lineage-tracing analyses spanning multiple generations54. In addition, the developers of MEMOIR have published an image-based barcode readout approach, termed ‘Zombie’55. In Zombie, DNA barcodes are transcribed post-fixation by phage RNA polymerases, amplifying the barcode sequence into an RNA cluster for subsequent analysis at single-nucleotide resolution. This allows in situ readout of CRISPR base edits post-fixation and circumvents any need for continuous barcode expression in live cells. Moreover, this approach allows the readout of dense barcode libraries and a much greater memory capacity than MEMOIR. Ultimately, these developments have culminated in the development of baseMEMOIR, which incorporates three-state memory, base-editing techniques, and Bayesian inference methods into the existing MEMOIR protocol56: In brief, a series of editable dinucleotide arrays were incorporated upstream of barcode labels, which undergo three-outcome stochastic mutagenesis at a rate proportional to cell division. These arrays provide each cell with a 792-bit phylogenetic memory (396 editable dinucleotides), which can then be amplified and read using an updated Zombie-FISH approach. Lastly, lineage trees are then constructed using a Bayesian phylogenetic reconstruction framework. These improvements have allowed for accurate lineage tracing of mouse embryonic stem cells over six generations. A remaining limitation of baseMEMOIR is that it cannot account for transient cell states, cell death, or other events that cannot be recorded at the experimental endpoint.
Fluorescence in situ hybridisation of cellular heterogeneity and gene expression programs, or FISHnCHIPs, is a recently developed method that boasts improved sensitivity of cell typing and gene expression over traditional FISH methods57. FISHnCHIPs uses scRNAseq-based gene expression profiles to design probes capable of targeting a set of co-localised genes, increasing fluorescence by up to 20 times in a given cell type. Leveraging co-expression patterns improves the efficiency of cell typing, enabling a user to image larger tissue sections at reduced magnification—a powerful tool when performing lineage tracing on elusive and rare cell populations. Moreover, applying co-localisation patterns to cell typing improves one’s accuracy in distinguishing closely related expression profiles, which can allow researchers to identify lineage hierarchies within previously uniform cell populations. Validated in mouse tissue(s) and human cancer tissue, this 2024 method shows broad applicability for future spatial omics and lineage-tracing studies.
Another modern approach to ISH, published in 2024, is decoding amplified targeted transcripts with fluorescence in situ hybridisation, or DART-FISH58. DART-FISH incorporates novel embedding and cytoplasmic staining protocols into a padlock probe-based ISH approach, which offers increased throughput and computational image deconvolution at the single-pixel level. The result is a high-throughput RNA mapping technique capable of multiplexing hundreds of genes in situ, including small molecules and human tissues. In the approach, target RNA is used to generate cDNA via reverse transcriptase, which is subsequently hybridised to padlock probes to undergo rolling circle amplification (RCA). RCA products are then efficiently decoded using an isothermal and enzyme-free decoding procedure, and an open-source computational method (SparseDeconvolution, SpD) performs image deconvolution at the single-pixel level. Notably, this method implements a polyacrylamide embedding protocol, which enhances feature retention (and therefore sensitivity) by 1.5-fold. This is facilitated by a 5’ acrydite addition to the padlock probe primers, enabling gel incorporation prior to RCA. This 5’ sequence is also co-opted for use in cytoplasmic labelling prior to barcode decoding (coined Ribosoma), in which fluorescent probes complementary to the universal 5’ sequence are used to visualise probe spatial distribution and are effective in visualising cell body morphology. As a proof-of-concept, DART-FISH was used to image and decode 121 genes in a 0.3 cm2 tissue section of human neocortex in less than 10 h. In summary, DART-FISH is a rapid, cost-effective approach to cell typing and morphology that introduces novel embedding and staining protocols without the need for high-magnification imaging (>20x) or specialised equipment. By employing cost and time-effective protocols using standard microscopy equipment, techniques such as DART-FISH simplify the implementation of modern ISH technologies and improve accessibility to next-gen lineage tracing.
In situ sequencing
In situ sequencing (ISS) was first developed in 201359, representing a major development for the spatial transcriptomics field and demonstrating great potential for lineage-tracing studies as an alternative methodology to ISH approaches in hypothesis-driven analyses. In contrast to ISH, ISS sequences individual transcripts directly in situ to preserve spatial information at a subcellular resolution. To accomplish this, ISS employs padlock probe, RCA, and (originally) sequencing-by-ligation technologies to detect and amplify endogenous RNA sequences. The capacity of ISS as a spatial gene expression technology was originally validated using human breast cancer tissue, in which ISS was successful in spatially quantifying the multiplexed expression for 31 transcripts with an average of 25 reads per cell using typical epifluorescent microscopy59. ISS techniques have since been applied in neural tissue in mice60,61 and Alzheimer’s disease in humans62 and cancer59,63.
Since its development, ISS has undergone several iterative improvements. In 2020, hybridisation-based in situ sequencing (HybISS) was developed64. A major limitation of ISS was the efficiency of sequencing-by-ligation, constrained by its design (a multi-step, enzyme-driven pathway). HybISS implemented sequencing-by-hybridisation afforded by a new probe design, which allows for non-competitive direct hybridisation and resulted in increased labelling intensity and signal-to-noise ratio. In 2022, ISS saw further improvement in methodology wherein direct RNA detection was achieved (dRNA-HybISS), avoiding the need for reverse transcription from cDNA65. Removal of cDNA synthesis and having padlock probes hybridise directly to mRNA improved transcript detection efficiency by more than fivefold. This improved transcript detection allows a user to investigate challenging genes-of-interest (i.e., low detection rate, limited expression) over larger areas (magnifications as low as 10X). ISS methodologies remain limited to a select number of tissues and experimental conditions (having only been around for a little more than a decade), however, they continue to improve in efficiency and applicability with each iteration.
ISS has also been implemented in multimodal lineage-tracing strategies, such as probabilistic cell typing by in situ sequencing (pciSeq)60. PciSeq is a technique in which scRNAseq data is used to identify a list of ~100 genes capable of distinguishing cell type by ISS. Once established, this list is used to generate a padlock probe library and implement ISS. Cell types are then determined via a probabilistic model based on the ISS expression level and scRNAseq profiling, providing insight into spatial organisation. Importantly, this synergistic method provides an opportunity to directly link imaging-based data (morphology, anatomical location, niche) to a given cell population’s complete transcriptomic profile, not otherwise available through imaging alone.
Another multimodal lineage-tracing strategy applying ISS is the base-specific in situ sequencing (BaSISS) workflow, which best exemplifies the potential ISS holds for imaging-based lineage tracing63. Herein, ISS technology is applied as a part of a multimodal strategy to identify sub-clonal lineages and their individual growth patterns in whole-tumour sections. In the BaSISS workflow, whole genome sequencing (WGS) is first performed to determine subclone populations and lineage hierarchy. Branch-specific mutation markers (capable of distinguishing individual phylogenetic branches) are then determined. BaSISS then applies ISS techniques to develop padlock probes capable of differentiating clonal populations. Once sequencing is complete, each cell’s ISS signals and corresponding genotypes (from WGS) are used to derive quantitative clonal maps using Bayesian modelling. These results can then be integrated with additional layers of spatial data (using additional tissue sections). By identifying mutations of interest prior to imaging, clones may be analysed across large tissue sections (>cm2) at relatively low cost. Given there is sufficient tissue for WGS, ISS, and any additional spatial sequencing, this iterative technique offers a comprehensive and affordable (per region of interest) approach to lineage tracing.
Computational approaches to data analysis and interpretation
As lineage-tracing datasets increase in size and complexity, so too must our approaches to analysis and interpretation. Even today, some imaging analyses are performed by traditional means (e.g., manual cell counting), but as technological advancements increase image volume, resolution, and multiplexing, imaging datasets are often pushed into the 100 + GB range. Thus, bulk processing methods, machine learning algorithms, and artificial intelligence become more commonplace (and often essential).
Image segmentation
Image segmentation is central to many image-based lineage-tracing workflows. Segmentation algorithms are commonly available as a feature in microscopy software, allowing users to process regions of interest that would otherwise be too large to quantify by traditional means. These features are beneficial in that they are widely available, easy to use, and help reduce experimenter bias. However, the incorporation of ultra-high resolution and multiplexed images into lineage-tracing workflows has created a demand for more sophisticated segmentation methods. These advanced methods must be able to handle complex, heterogeneous data that simpler algorithms, such as those based on thresholding, watershed, or simple feature-based classifiers, cannot.
StarDist, developed in 2018, is a deep-learning algorithm tailored for segmenting star-convex shapes such as cell nuclei66. It performs well in densely packed, overlapping samples and is built around a trainable CNN, with pre-trained models available for common tasks (e.g., H&E or DAPI-stained images). StarDist is open-source, Python-based, and integrates easily with platforms like QuPath, ImageJ/Fiji, and Napari. Its compatibility with Python libraries (e.g., Scikit-image, OpenCV, Dask, TensorFlow) allows for flexible and scalable workflows. However, StarDist focuses on nuclear segmentation, leaving cytoplasmic boundary determination for post-processing steps, such as nuclear expansion, which can introduce error. For more comprehensive segmentation in multiplexed imaging datasets, Mesmer employs Feature Pyramid Networks to determine both nuclei and whole-cell boundaries67, while Cellpose (2021) offers broader flexibility by adapting a modified U-Net architecture, which is capable of handling a wide variety of cell shapes and sizes without requiring star-convex assumptions68. To detect subcellular features such as RNA or protein puncta—common in lineage-tracing studies—PunctaFinder (2024) provides an efficient, Python-based solution that relies on traditional image-processing techniques, making it particularly effective in high-SNR, low-resolution regions68. PunctaFinder has been successfully applied, for example, to automate the quantification of Atg9-positive vesicles, whereas previous methods relied on manual counting of these structures.
Often, image size can exceed the reasonable capacity of standard software (longer processing times, computer crashes, etc.) and research groups are driven to develop their own software. Analysis of 3-D multiplexed confocal or light sheet imaging datasets in the 100 + GB range has led to unreachable hardware requirements during post-segmentation analysis (both in duration and rendering capacity). This spurred the development of XiT, an open-access software developed in 2018, which exports only the statistical parameters necessary for analysis, reducing workspace size by three orders of magnitude (100 GB to 100 MB)69. Developments such as these allow what otherwise would require state-of-the-art computing power to be accomplished on any modern laptop. Sharing open-access software within the research community provides opportunities for collaboration and knowledge dissemination while guiding development companies as they design future software releases.
Live-cell tracking
Another key step in various image-based lineage-tracing approaches is cell tracking, particularly in live-imaging experiments where cells are followed dynamically through division events. Unlike lineage-tracing methods that rely on fluorescent markers inherited by daughter cells to infer lineage relationships in later snapshots, live-imaging approaches use computational tracking to follow individual cells and their progeny over time70. For example, time-lapse microscopy and 3D imaging enable detailed tracking of cellular trajectories, however, the accuracy of tracking algorithms is often limited by the quality of segmentation. This prompted the development of TrackMate 7 in 2022, distributed as a Fiji plugin, which facilitates the integration of advanced segmentation algorithms, such as StarDist and Cellpose, into its built-in tracking pipelines71. TrackMate 7 has recently been used to automate the tracking of osteoblasts, replacing manual annotation, to study the dynamic cellular response following trypsin exposure72.
Mathematical modelling
Modern technologies often integrate mathematical modelling and statistical analysis to interpret complex biological data, such as in DART-FISH, where tools analyse multiplexed imaging data to identify mRNA transcripts within tissue contexts58. SparseDeconvolution (SpD) aids in resolving signal overlap in densely labelled samples73, and statistical methods ensure accurate decoding by normalising signal intensities and calculating confidence intervals. The processed data provides a spatial and quantitative representation of gene expression patterns, essential for understanding cellular hierarchies and lineage pathways.
Despite advances in segmentation and tracking, lineage tree reconstruction remains challenging due to incomplete data and limited temporal resolution. Mathematical models, such as MOLLUSC (2024), use spatial lineage tracing to estimate branch lengths, spatial diffusion, and mutation rates through maximum likelihood estimation (MLE)74. Similarly, LineageOT (2021) integrates lineage snapshots with scRNA-seq data to infer developmental trajectories by mapping both to a common space75. In addition to reconstructing lineage trees, recent methods aim to interpret patterns within them to gain biological insight. For instance, Lineage Motif Analysis (LMA) identifies statistically overrepresented cell fate patterns on lineage trees, revealing potential signatures of committed progenitor states or extrinsic interactions76. Applying LMA to datasets from zebrafish, rat retina, and mouse embryonic development revealed the spatial and temporal organisation in cell fate commitment and offered insights into the evolutionary variation of retinal cell type proportions.
Probabilistic approaches are particularly well-suited for handling the uncertainties inherent in biological systems, where experimental noise and variability in cell behaviour complicate inference. In the baseMEMOIR technique, targeted base edits accumulate in genomic sequences over time, and their random accumulation can be modelled as a stochastic process56. This randomness reflects both the intrinsic properties of CRISPR-based editing tools and the dynamic state of cellular metabolism and division, which influence editing efficiency. baseMEMOIR employs a Bayesian inference framework to estimate the most likely lineage trees from observed barcode distributions. This Bayesian approach integrates prior biological knowledge—such as expected mutation rates or cell-cycle characteristics—with the observed edits, iteratively updating lineage tree hypotheses by computing the likelihood of the observed patterns under each scenario.
Mathematical modelling not only enhances the efficiency of lineage tracing but also facilitates the integration of various hypotheses on tissue architecture, which can subsequently be validated or refuted by lineage-tracing experiments. For example, in 2021 a mathematical model was built to explore the developmental origins of the hepato-pancreato-biliary organ system77. By incorporating different hypotheses on the tissue architecture, these models predict possible outcomes, which are then directly compared with experimental lineage-tracing data to infer the most likely scenarios. This approach highlights the power of mathematical modelling to integrate diverse experimental observations into a common framework, enabling insights that may be inaccessible through purely experimental methods. Each of these tools serves a distinct purpose and addresses specific challenges in lineage tracing and tissue architecture, and it is essential to carefully select the appropriate method for each experiment. Collaboration between experimentalists and computational experts is often crucial to ensure the correct application and analysis of these techniques.
Next-generation lineage tracing begins with collaboration
Starting as a single cell, developing organisms undergo rounds of mitotic division and differentiation to achieve a diverse mosaic of tissue types and cell populations. In an adult human, development results in an estimated 30 trillion cells and over 200 cell types, all of which are genealogically connected to that first zygote through cell lineage78. Mapping these relationships from fertilised egg to mature organism—just as it was done for C. elegans in the 1983—is foundational to our understanding of development. As technologies improve at an ever-accelerating pace, each year brings new methods and techniques to improve the resolution at which we understand cell lineage. We have now seen the integration of next-generation sequencing technologies with traditional imaging-based lineage tracing (e.g., CloneSeq). Advancements in ISH and ISS continue to drive novel lineage-tracing approaches (e.g., DART-FISH, FISHnCHIPS,) and the underlying techniques are being integrated into increasingly sophisticated workflows (e.g., BaSISS). The increasing size and complexity of these applications, in turn, drives the development of high-throughput computational tools to process data (e.g., XiT, SpD), and predict relationships (e.g., MOLLUSC). These improvements in the breadth and specificity of lineage-tracing techniques forecast an optimistic future for developmental biology and the virtual human development (VHD) project (Fig. 2).

New VHD-related discoveries drive hypothesis formulation and inspire new lineage-tracing studies. The results of these studies are analysed by new or existing methods, and the resulting data is incorporated to improve and design mathematical models. In turn these results are added to the existing VHD umbrella, validating/refuting existing hypotheses and driving ongoing hypothesis formulation. Created using BioRender.
Collaboration has always been a pillar of high-quality research. As experimental methods become increasingly multimodal—even within a single experiment—interdisciplinary collaboration will be key not simply between but within research teams. Complex computational models are difficult to integrate without a strong mathematical background; likewise, developing suitable models is challenging without a deep understanding of the underlying biological systems they are based on. Collaboration must remain paramount in pursuit of the VHD project, as we seek to bridge the gap across tissue types, between fields of research, and merge findings spanning individual genes to population-level epidemiology.
Data availability
No datasets were generated or analysed during the current study.
References
Lefebvre, V., Angelozzi, M. & Haseeb, A. SOX9 in cartilage development and disease. Curr. Opin. Cell Biol. 61, 39–47 (2019).
Chen, C., Liao, Y. & Peng, G. Connecting past and present: single-cell lineage tracing. Protein Cell 13, 790–807 (2022).
Wagner, D. E. & Klein, A. M. Lineage tracing meets single-cell omics: opportunities and challenges. Nat. Rev. Genet. 21, 410–427 (2020).
Whitman, C. O. A contribution to the history of the germ-layers in Clepsine. J. Morphol. 1, 105–182 (1887).
Conklin, E. G. The embryology of crepidula, a contribution to the cell lineage and early development of some marine gasteropods. J. Morphol. 13, 1–226 (1897).
Vogt, W. Gestaltungsanalyse am Amphibienkeim mit Örtlicher Vitalfärbung. Wilhelm. Roux Arch. Entwickl. Mech. Org. 120, 384–706 (1929).
Miller, M. W. & Nowakowski, R. S. Use of bromodeoxyuridine-immunohistochemistry to examine the proliferation, migration and time of origin of cells in the central nervous system. Brain Res. 457, 44–52 (1988).
Nowakowski, R. S., Lewin, S. B. & Miller, M. W. Bromodeoxyuridine immunohistochemical determination of the lengths of the cell cycle and the DNA-synthetic phase for an anatomically defined population. J. Neurocytol. 18, 311–318 (1989).
Shuman, H. A., Silhavy, T. J. & Beckwith, J. R. Labeling of proteins with beta-galactosidase by gene fusion. Identification of a cytoplasmic membrane component of the Escherichia coli maltose transport system. J. Biol. Chem. 255, 168–174 (1980).
Pardy, K. in Methods in Molecular Biology 18 (eds. Murphy, D. & Carter, D. A.) 419–424 (Humana Press, Totowa, NJ, 1993).
Stuhlmann, H., Cone, R., Mulligan, R. C. & Jaenisch, R. Introduction of a selectable gene into different animal tissue by a retrovirus recombinant vector. Proc. Natl. Acad. Sci. USA 81, 7151–7155 (1984).
Holt, C. E., Garlick, N. & Cornel, E. Lipofection of cDNAs in the embryonic vertebrate central nervous system. Neuron 4, 203–214 (1990).
Sauer, B. & Henderson, N. Site-specific DNA recombination in mammalian cells by the Cre recombinase of bacteriophage P1. Proc. Natl. Acad. Sci. 85, 5166–5170 (1988).
Gu, H., Marth, J. D., Orban, P. C., Mossmann, H. & Rajewsky, K. Deletion of a DNA polymerase β gene segment in T cells using cell type-specific gene targeting. Science 265, 103–106 (1994).
Chalfie, M., Tu, Y., Euskirchen, G., Ward, W. W. & Prasher, D. C. Green fluorescent protein as a marker for gene expression. Science 263, 802–805 (1994).
Rodriguez, E. A. et al. The growing and glowing toolbox of fluorescent and photoactive proteins. Trends Biochem Sci. 42, 111–129 (2017).
Kim, H., Kim, M., Im, S.-K. & Fang, S. Mouse Cre-LoxP system: general principles to determine tissue-specific roles of target genes. Lab. Anim. Res. 34, 147 (2018).
Tan, S. H. & Nusse, R. In vivo lineage tracing reveals Axin2-expressing, long-lived cortical thymic epithelial progenitors in the postnatal thymus. PLoS ONE 12, e0184582 (2017).
Hsu, Y.-C., Pasolli, H. A. & Fuchs, E. Dynamics between Stem Cells, Niche, and Progeny in the Hair Follicle. Cell 144, 92–105 (2011).
Sauer, B. DNA recombination with a heterospecific Cre homolog identified from comparison of the pac-c1 regions of P1-related phages. Nucleic Acids Res. 32, 6086–6095 (2004).
Anastassiadis, K. et al. Dre recombinase, like Cre, is a highly efficient site-specific recombinase in E. coli, mammalian cells and mice. Dis. Model Mech. 2, 508–515 (2009).
Li, H., Weng, W. & Zhou, B. Perfect duet: dual recombinases improve genetic resolution. Cell Prolif. 56, e13446 (2023).
Liu, Y. L., Tang, X. T., Shu, H. S., Zou, W. & Zhou, B. O. Fibrous periosteum repairs bone fracture and maintains the healed bone throughout mouse adulthood. Dev. Cell 59, 1192–1209.e6 (2024).
Liu, K. et al. Tracing the origin of alveolar stem cells in lung repair and regeneration. Cell 187, 2428–2445.e20 (2024).
Zhao, H. et al. Identifying specific functional roles for senescence across cell types. Cell 187, 7314–7334.e21 (2024).
Livet, J. et al. Transgenic strategies for combinatorial expression of fluorescent proteins in the nervous system. Nature 450, 56–62 (2007).
Weissman, T. A. & Pan, Y. A. Brainbow: new resources and emerging biological applications for multicolor genetic labeling and analysis. Genetics 199, 293–306 (2015).
Dumas, L., Clavreul, S., Michon, F. & Loulier, K. Multicolor strategies for investigating clonal expansion and tissue plasticity. Cell. Mol. Life Sci. 79, 141 (2022).
Nicolai, L. et al. Single platelet and megakaryocyte morpho-dynamics uncovered by multicolor reporter mouse strains in vitro and in vivo. Haematologica 107, 1669–1680 (2021).
Ohmoto, M. et al. Maintenance and turnover of Sox2+ adult stem cells in the gustatory epithelium. PLoS ONE 17, e0267683 (2022).
Yin, J. et al. Fate mapping reveals compartment-specific clonal expansion of mononuclear phagocytes during kidney disease. Kidney Int. 104, 605–610 (2023).
Newton, P. T. et al. A radical switch in clonality reveals a stem cell niche in the epiphyseal growth plate. Nature 567, 234–238 (2019).
Dawson, C. A. et al. Tissue-resident ductal macrophages survey the mammary epithelium and facilitate tissue remodelling. Nat. Cell Biol. 22, 546–558 (2020).
Dawson, C. A., Mueller, S. N., Lindeman, G. J., Rios, A. C. & Visvader, J. E. Intravital microscopy of dynamic single-cell behavior in mouse mammary tissue. Nat. Protoc. 16, 1907–1935 (2021).
Lee, T. & Luo, L. Mosaic analysis with a repressible cell marker for studies of gene function in neuronal morphogenesis. Neuron 22, 451–461 (1999).
Yu, H.-H., Chen, C.-H., Shi, L., Huang, Y. & Lee, T. Twin-spot MARCM to reveal the developmental origin and identity of neurons. Nat. Neurosci. 12, 947–953 (2009).
Zong, H., Espinosa, J. S., Su, H. H., Muzumdar, M. D. & Luo, L. Mosaic analysis with double markers in mice. Cell 121, 479–492 (2005).
Contreras, X. et al. A genome-wide library of MADM mice for single-cell genetic mosaic analysis. Cell Rep. 35, 109274 (2021).
Laukoter, S. et al. Imprinted Cdkn1c genomic locus cell-autonomously promotes cell survival in cerebral cortex development. Nat. Commun. 11, 195 (2020).
Amberg, N., Pauler, F. M., Streicher, C. & Hippenmeyer, S. Tissue-wide genetic and cellular landscape shapes the execution of sequential PRC2 functions in neural stem cell lineage progression. Sci. Adv. 8, eabq1263 (2022).
Zeng, J. et al. Dichotomous ovarian cancer-initiating potential of Pax8+ cells revealed by a mouse genetic mosaic model. iScience 26, 106742 (2023).
Zeng, J. et al. A genetic mosaic mouse model illuminates the pre-malignant progression of basal-like breast cancer. Dis. Model Mech. 16, dmm05019 (2023).
Cheung, G. et al. Multipotent progenitors instruct ontogeny of the superior colliculus. Neuron 112, 230–246.e11 (2024).
Gall, J. G. & Pardue, M. L. Formation and detection of RNA-DNA hybrid molecules in cytological preparations. Proc. Natl. Acad. Sci. 63, 378–383 (1969).
Langer-Safer, P. R., Levine, M. & Ward, D. C. Immunological method for mapping genes on Drosophila polytene chromosomes. Proc. Natl. Acad. Sci. 79, 4381–4385 (1982).
Bauman, J. G. J., Wiegant, J., Borst, P. & van Duijn, P. A new method for fluorescence microscopical localization of specific DNA sequences by in situ hybridization of fluorochrome-labelled RNA. Exp. Cell Res. 128, 485–490 (1980).
Femino, A. M., Fay, F. S., Fogarty, K. & Singer, R. H. Visualization of single RNA transcripts in situ. Science (1979) 280, 585–590 (1998).
Raj, A., van den Bogaard, P., Rifkin, S. A., van Oudenaarden, A. & Tyagi, S. Imaging individual mRNA molecules using multiple singly labeled probes. Nat. Methods 5, 877–879 (2008).
Shah, S., Lubeck, E., Zhou, W. & Cai, L. In situ transcription profiling of single cells reveals spatial organization of cells in the mouse hippocampus. Neuron 92, 342–357 (2016).
Lubeck, E., Coskun, A. F., Zhiyentayev, T., Ahmad, M. & Cai, L. Single-cell in situ RNA profiling by sequential hybridization. Nat. Methods 11, 360–361 (2014).
Lignell, A., Kerosuo, L., Streichan, S. J., Cai, L. & Bronner, M. E. Identification of a neural crest stem cell niche by spatial genomic analysis. Nat. Commun. 8, 1830 (2017).
Frieda, K. L. et al. Synthetic recording and in situ readout of lineage information in single cells. Nature 541, 107–111 (2017).
Askary, A. et al. The lives of cells, recorded. Nat. Rev. Genet. 26, 203–222 (2025).
Chow, K.-H. K. et al. Imaging cell lineage with a synthetic digital recording system. Science 372, eabb3099 (2021).
Askary, A. et al. In situ readout of DNA barcodes and single base edits facilitated by in vitro transcription. Nat. Biotechnol. 38, 66–75 (2020).
Chadly, D. M. et al. Reconstructing cell histories in space with image-readable base editor recording. bioRxiv. https://doi.org/10.1101/2024.01.03.573434 (2024).
Zhou, X. et al. Highly sensitive spatial transcriptomics using FISHnCHIPs of multiple co-expressed genes. Nat. Commun. 15, 2342 (2024).
Kalhor, K. et al. Mapping human tissues with highly multiplexed RNA in situ hybridization. Nat. Commun. 15, 2511 (2024).
Ke, R. et al. In situ sequencing for RNA analysis in preserved tissue and cells. Nat. Methods 10, 857–860 (2013).
Qian, X. et al. Probabilistic cell typing enables fine mapping of closely related cell types in situ. Nat. Methods 17, 101–106 (2020).
Lundin, E. et al. Spatiotemporal mapping of RNA editing in the developing mouse brain using in situ sequencing reveals regional and cell-type-specific regulation. BMC Biol. 18, 6 (2020).
Chen, W.-T. et al. Spatial transcriptomics and in situ sequencing to study Alzheimer’s Disease. Cell 182, 976–991.e19 (2020).
Lomakin, A. et al. Spatial genomics maps the structure, nature and evolution of cancer clones. Nature 611, 594–602 (2022).
Gyllborg, D. et al. Hybridization-based in situ sequencing (HybISS) for spatially resolved transcriptomics in human and mouse brain tissue. Nucleic Acids Res. 48, e112 (2020).
Lee, H., Marco, Salas, S., Gyllborg, D. & Nilsson, M. Direct RNA targeted in situ sequencing for transcriptomic profiling in tissue. Sci. Rep. 12, 7976 (2022).
Schmidt, U., Weigert, M., Broaddus, C. & Myers, G. Cell detection with star-convex polygons. 265–273. https://doi.org/10.1007/978-3-030-00934-2_30 (2018).
Greenwald, N. F. et al. Whole-cell segmentation of tissue images with human-level performance using large-scale data annotation and deep learning. Nat. Biotechnol. 40, 555–565 (2022).
Terpstra, H. M. et al. PunctaFinder: An algorithm for automated spot detection in fluorescence microscopy images. Mol. Biol. Cell 35, mr9 (2024).
Coutu, D. L., Kokkaliaris, K. D., Kunz, L. & Schroeder, T. Multicolor quantitative confocal imaging cytometry. Nat. Methods 15, 39–46 (2018).
Maruno, T. et al. Visualization of stem cell activity in pancreatic cancer expansion by direct lineage tracing with live imaging. Elife 10, e55117 (2021).
Ershov, D. et al. TrackMate 7: integrating state-of-the-art segmentation algorithms into tracking pipelines. Nat. Methods 19, 829–832 (2022).
Hassanzadeh, A., Elyasi, S. N., Salih, S., Abdulkareem, S. S. & Saeed, S. R. Waveguide evanescent field fluorescence microscopy images of osteoblast cells: the effect of trypsin and image processing using TrackMate. Microsc Res Tech. 88, 1326–1334 (2025).
Zhao, W. et al. Sparse deconvolution improves the resolution of live-cell super-resolution fluorescence microscopy. Nat. Biotechnol. 40, 606–617 (2022).
Mai, U., Hu, G. & Raphael, B. J. Maximum likelihood phylogeographic inference of cell motility and cell division from spatial lineage tracing data. Bioinformatics 40, i228–i236 (2024).
Forrow, A. & Schiebinger, G. LineageOT is a unified framework for lineage tracing and trajectory inference. Nat. Commun. 12, 4940 (2021).
Tran, M., Askary, A. & Elowitz, M. B. Lineage motifs as developmental modules for control of cell type proportions. Dev. Cell 59, 812–826.e3 (2024).
Willnow, D. et al. Quantitative lineage analysis identifies a hepato-pancreato-biliary progenitor niche. Nature 597, 87–91 (2021).
Sender, R., Fuchs, S. & Milo, R. Revised estimates for the number of human and bacteria cells in the body. PLoS Biol. 14, 1–14 (2016).
Xu, B., Kucenas, S. & Zong, H. zMADM (zebrafish mosaic analysis with double markers) for single-cell gene knockout and dual-lineage tracing. Proc. Natl. Acad. Sci. 119, e2122529119 (2022).
ARAKI, T. The history of optical microscope. Mech. Eng. Rev. 4, 16-00242–16-00242 (2017).
Minsky, M. Memoir on inventing the confocal scanning microscope. Scanning 10, 128–138 (1988).
Acknowledgements
The authors would like to acknowledge the Stem Cell Network, whose support facilitated the preparation of this manuscript and Maria Abou Chakra for approving the use of the VHD logo. S.S. also acknowledges CIHR for their support via the doctoral research award (CGS-D).
Author information
Authors and Affiliations
Contributions
S.S. and R.A.G.T. wrote the manuscript. S.S., R.A.G.T., D.L.C., and L.J.S. were involved in editing the manuscript. All authors have read and approved the manuscript.
Corresponding author
Ethics declarations
Competing interests
S.S., R.G., and L.S. declare no financial or non-financial competing interests. D.C. serves as editor of this journal and had no role in the peer review or decision to publish this manuscript. D.C. declares no financial competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Short, S., García-Tejera, R., Schumacher, L.J. et al. Next generation lineage tracing and its applications to unravel development. npj Syst Biol Appl 11, 60 (2025). https://doi.org/10.1038/s41540-025-00542-w
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41540-025-00542-w
