
Abstract
The recent emergence and global spread of the human Monkeypox virus (MPXV), including its transmission to non-endemic regions, have raised significant global health concerns. In this proof-of-concept study, we developed a recombinant protein-based MPXV vaccine candidate, employing an innovative and versatile multivalent, self-assembled nanocage protein scaffold. Two immunogenic antigens derived from the contemporary circulating MPXV strain have been incorporated into a self-assembled non-structural protein-10 (NSP-10) scaffold, expressed, and purified using an Escherichia coli expression system without a purification tag. The vaccine candidate elicited strong antibody responses in mice and conferred protection against the lethal Vaccinia virus in an intranasal and skin pock in vivo study. Additionally, an intranasal challenge with the MPXV strain clade IIb in immunized mice demonstrated promising outcomes, including a significant reduction in viral titres and eliciting a robust neutralizing antibody response. This study demonstrates a feasible, scalable, and cost-effective approach for the development of the MPXV vaccine.
Similar content being viewed by others

Introduction
In the recent past, the world has been witnessing the outbreak of emerging and reemerging viruses of zoonotic and pandemic potential1. One such case was the outbreak of Monkeypox virus (MPXV) in early May 2022, which extended beyond the endemic tropical rainforest African Region2,3 and transmitted to 22 countries across the European Region, the Region of the Americas, the Western Pacific Region, and the South-East Asia Region4. The emergence of MPXV is concerning, despite its slow transmission, due to the widespread occurrence of new cases in non-endemic countries outside of Africa, along with a continued presence in Africa5,6. MPXV is classified within the Orthopoxvirus genus, which is part of the extensive family of DNA viruses encompassing poxviruses that affect both humans and animals7,8. MPXV exhibits numerous structural and functional resemblances to smallpox, which is caused by the variola virus, although the disease caused by MPXV tends to be milder and more self-limiting9. Due to the high antigenic similarity between these two viruses, the approved vaccines for smallpox vaccine JYNNEOSTM (Imvanex in the UK and Europe and Imvamune in Canada), ACAM20000, and LC16m8 (licensed in Japan) are expected to cross-protect against MPXV10,11. Since the 1980s, the unvaccinated population against smallpox, coupled with the global emergence and spread of MPXV, has spurred the research community to focus on developing and evaluating other novel vaccine platforms for the MPXV vaccine development12,13,14. However, another significant factor that needs to be considered is the availability, equity, and accessibility of the MPXV vaccine to the populations of low- and middle-income countries (LMICs), where large segments of the population have remained under-immunized.
The MPXV is an enveloped virus with a complex life cycle and produces two forms with surface proteins that are required for infectivity15. The virus is categorized into two distinct clades: clade I, previously referred to as the Congo Basin (Central Africa) clade, and clade II, previously known as the West African clade. Clade II is divided further into two subclades, IIa and IIb16. The MPXV virus sequence isolated in the 2022 outbreak was closely related to the clade II MPXV sequence. There are two infectious forms of the virus: intracellular mature virion (IMV), and extracellular enveloped virion (EEV), which has an additional membrane surrounding the IMV particle with another set of unique membrane proteins17,18. The envelope and membrane surface proteins of pox virus family have shown good immunogenicity and induce protective immune responses in non-human primates15. The MPXV surface antigens A35R and B6R from the EEV, and M1R, A29L, E8L, and H3L from the IMV are considered important for eliciting protective immune responses19. Their orthologs in the Vaccinia virus (VACV) are A27L, A33R, B5R, L1R, D8L, and H3L, respectively20, several of which (such as L1R, A33R, and B5R) have been shown to confer protection in the previous animal studies21,22,23,24. A29L (ortholog: A27L) and H3L facilitate viral attachment to host cells via heparan sulfate and are important for eliciting neutralizing antibody responses. E8L (ortholog: D8L) binds chondroitin sulfate and is a major target of the humoral immune response. M1R (ortholog: L1R) is essential for viral entry and is known for its strong immunogenicity and protective capacity. The EEV-associated antigens A35R (ortholog: A33R) and B6R (ortholog: B5R) play crucial roles in virion release and intercellular spread; both have been associated with robust protection in previous orthopoxvirus studies. H3L is a surface membrane protein of the IMV that facilitates viral attachment to host cells and is a key target for neutralizing antibodies20. Together, these antigens have been widely explored for multivalent vaccine formulations due to their ability to induce potent and protective immune responses13,25,26. These antigens were selected based on their surface accessibility, immunogenicity, and conservation across orthopoxviruses. The VACV, a well-characterized member of the pox virus family, has historically been used as a live-virus vaccine for smallpox eradication and currently serves as a widely used platform for the development of recombinant vaccines against various infectious diseases, including emerging orthopoxviruses such as monkeypox12,27,28,29.
In this study, we have designed three antigens (Mpx-V1, Mpx-V2, and Mpx-V3) using different combinations of MPXV surface antigens in a novel Multivalent Self-assembled Nanocage (MSN) scaffold protein as a nanocarrier. The technology is based on exploiting the inherent biological property of a viral non-structural protein (NSP-10) of the Coronavirus family, which normally self-assembles into a spherical multimeric structure and could display the foreign epitopes or domains stapled to the N and C terminal ends and an inner hollow hydrophobic chamber, which stabilizes the nanocage structure30,31. This is a 17 kDa protein that can display monomeric antigens, each set in the N- or C-terminals and self-assembled into a spherical dodecahedral nanoparticle. A dodecahedral structure will display up to 12 epitopes in the N terminus and 12 epitopes in the C terminus; however, depending on the antigen type and its length when fused to NSP-10, the final recombinant protein antigen product can form oligomers ranging from trimers to dodecamers. This protein scaffold does not share sequence homology with any other known proteins and is highly conserved within the Coronavirus genus, hence, it has versatile applications as a nanoparticle-based vaccine development. The recombinant Mpx immunogen protein is expressed in a prokaryotic expression system, thus allowing the development of highly scalable, cost-effective subunit protein-based immunogens. The key features of using this plug-and-play NSP-10 scaffold or vehicle are i. multivalence. ii. ease in development with high scalability and iii. high adaptability, allowing it to be tailored to different pathogens. Out of three designed immunogens (Mpx-V1, V2, and V3), Mpx-V3 has been studied further based on the expression, ease of purification, and yield. The immunogen Mpx-V3 has been designed by tethering the surface antigens of MPXV, both the N and C terminal of NSP-10, and comprehensively characterized for its biochemical and biophysical properties. The nanocage adorned with E8L and H3L antigens elicited robust antigen-specific antibody responses and provided protection against both intranasal and skin pock in vivo preclinical challenge models against the VACV, as well as intranasal challenge against the Mpox virus strain (clade IIb).
In summary, our findings underscore the development of a cost-effective, scalable Mpox protein-based vaccine candidate based on an innovative protein design approach. This recombinant protein platform enables rapid, cost-effective production and is well-suited for industrial-scale manufacturing to support the generation of millions of vaccine doses.
Results
Designing multivalent immunogens through plug-and-play combinations of Mpx antigens in NSP-10 vehicle, expression, and purification
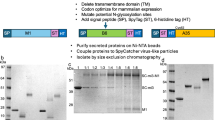
In the present study, the antigenic epitopes of Mpx surface antigens were fused to the NSP-10 nanocage forming the backbone (Supplementary Fig. 1a) using a G-S linker both at the N- and C-terminals as presented schematically in Fig. 1a. The E. coli codon-optimized genes for each construct were synthesized commercially (ThermoFisher Scientific, USA) and cloned into a pET28b (+) vector using standard molecular biology techniques. The antigen Mpx-V1 consists of four Mpx antigens (B6R, A29L stapled at the N terminal, and M1R, A35R at the C terminal of NSP-10), with a theoretical molecular weight of ~96 kDa, and was expressed in the soluble fraction upon IPTG induction. However, due to feeble expression, it was excluded from further downstream processing (Fig. 1a, Supplementary Fig. 2a, b). Similarly, Mpx-V2, containing B6R and A35R antigens with a theoretical molecular weight of ~61 kDa, was expressed in the insoluble fraction but contained multiple contaminating proteins, and we were unable to recover the pure protein from the inclusion bodies (Fig. 1a, Supplementary Fig. 2c, d). Mpx-V3 contains the E8L and H3L antigens with a theoretical molecular weight of ~79 kDa, it was abundantly expressed upon IPTG induction and primarily accumulated intracellularly as inclusion bodies (Fig. 1a, b). Conventionally considered as junk protein, inclusion bodies offer the advantage of yielding >90% pure protein without the need for multiple chromatographic steps, thereby increasing the recovery of the expressed proteins, w.r.t. similar proteins that tend to aggregate during the expression. Here, the extracted inclusion bodies were washed stringently with ionic and non-ionic detergents containing buffer solutions to recover >90% pure Mpx-V3 antigen. The obtained Mpx-V3 was then solubilized using a mild solubilization technique. The urea concentration during solubilization was kept to a minimum (~2 M) to reduce excessive denaturation and lower the cost when scaling up the production of vaccine drug substance32,33,34,35,36,37,38. The Mpx-V3 protein was finally folded via the pulsatile dilution method extensively used by the industry38,39 in a refolding buffer containing 20 mM Tris and 5% sucrose as a stabilizer for 12–16 h. The protein yield averaged ~100 mg/L at pH 8.5, as observed in the laboratory shake flask culture. This could easily be scaled up to >1 g/L in a bioreactor40,41. The expressed and refolded Mpx-V3 protein was subjected to anion exchange chromatography in flow-through mode and eluted at ~9 mL volume in the Superdex 200 column, indicating the formation of oligomers of higher-order structure (Supplementary Fig. 3a). We performed a calibration run on the Superdex 200 column using reference proteins of known molecular weights to assess column performance and verify separation accuracy. To further refine the elution profile of the synthesized Mpx-V3 immunogen, we used a TSKgel G4000PW 500 Å column, which has a more defined pore size of 50 nm. The elution profile of Mpx-V3 indicates a major peak at 8.4 retention time covering 83.1% peak area along with five minor peaks at 5.1, 10.2, 10.7, 10.8, and 13.2 covering a peak area of 0.3, 4.1, 7.9, 1.8, and 4.6, respectively (Fig. 1d). The early eluting peak at 5.1 min (retention time) appears to correspond to higher multimeric forms (∼dodecamer) while four eluting peaks after the main peak represent lower molecular weight oligomeric forms (trimers, tetramers etc.). Furthermore, we also verified the void volume of the column by running blue dextran using the same mobile phase. The void volume of the column was calculated to be ~7.7 mL, indicating that the peaks appeared due to molecular sieving of the oligomeric forms present in the Mpx-V3 recombinant protein (Supplementary Fig. 3c). The elution profile of Mpx-V3 on a Tskgel G4000PW 500 Å column predominantly represents a high-order oligomeric structure with the presence of some low-order oligomers (Fig. 1d).

a Schematic representation of the screening of MPXV candidates to process for downstream applications. Mpx-V1 failed to express properly. Mpx-V2 expressed as inclusion bodies, but these were heavily associated with contaminating bacterial proteins, hence the challenge associated with purification. Mpx-V3 expressed as inclusion bodies and bacterial contaminating proteins were very low, which were washed off, enabling recovery of highly pure inclusion bodies. Mpx-V3 was processed further for active protein purification. b The effect of IPTG induction on the expression profile of Mpx-V3 immunogen. Lane 1: Marker, Lane 2: Uninduced, Lane 3: Induced. c The Anion exchange chromatogram of the purified Mpx-V3 immunogen. The shown chromatogram was obtained using the Anion exchange chromatography performed at flow-through (FT) mode. d The elution profile of Mpx-V3 immunogen on a Tskgel G4000PW 500 Å column shows a single major peak (83.1% Area) at 8.4 retention time corresponding to oligomer formation whereas, five minor peaks at 5.1, 10.3, 10.7, 11.8, and 13.2 covering a percentage area of 0.5, 2.3, 7.9, 1.7, and 4.6, respectively. e The Mpx-V3 immunogen showed a single intense band, which resolved at an apparent molecular weight of ~79 kDa. The western blots confirm the specificity of the Mpx-V3 immunogen against the anti-NSP-10, anti-E8L, and anti-H3L commercial antibodies.
The Mpx-V3 immunogen was resolved at an apparent molecular weight of ~79 kDa on SDS-PAGE and further transferred onto a PVDF membrane for Western blotting. The blot was reactive against anti-NSP-10 (in-house developed polyclonal mouse antibody), anti-E8L, and anti-H3L commercial antibodies as observed by a prominent band on development with ECL reagent (Fig. 1e). This conformed to the specificity, corroborating with the theoretically calculated molecular weight of the polypeptide chain of the Mpx-V3 antigen. Both E8L and H3L are immunogenic regions on the membranes of IMVs and are potential targets for vaccine design20. However, the combined use of only E8L and H3L as a potential vaccine target has not been extensively investigated to date. We further processed the Mpx-V3 antigen and observed no batch-to-batch variation either in terms of expression or yield in the shake flask culture.
To evaluate the scalability of Mpx-V3 production, a batch fermentation was conducted in a 5-L fermenter at the Vaccine Design and Development Centre (VDDC), THSTI. Following induction with 1 mM IPTG, the transformed E. coli expressed Mpx-V3 predominantly as inclusion bodies, as shown previously, with maximal expression observed within 9 h in shaker flask culture (Supplementary Fig. 1b). After biomass harvesting, a total of 349 g was obtained. From this, 80 g of cell pellet was suspended in 50 mM Tris-HCl buffer (pH 8.0) containing 10 mM EDTA, 2 mM DTT, 300 mM NaCl, and 1 mM PMSF. Cell lysis was carried out using a Panda Plus high-pressure homogenizer at 1000 bar for six cycles. The lysate was then centrifuged at 10,000 rpm for 30 min at 4 °C to separate inclusion bodies from cellular debris. The resulting inclusion bodies were washed with 50 mM Tris-HCl buffer supplemented with 10 mM EDTA, 2% Triton-X-100, and 1 mM PMSF to remove contaminants, followed by additional washing with Milli-Q water to eliminate residual salts and detergents. A final centrifugation at 10,000 rpm for 30 min at 4 °C yielded 30 g of purified inclusion bodies containing the target protein. The presence of Mpx-V3 inclusion bodies was confirmed by SDS-PAGE analysis (Supplementary Fig. 1c, d). These inclusion bodies were solubilized in 60 mL of solubilization IB buffer (20 mM Tris-HCl) containing 2 M urea, resulting in a protein concentration of 8.5 mg/mL, as determined by absorbance. The solubilized protein was refolded by dilution into 600 mL of refolding buffer (20 mM Tris-HCl, pH 8.5) containing 5% sucrose, and the mixture was gently stirred at 4 °C for 4–6 h using a magnetic stirrer (Supplementary Fig. 1e). In the 9 h of fed-batch fermentation, 8.5 g/L of Mpx-V3 was produced at a cell OD of 74. The refolded protein was further analyzed using SDS-PAGE to assess purity and integrity (Supplementary Fig. 1f).
Biophysical characterization of Mpx-V3 nanocage protein antigen and stability assessment
The cellular uptake of proteins is strongly influenced by their size, shape, and surface charge, with zeta potential playing a key role in modulating interactions with cell membranes and binding efficiency. Higher zeta potential values contribute to strong membrane binding and enhanced cellular uptake42. In this study, we have observed the effect of storage temperature on the zeta potential of the Mpx-V3 immunogen. While optimizing the process parameters, we observed when the Mpx-V3 protein was stored at −20 °C, the surface Zeta potential was obtained of −11 mV, in contrast when the Mpx-V3 was stored at 4 °C and then evaluated for the Zeta potential, it was obtained to be −27 mV (Fig. 2a). Protein storage temperature can significantly influence zeta potential values by altering protein conformation, surface charge distribution, and aggregation state43,44. Since the storage buffer (Tris-HCl) has low ionic strength and relatively weak buffering capacity at such a low temperature, therefore, the change in pH value is relatively high. Additionally, low-concentration Tris-HCl solutions are more susceptible to environmental factors like temperature during storage, resulting in a change in pH value. This decrease in pH value may favor the agglomeration of the Mpx-V3 immunogen (stored at −20 °C), leading to a reduced zeta potential due to the hindered steric stabilization effect44,45,46,47.

a The effect of storage temperature on the stability as measured by the Zeta potential of Mpx-V3 immunogen. (i) The Apparent Zeta Potential of Mpx-V3 immunogen stored at pH 7.0 and a temperature of −20 °C depicts the Zeta value of −11 mV corresponding to the low stability of the prepared formulation. (ii) The Apparent Zeta Potential of Mpx-V3 immunogen stored at a temperature of 4 °C depicts the Zeta value of −27 mV corresponding to the good stability of the synthesized Mpx-V3 immunogen. b Intrinsic tryptophan fluorescence measurement of Mpx-V3 immunogen. The emission maxima of fluorescence for Mpx-V3 were recorded at a wavelength of 335 nm. c Far-UV circular dichroism spectroscopy of Mpx-V3 immunogen. d Negative staining-TEM (Ns-TEM) micrographs of MPXV antigen sample at 57KX magnification. Few representative of Mpx-V3 dodecamer-sized particles have been shown using red circles. Scale bar is 20 nm in length. e 2D class averages of particles picked from Ns-TEM images. The scale bar is 20 nm in length, and dodecamer-like classes (15–20 nm size) have been marked with red squares. Particles ranging from 10 to 15 nm have been marked in yellow colored box, and particles ranging from 5 to 10 nm have been marked in blue colored box. f Computationally generated density maps (at 20 Å resolution) of predicted Mpx-V3 protein structures have been shown. Scale bar is 10 nm in length.
The intrinsic fluorescence of a protein is representative of fluorescence from all the aromatic amino acids (mostly tryptophan and partially tyrosine residues) and is highly sensitive to the hydrogen bonding, polarity of the protein’s microenvironment, as well as other non-covalent interactions. The designed Mpx-V3 antigen comprised of 8 Trp, 35 Tyr, and 32 Phe residues. In aqueous solution, the Mpx-V3 protein exhibited an emission-maximum (λmax) of ~335 nm (Fig. 2b), which is believed to be mainly contributed from the tryptophan residues and slightly from the tyrosine residues (λmax = ~303 nm in water and insensitive to solvent polarity). A λmax of 335 nm usually indicates that tryptophan residues are well inside the core structure, indicating the proper folding of the Mpx-V3 protein48. Further, Mpx-V3 was assessed by Far-UV spectroscopy for the prediction of secondary structure. We used Jasco Spectra Manager software to deduce the secondary structure of the purified protein. The spectra were analyzed using Spectra Manager software and compared to Yang’s reference spectra. Assessment by Spectra Manager software showed a presence of 17.0% helix, 32.8% beta, 16.9% turn, and 33.2% random coil structure (Fig. 2c).
Furthermore, visual inspection of the morphology of Mpx-V3 was assessed by negative staining transmission electron microscopy (Ns-TEM), and it was observed to consist of particles ranging in size from ~5 to 30 nm (Fig. 2d, e). The percentage distribution of the varied-size particles was also plotted based on the Ns-TEM analysis which shows the cumulative percentage of particles ranging from size (nm) ≥5–10, ≥10–15, ≥15–20, and ≥20–30 are 7.32, 63.53, 29.15, and 2.66, respectively. The majority of the particles come in the range of ≥10–15 nm (Fig. 2e). Further, the picked particles from these images were subjected to 2D classification, which produced multiple classes with particles that matched the shape and size of a predicted Mpx-V3 dodecamer model (Fig. 2f). Smaller oligomers that may resemble trimers or tetramers were also observed. The larger dodecameric particles formed ~29.15% of the total particles, with smaller oligomers making up the remaining particle set. The predicted model has been generated using the Molmap, and a model of Mpx-V3 dodecameric structure has been shown in Supplementary Fig. 2f. The hydrodynamic size of the expressed and purified Mpx-V3 protein was measured by dynamic light scattering (DLS). As shown in the graph (Supplementary Fig. 3d), the size of particles measured in DLS ranges from 10 nm to ~60 nm, with the average size reported as 29 ± 0.95 nm and PDI of 0.4, whereas the measurement in a volume percentage vs size (d.nm) interpretation showed size of ~17.89 nm (Supplementary Fig. 3dii) with major coverage of 99.9% peak area. In the academic R&D set up, the DLS and Ns-TEM analyses revealed a degree of polydispersity in the nanocage particles, indicating variability in particle size and assembly. Such heterogeneity may impact consistency and regulatory hurdles. However, in an industrial setting, this can be effectively addressed through optimized process control strategies—such as refined expression conditions, controlled refolding protocols, and advanced purification techniques (SEC with process-scale columns, microfluidics-based sorting and formulation, HPLC etc) to ensure uniform particle size distribution and these approaches, often used in combination and integrated with in-line process analytical tools, allow for maintaining narrow quality parameters in GMP settings and reducing batch-to-batch variation to meet the regulatory standards for quality and reproducibility. We further determined the stability of the synthesized protein antigen by storing Mpx-V3 antigen at different temperatures, and it was observed that Mpx-V3 was stable at 25 °C for up to 1 week without any excipient (Supplementary Fig. 2e).
The prophylactic Mpx-V3 immunization in BALB/c mice induced high levels of immune responses
To assess the immunogenicity of Mpx-V3 antigen, we immunized BALB/c mice at a dose of 25 µg either with AddaVaxTM (<6% Squalene-based adjuvant, given in 1:1 v/v) or Alhydrogel (2% Aluminum hydroxide-based adjuvant, given in 1:1 v/v) in one prime (0-day) and one boost (28-day) regimen via the intramuscular route. Naïve mice were immunized with the adjuvant alone as a control. All Mpx-V3+AddaVaxTM vaccinated mice induced Mpx-V3 specific IgG response, 14 days post-prime immunization with endpoint titers of >1:12150 and post-boost immunization the endpoint titers increased to around 4-logs as compared to pre-bleed <1:50, indicating an anamnestic response to Mpx-V3 immunization (Fig. 3a). Similarly, Mpx-V3+Alhydrogel immunized mice sera showed a ~4-fold increase in the dilutions and elicited higher antibody responses after the booster dose. The antibody responses to the backbone NSP-10 were insignificant when comparing the prime immunized sera to the boost immunized sera (Supplementary Fig. 4a). The boost serum collected was also subjected to IgG isotyping to determine the representation of Th1 and Th2-type responses. Both the AddaVaxTM-adjuvanted Mpx-V3 and Alhydrogel-adjuvanted Mpx-V3 vaccines elicited an immune response with a Th2 bias, as demonstrated by an IgG1 to IgG2a ratio exceeding 2, which was more prominent in the Alhydrogel-adjuvanted immunized mice (Fig. 3b, c). We further assessed the neutralization titer of the immunized sera against the VACV Western Reserve (WR) strain as shown in Fig. 3d. Both the adjuvanted immunized mice sera showed elicitation of neutralization titer as measured against the VACV.

Six to eight weeks old BALB/c mice (n = 6) were immunized with 25 µg of Mpx-V3 either with AddaVaxTM or Alhydrogel adjuvant and with respective adjuvants alone. The blood sample was drawn from mice before and after prime and boost immunization, serum was separated, heat-inactivated, and stored at −20 °C. Individual mice values were plotted. a The test serum samples (i.e, pre-bleed, prime, and boost) obtained from the immunized animals were used to determine the IgG level using ELISA by endpoint titer of anti-Mpx-V3 antibodies using the Mpx-V3 antigen-coated plates. Individual mice (n = 6) values were plotted, and error bars represent SEM. Statistical analysis between the two groups was performed by unpaired, two-tailed Student’s t-test. b The level of IgG subtypes (IgG1, IgG2a, IgG2b, IgG2c and IgG3) in the boost serum from Mpx-V3 with AddaVaxTM and Mpx-V3 with Alhydrogel group was estimated in boost serum samples. The individual mice (n = 6) endpoint titer was plotted. c The ratio of endpoint titer of IgG1 to IgG2a was calculated for Mpx-V3+AddaVaxTM and Mpx-V3+Alhydrogel groups after averaging the individual values, and mean values are plotted on a graph. d Neutralizing antibody titers of day 35 anti-Mpx-V3 sera against VACV were determined by the PRNT50 method. PRNT50 value from individual mice is plotted; the error bar represents the SEM. GraphPad Prism 9 was used for calculating statistical analysis. One-way ANOVA was used for comparing multiple groups, and Dunnett’s multiple comparison tests were applied to calculate the p values. e Splenic T cell response of naïve, adjuvant control, and Mpx-V3 along with adjuvanted immunized mice against the E8L and H3L stimulation. The data shown here is from three mice and was analyzed using the FlowJo software, and graphs were plotted using the GraphPad Prism 9. One-way ANOVA was used for comparing multiple groups, and Dunnett’s multiple comparison tests were applied to calculate the p values.
As T cell immune response also plays an important role in the clearance of viruses, we analyzed the T cell immune responses of Mpx-V3-immunized mice (as described in “Materials and Methods”) by in vitro stimulating the splenocytes with recombinant H3L and E8L soluble commercial proteins. The frequency of intracellular IL-2 and IFN-γ cells was measured using flow cytometry analysis as shown in Fig. S6. In both the adjuvanted Mpx-V3 immunized mice group, IFN-γ and IL-2 positive cells were present in significantly higher frequency (Fig. 3e), although the E8L-specific induction is comparatively higher than H3L soluble proteins. The frequency of the cytokine-producing CD4+ and CD8+ T cells among both the protein stimulations was also measured and presented in Fig. 3e. The observations inferred from the splenic T cell responses strongly support the induction of Th1 biased cellular immunity for both the antigens (E8L and H3L). This elevated response regulates a Th1-driven cascade of pro-inflammatory mediators, which might result in the recruitment of immune cells for rapid elimination of the viral infection.
We quantified the serum concentration (pg/mL) of IFN-γ and IL-2 cytokines by the ELISA method. The administration of Mpx-V3 adjuvanted with AddaVaxTM renders an increased level of IFN-γ and IL-2 cytokines with concentrations of 694.3 pg/mL and 240 pg/mL, respectively (Supplementary Fig. 4b). In contrast, the observed value for IFN-γ and IL-2 cytokines was 430.4 pg/mL and 84.2 pg/mL in the group immunized with Mpx-V3 and Alhydrogel, respectively (Supplementary Fig. 4b). Our studies suggest that although the adjuvanted antigens primarily induced Th2-biased responses, they also have the ability to shift an established Th2 response toward a more balanced immune profile by promoting Th1-mediated cell-mediated immunity. This is advantageous for a vaccine capable of eliciting both humoral and T cell-mediated immune responses. We further determined whether Mpx-V3 immunized serum could detect the VACV infection. We infected the Vero cells with the VACV WR strain, and after fixing and permeabilizing, the cells were incubated with Mpx-V3 boost serum and anti-VACV (WR) B5R rabbit polyclonal antiserum (Supplementary Fig. 4c); in other set of experiment, the infected cells were probed with commercial anti-E8L and anti-H3L antibodies, and all were probed with anti-mouse Alexa-fluor-488 secondary antibody (Supplementary Fig. 4d). The results suggested the immunized MpX-V3 mice sera were strongly cross-reactive to VACV.
The immunization of Mpx-V3 antigen in BALB/c mice conferred protection against the intranasal VACV challenge
Next, we evaluated the protective efficacy of the immunized BALB/c mice by challenging them intranasally with the VACV WR strain. The Mpx-V3 vaccinated BALB/c mice initially had a similar trend of weight loss as that of AddaVaxTM and Alhydrogel immunized mice up to 7th dpi, but after 8th dpi, the Mpx-V3 vaccinated animals started to recover with overall general status of healthy appearance (no fur erection or weight loss), proper food intake, and fine mobility (Fig. 4b). Out of six animals in Mpx-V3 vaccinated along with AddaVaxTM and Alhydrogel adjuvant group, those were monitored up to 14 days post-challenge, one mortality was witnessed on 8th dpi (80% survival rate), as compared to the adjuvanted control groups where all mice succumbed to death by 8th dpi (zero survival) (Fig. 4c). All the adjuvanted control group animals showed >25% weight loss (were considered dead and hence euthanized) with gross diseased appearance showing altered mobility, weight loss, piloerection, reduced food intake, shivering, and huddling (Fig. 4d). We further monitored the temperature changes post-challenge. It was witnessed that adjuvant control animals experienced hypothermia post day 3 infection, and Mpx-V3 antigen vaccinated animals either showed hyperthermia or normal body temperature. However, the temperature fluctuations were not more than ±2 °C of 37 °C (Fig. 4e). Organs harvested at 5 days post-infection (dpi) were analyzed for viral load using the plaque-forming unit (PFU) assay, with samples from two mice tested in triplicates. Control animals receiving AddaVax™ or Alhydrogel alone exhibited significantly higher viral titers in both lungs (Fig. 4f) and trachea (Fig. 4g). In contrast, the Mpx-V3 vaccinated groups showed marked viral suppression. Notably, the Alhydrogel + Mpx-V3 group exhibited complete viral clearance in the lungs and trachea, with no detectable plaques. The AddaVax™ + Mpx-V3 group also showed a substantial reduction in lung viral titers, with only 4–5 plaques compared to 18–20 plaques in the adjuvant-only controls (p < 0.0001). The presence of a few plaques in the lungs of the AddaVax™ + Mpx-V3 group, compared to the absence of plaques in the other vaccinated groups, was reflected as a slightly elevated bar in the graph. Further histopathological studies of major organs in the adjuvanted control groups showed severe inflammation as seen by the presence of perivascular cuffing, pneumonitis, and infiltration of monocytes and macrophages (black arrow) (Supplementary Fig. 5a). Immunohistochemistry revealed the presence of VACV in the adjuvanted-only control group, indicated by reactivity to the anti-E8L monoclonal antibody, in contrast to the vaccinated group (Supplementary Fig. 5a). These data demonstrated that Mpx-V3 expressing multiple epitopes of E8L and H3L is a potent immunogen and able to elicit significant immune responses for virus clearance and protection. Bioinformatics analysis between different clades of MPXV and VACV showed a high degree of homology (>90%) and 99–100% homology of the E8L and H3L antigens between the different MPXV clade strains (Supplementary Table 1), thus indicating the Mpx-V3 immunogen cross-protectiveness to the VACV.

a Schematic of an intranasal challenge study with VACV Western Reserve strain (n = 8) for all the groups (n = 6 for body weight, survival, and n = 2 for titer). b, c The prime-boost immunized mice were intranasally challenged with 50 μL of 1 × 107 PFU/mL of vaccinia virus. After the challenge, changes in b body weight and c survival were recorded every day for 2 weeks. The data shown here is mean ± SD from one challenge study, having n = 6 in each group’s and for survival, individual animal record was plotted. d Graphical representation of behavior scores of infected mice. Animals were scored based on the clinical symptoms exhibited by them into 1–10. Scoring with 0 = no alteration in behavior, 1 = no restriction of movement; blink frequently; no body stiffening; no hind limb paralysis; 2 = dull; 3 = piloerection; 4 = shivering; 5 = hunched posture; 6 = restriction of movement; 7 = blink frequently; no body stiffening; no hind limb paralysis; 8 = restriction of movement; body stiffening; no hind limb paralysis; 9 = restriction of movement; eyes closed; 10 = body stiffening; hind limb paralysis, sometimes tremor. The data shown here is each group’s mean ± standard deviation. e Heatmap of temperature changes observed after infection up to 14 days. All the groups, including normal control, adjuvant control, and Mpx-V3-immunized groups, were assessed for change in body temperature up to 14 days post-infection by the VACV. The data shown here is each group’s mean ± standard error of mean (SEM). f, g Vaccinia virus titer was determined in the lung and trachea samples harvested at 5 days post-infection, respectively (n = 2 mice). Upon harvesting, organ samples were weighed, homogenized, sonicated, and centrifuged. Later, aliquots of the supernatant from the samples were stored for titer calculation in Vero cells. Confluent Vero cells were infected with different dilutions of organ supernatant for 2 h, and later the cells were washed and layered with 2% CMC in 2% FBS containing DMEM for 48 h. Then, the media was removed, cells were fixed with 4% paraformaldehyde, and stained with 0.5% crystal violet. Plaques were counted from the plates to calculate virus titer in PFU/g. The values plotted are from biological replicates as mean ± SEM, statistical analysis between the two groups was performed by unpaired, two-tailed Student’s t-test.
Evaluation of the protective efficacy of Mpx-V3 antigen against the VACV via the tail scarification method
To study whether the Mpx-V3 immunogen could protect against the skin pock lesions, we developed a tail scarification model using BALB/c mouse49. Immunized BALB/c were challenged with 10 μL of 1 × 107 PFU/mL of the VACV (WR) using the tail scarification technique (Fig. 5a)49. The temperature variations among the adjuvant controls and Mpx-V3 + adjuvants were not more than ±3 °C of 37 °C (Fig. 5b). Following tail scarification, a bloody wound on the scarification site and a clot appeared in all groups as the disease progressed. After 6 days of post-tail scarification, the clot started fading in the normal control group inoculated with PBS only (Fig. 5c). The skin lesions in the groups AddaVaxTM control and Alhydrogel control tend to appear 7–10 days post-tail scarification. The skin lesions eventually developed into scabs on day 12 that peeled off between 18 and 21 days post-scarification, leaving a scar at the scarification site. The group treated with Mpx-V3 antigen adjuvanted with Alhydrogel didn’t show any significant skin lesions and scabs after the fading of blood clots and recovered completely after 12–14 days of the VACV infection through scarification (Fig. 5c). In the group treated with Mpx-V3 antigen along with AddaVaxTM, we observed skin lesions and scabs from day 8 onwards lasting up to 17–19 days, the recovery in this group is delayed as compared to the group treated with Mpx-V3 antigen with Alhydrogel. To reduce variations, a “clinical take” assessment was performed on day 12 post-scarification when all scabs reached their maximal size (Fig. 5c). The tail scarification in the control group with PBS resulted in a bloody wound on the site of scarification that was clotted, it did not develop further and disappeared after 5–6 days. Apart from the local reaction described above, all vaccinated mice survived and showed no signs of morbidity. Thermal profiling of the body temperature was also measured following the post-tail scarification for 21 days. In the case of VACV-infected animals, the development of skin lesions and scabs was accompanied by a rise in body temperature, which was one of the crucial symptoms. Similar observations have also been reported in our study, the adjuvant control groups showed a high fever between 8 and 12 days. In contrast, the Mpx-V3 antigen-treated groups, along with both the adjuvants, showed a low rise in body temperature similar to the normal control group.

a Schematic of skin pock lesion challenge study with VACV Western Reserve strain. b Thermal profiling of the immunized mice following the tail scarification technique. All the groups, including normal control, adjuvant control, and Mpx-V3-immunized groups, were assessed for change in body temperature up to 21 days post-tail scarification by the VACV. The data shown here is each group’s mean ± standard error of the mean (SEM). c In the tail pock lesion challenge experiment, BALB/c mice were immunized with 25 μg of Mpx-V3 with AddaVaxTM or Alhydrogel. After prime and boost immunization, 21 days after the boost, mice were challenged with VACV (10 μL of 1 × 107 PFU/mL) vial tail scarification method as mentioned in the methods section. The appearance of “clinical signs” was evaluated on 6, 12, and 21 days post-tail scarification. The pictures shown in a red box represent the maximum intensity of skin lesions and scabs in adjuvant control groups, whereas the green box indicates the complete recovery in the Mpx-V3 immunized mice from the skin lesions and scabs.
Mpx-V3-vaccinated mice are protected from a clade II MPXV challenge
We further performed the challenge study of the Mpx-V3 immunized mice with the MPXV clade IIb A.2.1 strain to evaluate the protective potential of the Mpx-V3 immunogen. Upon receiving the MPXV clade IIb A.2.1 virus from the National Institute of Virology (NIV, ICMR), the virus was cultured in Vero cells, and the titers were determined. The challenge model was established in BALB/c mice using different doses of viruses (data not shown). Briefly, 6–8 weeks old BALB/c mice infected with 50 μL of 1 × 106 PFU/mL of virus intranasally showed up to 18–20% loss of body weight and developed clinical signs of weight loss up to 20%, including piloerection, dullness and shivering however, no mortality was observed in these mice up to 21 dpi. To check the protective efficacy of Mpx-V3 immunogen, we immunized 6–8-week-old mice with 25 μg of Mpx-V3 along with AddaVaxTM or Alhydrogel, in a prime-boost strategy via intramuscular immunizations (Fig. 6a). We determined the serum neutralization titers in the boost sera collected from immunized mice. Both the adjuvanted immunized sera showed a significant neutralization against MPXV strains obtained from the Clade IIb strain (isolated from India by the National Institute of Virology, ICMR) and NR-27 (BEI resources, NIAID, NIH) (Fig. 6b). 21 days after the booster dose, mice were shifted to THSTI’s ABSL3 facility and challenged via an intranasal route with MPXV clade IIb A.2.1 (NIV, ICMR), and animals were monitored closely for 14 dpi. The blood sample was collected 2 weeks post every immunization and 6th day post-viral challenge, and the lung and trachea were harvested to determine the viral load. The Mpx-V3 immunized mice did not show any significant signs of disease or weight loss, however, the AddaVaxTM and Alhydrogel immunized mice exhibited dullness, piloerection and hunched posture and loss in body weight up to 14 days post-challenge observation (Fig. 6c). Previous study related to BALB/c mice as a challenge model infected intranasally (i.n.) with Zaire-79 also showed the same set of clinical signs like loss of appetite, hunched posture, piloerection (Fig. 6c) and transient weight loss of approximately <20% without mortality50.

35 female BALB/c mice of 6–8 weeks of age were randomly divided into five groups, having n = 7 mice in each group. a Following the previous immunization scheme and dose, mice were immunized in a prime and boost dose of 25 μg along with AddaVaxTM and Alhydrogel as shown in the schematic. b Neutralizing antibody titers (n = 6) of day 35 Mpx-V3 sera against VACV (WR), MPXV (N-27, from BeI resources), and MPXV (Clade IIb, from NIV Pune) were determined by the PRNT50 method. PRNT50 value from individual mice is plotted, and the error bar represents the SEM. c The immunized mice (n = 7) were intranasally challenged with 50 μL 1 × 106 PFU/mL of MPXV. Post-infection changes in body weight and clinical scores were recorded every day for 2 weeks. Animals were scored based on the clinical symptoms exhibited by them into 1–10. Scoring with 0 = no alteration in behavior, 1 = no restriction of movement; blink frequently; no body stiffening; no hind limb paralysis; 2 = dull; 3 = piloerection; 4 = shivering; 5 = hunched posture; 6 = restriction of movement; 7 = blink frequently; no body stiffening; no hind limb paralysis; 8 = restriction of movement; body stiffening; no hind limb paralysis; 9 = restriction of movement; eyes closed; 10 = body stiffening; hind limb paralysis, sometimes tremor. The data shown here is each group’s mean ± standard deviation. d The virus titer of MPXV was determined in the lung and trachea samples (n = 6) harvested at 6 days post-infection, respectively. Upon harvesting, organ samples were weighed, homogenized, sonicated, and centrifuged. Later, the aliquots of the supernatant from the samples were stored for titer calculation in Vero cells. Confluent Vero cells were infected with different dilutions of organ supernatant for 2 h, and later the cells were washed and layered with 2% CMC in 2% FBS containing DMEM for 48 h. Then, the media was removed, cells were fixed with 4% paraformaldehyde, and stained with 0.5% crystal violet. Plaques were counted from the plates to calculate virus titer in PFU/g. The values are plotted as mean ± SEM, and statistical analysis between the two groups was performed by unpaired, two-tailed Student’s t-test. GraphPad Prism 9 was used for calculating statistical analysis. One-way ANOVA was used for comparing multiple groups, and Dunnett’s multiple comparison tests were applied to calculate the p values.
We determined the viral load in the lungs and the trachea tissue collected on day 6 post-viral challenge. The tissue samples from Mpx-V3 immunized mice had developed fewer or negligible plaques as compared to adjuvant controls (Fig. 6d). There was a significant reduction in lung and trachea virus titers in Mpx-V3 immunized mice as compared to AddaVaxTM and Alhydrogel control groups. When Mpx-V3 adjuvanted groups were compared between Mpx-V3+AddaVaxTM and Mpx-V3+Alhydrogel groups, it was found that alum-based adjuvant Alhydrogel was augmenting a more significant viral reduction (p < 0.0024). In summary, Mpx-V3 was found to be immunogenic and capable of protecting immunized mice from MPXV challenge. Furthermore, the lung samples collected on day 6 post-infection were assessed for histopathological analysis, and we found infiltration of monocytes and macrophages along with perivascular cuffing (pointed in black arrows) in the adjuvant control groups, whereas the adjuvanted Mpx-V3 group showed comparatively better morphology (Supplementary Fig. 5b).
Discussion
In this study, we introduce a novel approach utilizing an MSN-based subunit protein scaffold platform for the development of a multivalent MPXV recombinant subunit vaccine. This approach is characterized by its seamless integration, allowing for easy customization and adaptation to different vaccine targets. The vehicle is the SARS-CoV-2 non-structural protein-10 (NSP-10), which is a part of the Coronavirus replication machinery51. The NSP-10, consisting of 148 residues and containing two zinc fingers-like motifs, which is in complex with NSP-14-NSP-16 forms the CoV replication-transcription complex52. The usage of NSP-10 for vaccine development was first described by Carter et al.53, NSP-10 consists of 12 monomers that assemble into a spherical particle, resembling a dodecamer with twelve faces. The N- and C-termini are surface-exposed and located on separate threefold axes, facilitating the attachment of foreign antigens31. We have exploited this property of NSP-10 to act as a vehicle expressing the MPXV surface antigens. We employed the E. coli expression system for antigen production, a widely used and well-established platform for the manufacturing of biologics38. The antigens have been designed containing promising immunogenic surface proteins such as B6R, A29L M1R, A35R, E8L, and H3L54, however, based on expression and purification strategy, only Mpx-V3 antigen was further characterized which contains two immunogenic epitopes E8L and H3L (Fig. 1). E8L and H3L are the surface-exposed antigens from the IMV form of MPXV that play key roles in viral entry and immune recognition. Their high immunogenicity and conservation make them strong candidates for inclusion in vaccine development capable of inducing robust neutralizing antibody responses55,56. All the antigens were tagless and produced using a low-cost and widely used E. coli expression system, which is suitable for large-scale fermentation and can be readily manufactured in LMICs. The Mpx-V3 immunogen which was taken up for further evaluation was recovered in the inclusion bodies. Functional proteins have been recovered from inclusion bodies since 1980s, the first being insulin57. Several functional proteins like growth hormone, interferon alpha 2b58,59 extracellular domain PDL-160, anti-PDL-1 single domain antibodies61, lysozyme, ovine growth hormone, yellow fluorescent protein and anti-cancer target MDM-262 have been recovered at a yield as high as 20 g/L63,64,65,66,67,68,69. In this study, mild solubilization was utilized to recover Mpx-V3. Mild solubilization has been reported to improve yield and retain activity of human growth hormone, ovalbumin and recently SARS-CoV-2 RBD32,34,35,36,37,70,71,72,73,74 Mpx-V3 was then refolded by pulsatile dilution method owing to its ease and being wide acceptability in the industries38,39,75,76,77. However, inclusion body-derived expression systems often require careful optimization of refolding conditions. In our study, a reproducible refolding protocol was established that maintained structural integrity and antigenicity of the nanocage constructs. While this adds a step in the production pipeline, our pilot-scale fermentation in a 5 L fermenter yielded ~8.5 g/L of purified protein, demonstrating both feasibility and scalability of the process. The ~79 kDa protein is multimerized in the NSP-10 nanocage to form the oligomeric higher-order structure protein as evidenced by SEC purification and Ns-TEM analysis (Figs. 1 and 2). The Ns-TEM analysis revealed a mixture of oligomers, with particles measuring ~5–30 nm in size. Notably, the 2D classification indicated the presence of distinct oligomeric structures, including tetramers, pentamers, and dodecamers. Among these, dodecameric structures accounted for ~31.2% of the total particles observed (Fig. 2d, e). This distribution suggests that NSP-10 retains its inherent biological properties, folding naturally to form nanoparticle structures. These findings underscore the structural integrity and self-assembling capability of NSP-10, essential for its role as a scaffold in the antigen designing and development. Several bioinformatics and molecular biology studies have been conducted to develop a multiepitope-based vaccine against MPXV to identify and assemble these epitopes into a single vaccine construct78,79,80. However, we observed polydispersity with mixture of different oligomeric structures, as shown in Ns-TEM and DLS. The size of particles measured in DLS range from 10 nm to ~60 nm, a broad distribution with multiple sizes of particles detected. In the TEM data, we also see a similar size range distribution, but with a larger proportion of particles between 10 and 15 nm in size. It is possible, that the largest oligomeric sizes (~29–30 nm) are heterogeneous in shape, this would cause them to not be picked up by 2D class averaging in Ns-TEM, but DLS could show it as it is based on average hydrodynamic radius alone. This indicated presence of different multimeric forms with few predominant species with some other proteoforms having different degree of multimerization. As per our knowledge, it is not uncommon for nanoparticle vaccines to exhibit some particle size heterogeneity and this does not necessarily compromise their safety or efficacy. For example, characterization of VLPs in FDA approved multivalent Human Papilloma Virus (HPV) vaccine GARDASIL® by cryo transmission electron microscopy revealed presence of VLPs with average size distribution of 54 ± 3 nm81. Furthermore, Jerajani et al. in their attempt to develop quadrivalent Multi-Dose VLP based HPV Vaccine showed formation of 35–75 nm monovalent AH-adsorbed HPV16 VLPs82. However, the self-assembly process of this nanocage is influenced by factors such as buffer composition, pH, ionic strength, and protein concentration. By carefully optimizing these parameters, we could achieve relatively uniform nanocage particle sizes. We anticipate that further refinement of process controls, in collaboration with industry partner, will help maintain consistency at scale to align with regulatory requirements, and will enable consistent maintenance of particle size distribution within a narrow and defined quality range.
Briefly, this nanocage methodology offer several advantages, including multivalent antigen display, enhanced immunogenicity, and the potential for dose sparing. They also enable geometric presentation that mimics the native viral structure, potentially improving immune recognition. However, challenges include the need for precise assembly, homogeneous preparation and stability optimization. In our study, the higher molecular weight protein Mpx-V1 (~96 kDa) was difficult to express and purify, suggesting that antigens length may be playing an important role when tethered into the NSP-10 nanocage system, while the final recombinant protein with a lower molecular weight, is more suitable for efficient production. Additionally, the final yield and purity were influenced by intrinsic antigen properties such as surface charge and assembly behavior, which played a key role in minimizing host protein contaminants, as seen with Mpx-V2. Despite these challenges, this strategy represents a novel and promising approach for vaccine development, with strong potential for further optimization to improve process efficiency and industrial scalability.
Zeng et al. have developed two multi-antigen mRNA vaccine candidates, consisting of four (M1, A29, B6, A35) or six (M1, H3L, A29, E8L, B6, A35) antigens, where the mixed MPXV multi-antigen mRNA vaccine candidates showed effective protection against VACV challenge83. Two multivalent mRNA vaccine candidates consisting of quadrivalent vaccine encoding the MPXV antigens A35, B6, M1, H3L and a trivalent vaccine (without H3L) showed complete protection from vaccinia, clade I, and clade IIb MPXV84. While previous studies, have used multi-antigen mRNA vaccines encoding up to six or multivalent MPXV antigens for protection, our study shows that a bivalent formulation with just E8L and H3L, displayed on an NSP-10 nanocage and delivered with squalene- or alum-based adjuvants, is sufficient to induce strong humoral and neutralizing antibody responses. Upon determining the Th1:Th2 ratio in the in vivo study, it was observed that both the AddaVax™-and Alhydrogel-adjuvanted Mpx-V3 vaccine exhibited a Th2-type skewness. The elevation of Th2 cells stimulates B cells to produce antibodies, crucial for neutralizing viruses and preventing their entry into host cells. The Th2-dominated immune response observed with the AddaVaxTM- and Alhydrogel-adjuvanted Mpx-V3 vaccine suggests a stronger focus on antibody production, which may be effective in generating a strong initial antibody response Further analysis of antigen-specific T cell responses, showed induction of CD4 and CD8-mediated T cell responses and secretion of cytokines IFN-γ and IL-2 which are critical for virus clearance85.
Notably, studies have reported mixed Th1/Th2 responses in similar contexts, highlighting the complexity of adjuvant effects on immune modulation85,86. Our findings indicate that while the adjuvanted antigens predominantly triggered Th2-biased responses, they also demonstrated the capacity to modulate an established Th2 response toward a more balanced immune profile by enhancing Th1-mediated cell-mediated immunity. Such a response is highly advantageous for a vaccine, as it supports the induction of both robust humoral and T cell-mediated immunity. Here, alum-adjuvanted formulation induced a robust humoral response along with evidence of Th1-type cellular responses and showed significant virus clearance in lungs and trachea as compared to the AddaVaxTM-adjuvanted formulations. Alum’s well-established safety profile, regulatory acceptability, and ability to support antibody-driven protection make it a suitable adjuvant for early-stage development for this Mpx-V3 immunogen.
Further protective efficacy study by intranasal challenge with VACV showed five out of six immunized mice were protected against the lethal virus challenge in both the adjuvanted immunized group (Fig. 4), suggesting the induction of neutralizing E8L and H3L-specific antibodies are highly protective in nature, which supports the notion of E8L and H3L are promising protective antigens and multivalent expression of these antigens might be stimulating the generation of strong and high antibody titers. While the cause of one death out of six animals could not be conclusively determined, we acknowledge that variability in individual immune responses and biological factors or the heterologous VACV used for the challenge can contribute to such outcomes. It is important to note that the remaining five mice in the group showed no signs of severe illness, and the overall protection observed in the group indicates that the immune response was effective. Moreover, in the skin lesion challenge experiment, both the adjuvanted immunized mice showed complete recovery from VACV skin scab and lesion formations.
Furthermore, the challenge study using the MPXV clade IIb A.2.1 strain (a non-lethal model) demonstrated lower body weight loss, absence of clinical signs, and a significant reduction in viral titers compared to the adjuvant-alone virus control (Fig. 6). Additionally, the immunized sera exhibited high neutralizing antibody responses against MPXV strains. Testing neutralizing antibody titers against two viruses from the same clade (Clade IIb), but sourced from different repositories, indicated that the immune response was not strain-specific within the clade. This finding supports the broader applicability and cross-strain effectiveness of the immunogen. Collectively, these findings highlight Mpx-V3 as a promising vaccine candidate with robust protective efficacy. One of the limitations of our study is the unavailability of the NHP model, for which the studies are restricted to mouse preclinical models. However, we are committed to collaborating globally to facilitate additional studies in the designated sites together with our industry partner. Future studies are planned to structurally define the Mpx-V3 antigen to understand how the antigens are displayed on the surface of NSP-10 and the distribution pattern of E8L and H3L epitopes.
Another limitation of the current study is the absence of a group immunized with the Mpx-V3 antigen alone, without adjuvant. This omission restricts our ability to fully delineate the extent of immune enhancement specifically attributable to the adjuvants. Given the focus on evaluating clinically relevant adjuvant formulations and the ethical constraints under the 3Rs (Replacement, Reduction, and Refinement) framework, the study design prioritized groups with the highest translational value. Future studies will include an antigen-alone control to better define the intrinsic immunogenicity of the antigen and more clearly assess adjuvant-dependent modulation of immune responses. A direct comparison with existing Mpox vaccines or licensed live-virus-based vaccines, such as ACAM2000 or Modified Vaccinia Ankara, would have provided valuable insights; however, this was not feasible in the current study due to the unavailability of licensed vaccines in India and the limitations of our current resources. However, the observed 5/6 survival rate against the highly lethal heterologous VACV strain and significant virus clearance, protection against the MPXV challenge is noteworthy, especially given that this is a first-generation recombinant protein subunit vaccine utilizing a nanocage-based delivery system. We acknowledge that further optimization—especially in adjuvant formulation, antigen stability, and dosing schedule—could enhance the vaccine efficacy.
Nevertheless, the study highlights a novel and potent Mpx immunogen consisting of two MPXV surface antigens E8L and H3L, both of which can be utilized to develop a highly scalable and cost-effective multivalent vaccine against MPXV. Developing a multivalent MPXV vaccine using an E. coli expression system offers significant advantages in terms of efficiency, cost-effectiveness, and scalability compared to other expression platforms. E. coli, a well-established workhorse in biotechnology, provides a robust platform for producing recombinant proteins at large scales. However, the E. coli expression system may not be suitable for antigens that are heavily glycosylated or rely on glycosylation for eliciting effective neutralizing antibody responses. Bioinformatics analysis of the E8L protein revealed the presence of only a single predicted N-glycosylation site at position N29 (data not shown), suggesting that N-glycosylation may not be critical for eliciting protective immune responses. This supports the suitability of the bacterial expression system for producing this immunogen.
Taken together, this approach offers a highly scalable, thermostable and cost-effective platform for MPXV vaccine development, simplifying distribution logistics and enhancing accessibility and efficacy across diverse geographic and environmental settings.
Materials and methods
Materials
The reagents and tools utilized in this study included a comprehensive range of antibodies, viral and bacterial strains, chemical reagents, cell lines, commercial kits, and software essential for analysis. Various subclasses of anti-mouse immunoglobulin G (IgG) antibodies, including IgG1, IgG2a, IgG2b, IgG2c, and IgG3, were procured from Jackson ImmunoResearch. Specific monoclonal antibodies such as E8L and H3L were obtained from Abexxa Ltd, while fluorescently labeled antibodies, including Alexa Fluor 488 Rat anti-mouse CD8a, PE Rat anti-mouse CD4, APC-CY7 Rat anti-mouse IFN-γ, and PE-CY7 Rat anti-mouse IL-2 were sourced from BD Pharmigen™. Viral strains, including the VACV (NR-56, BEI Resources, NIH) and two MPXV strains (MCL-22-H-5317 from ICMR-NIV Pune and NR-27 from BEI Resources, NIH), were used, along with the E. coli BL-21 strain for recombinant protein expression and the pET28b expression vector from Thermo Fisher. Adjuvants such as Addavax™ (Invivogen) and Alhydrogel (Superfos) were employed in immunization protocols. The following reagent was obtained through BEI Resources, NIAID, NIH: MPXV, WRAIR 7-61, NR-27. Each vial contains ~1 mL of cell lysate and supernatant from African green monkey kidney cells (Vero; ATCC ® CCL-81) infected with Mpox virus, WRAIR 7-61. NR-56, BEI resources VACV, WR was derived from the original New York City Board of Health (NYCBH) strain by intracerebral passages in mice. VACV, WR (mouse adapted) was deposited at ATCC ® as VR-119™ in 1949 by Walter Reed Army Institute of Research (WRAIR). Additional chemicals included TMB (Sigma), PBS (Sisco Research Laboratories), sulfuric acid (Merck), and formaldehyde (Merck). Vero and Vero E6 cell lines were obtained from ATCC for virus propagation and in vitro assays. For protein purification and cytokine analysis, the HiTrap Q FF and Superdex 200 Increase columns (Cytiva), along with the BD Biosciences Th1/Th2 Cytokine Bead Array (CBA) kit, were utilized. Experimental mice of the BALB/c strain were procured from the Small Animal Facility. Data analysis and visualization were conducted using GraphPad Prism (v9.0), FlowJo (v9.0), Unicorn (v7.1), and Spectra Manager (Jasco). These materials formed the foundation for conducting immunological, virological, and biochemical experiments throughout the study.
Cloning, expression, and cell lysis
The construct Mpx-V1 was designed by fusing B6R, A29L at the N terminus and M1R, A35R at the C terminus of NSP-10, whereas construct Mpx-V2 was designed by fusing B6R site at the N terminus and A35R site at C terminus of NSP-10. For Mpx-V3, the synthetic gene coding for E8L and H3L was fused to the NSP-10 nanocage backbone at the N terminus and C terminus, respectively, using glycine-serine linker combinations. The Mpox antigens (Mpx-V1, V2, and V3) in this study were derived from MPXV, complete genome NCBI Reference Sequence: NC_063383.1. The designed constructs were codon optimized and commercially cloned in a pET28b (+) vector, Thermo Fisher Scientific, USA. The vector was transformed into competent BL-21 E. coli cells. About 1–3 µL of 100 ng plasmid was mixed with 100 μL of freshly thawed competent cells and incubated for 30 min on ice. The mixture was subjected to heat shock at 42 °C for 15–30 s, and an appropriate dilution was spread on LB agar from which a single colony was screened and processed further. E. coli cells were cultured in Luria-Bertani (LB) media, and induction was carried out for 4 h (1 mM IPTG). The cell pellet was lysed in lysis buffer (50 mM Tris-HCl, pH 8.5, 100 mM NaCl, 5 mM EDTA) and homogenized for 10 min at 8000 × g followed by sonication for 10 cycles at 50% amplitude (QSonica, Q700). The lysate was then centrifuged at 12,000 × g, 30 min, 4 °C (Sorvall RC 6+, USA). The pellet containing the inclusion bodies was further processed for inclusion body isolation.
Mini-Scale (5 L batch) fermentation using Eppendorf DASGIP Bioblock for microbial culture system
Mini-scale fermentation was performed in a 5 L Eppendorf fermenter at VDDC lab, THSTI, using a chemically defined (CD) medium41,77. An overnight pre-culture was prepared by inoculating 100 mL of CD medium supplemented with kanamycin and incubated at 37 °C with agitation. This culture was subsequently transferred to a 500 mL Erlenmeyer flask containing fresh CD medium and grown until the optical density at 600 nm (OD600) reached ~3.0. The culture was then used to inoculate 2.8 L of CD medium in a 5 L fermenter, which had been previously sterilized at 121 °C for 20 min and cooled to 37 °C prior to inoculation. During fermentation, critical process parameters, including OD600, pH, aeration, antifoam addition, carbon source concentration, and agitation speed, were continuously monitored. The fermentation was initiated in batch mode, and dissolved oxygen (DO) levels were maintained at desired setpoints using a cascade control system that regulated the stirrer speed in response to DO concentration. Foam formation was suppressed by the addition of a 10% antifoam solution (Sigma-Aldrich), and the cultivation temperature was maintained at 37 °C. pH was regulated at 6.8 using a standard pH electrode (Mettler Toledo), with 5 M NaOH added as necessary for pH control. Prior to transitioning to fed-batch mode, DO-stat control was employed to determine the appropriate timing and volume of feed additions. A calibrated peristaltic pump was used to deliver feed media at controlled rates. The feeding strategy included: feed Medium 1: 50% (w/v) glucose, 30% (w/v) yeast extract, and 2.4 g MgSO4 per 100 mL. Feed Medium 2: A buffer solution comprising K2HPO4, KH2PO4, Na2HPO4·12H2O, (NH4)2SO4, NH4Cl, and trace elements. Feed rates were adjusted based on the metabolic activity of the culture. Hourly sampling was conducted throughout the fermentation process, with OD600 measurements used to monitor cell growth. When the culture reached the late logarithmic phase (OD600 ≈ 45), protein expression was induced by the addition of 1 mM isopropyl β-D-1-thiogalactopyranoside (IPTG). Following 4 h of induction, the culture was harvested at an OD600 of 74. Cell harvesting was carried out by centrifugation at 10,000 rpm for 20 min at 4 °C using a Thermo Scientific centrifuge. A total of 349 g of E. coli cell biomass was recovered from the 5 L fermentation.
Isolation and purification of Mpx-V3 from inclusion bodies
The inclusion bodies were washed with a combination of ionic (1% sodium deoxycholate) and non-ionic (1% Triton-X-100) detergents to remove the contaminants. The washed inclusion bodies were solubilized in mild conditions utilizing a low concentration of urea (~2 M), followed by pulsatile refolding in ice-cold refolding buffer 20 mM Tris pH 8.5 with 5% sucrose as a stabilizer for 12–16 h. The refolded Mpx-V3 was then subjected to Anion Exchange chromatography in flow-through mode. The column was equilibrated with 5 column volume (CV) of refolding buffer (20 mM Tris, 5% sucrose) and loaded with Mpx-V3. Elution was carried out with a gradient of 0–1 M NaCl spanning 20 CV (flow rate of 0.500 mL/min). The eluted protein was subjected to gel filtration chromatography (Superdex 200 Increase). The column was pre-equilibrated with refolding buffer, and Mpx-V3 was injected. Elution was carried out for 1.5 CV (flow rate of 0.750 mL/min). We also run the calibration standard of Blue Dextran (Mr 2000,000), Thyroglobulin (Mr 669,000), Ferritin (Mr 440,000), Aldolase (Mr 158,000), Conalbumin (Mr 75,000), Ovalbumin (Mr 44,000), Carbonic Anhydrase (Mr 29,000), and Ribonuclease A (Mr 137000) at the concentration of 1.5 mg (Thyroglobulin, Aldolase, Conalbumin, Ovalbumin, and Ribonuclease A) and 0.2 mg (Ferritin and Carbonic Anhydrase) at the flow rate of 0.5 mL/min to evaluate the performance of Superdex 200 column. To further refine the elution profile of the synthesized Mpx-V3 immunogen, we used a Tskgel G4000PW 500 Å column, which has a more defined pore size (50 nm) with all the remaining experimental conditions as same as for the Superdex 200 column. The reference Blue Dextran was also run on the same column to verify the void volume using the same mobile phase and experimental conditions. The UNICORN 7.0 software was used to interpret the results. The NSP-10 obtained from BEI Resources, USA, was also purified similarly (data not shown). The following reagent was obtained through BEI Resources, NIAID, NIH: Vector pET-28a (+) Containing the SARS-Related Coronavirus 2, Wuhan-Hu-1 Non-Structural Protein-10 Gene, NR-53502.
Biophysical and biochemical characterization of Mpx-V3
Mpx-V3 was characterized for size and zeta potential by DLS through Malvern Zetasizer UK (Nano ZS). Briefly, 1 mg/mL of Mpx-V3 protein was prepared in the subsequent buffer and sonicated for 5–10 min before measurement87,88. The prepared samples were then monitored for hydrodynamic size and zeta potential in a triplicate manner, and the observations were interpreted as Mean ± SD. The effect of temperature and pH fluctuation was also observed on the long-term stability of the Mpx-V3 immunogen by measuring the surface Zeta Potential charge. To evaluate the temperature influence, the Mpx-V3 stored at two different storage temperatures (4 °C and −20 °C) was used at a concentration of 1 mg/mL to determine the Zeta Potential. The intrinsic tryptophan fluorescence maxima were estimated by fluorescence spectroscopy. The fluorescence of Mpx-V3 was measured using a Varian Fluorescence Spectrophotometer. The emission spectrum of Mpx-V3 (1 mg/mL) was acquired at 290 to 450 nm by excitation at 280 nm wavelength. The temperature was maintained at 4 °C by the Peltier system. Next, the secondary structure was estimated by Far-UV Circular Dichroism. A JASCO J-815 spectrometer (Japan) was used to acquire the Far-UV spectrum of Mpx-V3. The spectrum was acquired at a bandwidth of 1 nm with a step size of 1 nm. Spectra Manager software was used to analyze the spectrum with Yang’s reference spectra and estimate the secondary structure.
For room temperature (RT) negative staining-TEM (Ns-TEM), 3ul Mpx-V3 sample was added on 300-mesh Cu TEM grids (Ted Pella, Inc.), which were glow-discharged (negative polarity) for 30 s in a PELCO easiGlowTM Glow-Discharge Cleaning System. The sample was incubated at RT for 1 min. The excess sample solution was carefully blotted off using Whatman Filter paper. This was followed by touching the grid to a drop of 1% freshly prepared uranyl-acetate solution and blotting the excess liquid. Ns-TEM data were acquired on a 120 kV Talos L120C RT electron microscope with a bottom-mounted Ceta camera (4k × 4k) at magnification 57KX. Fifty-seven micrographs were collected, and 87000 particles were picked by LoG-based auto-picking in Relion 4.089, followed by multiple rounds of 2D classification. Due to the heterogeneity in the sample, confident 3D structure determination was not possible. To further analyze the data, we predicted the individual antigen structures, H3L using Alpha-Fold software90 and E8L via the Uniprot database, respectively. The monomeric Mpx-V3 fusion protein structure was then used to model the dodecameric structure based on the known NSP-10 dodecamer structure (PDB ID: 2G9T) with minimum clashes. Using UCSF ChimeraX91, a molmap of the predicted dodecameric Mpx-V3 structure was generated at 20 Å resolution. These models were used for size and shape comparisons with the 2D classes obtained from the Ns-TEM data. 2D classes consisting of particles with sizes close to 18 nm and had an overall globular shape, thus, estimated to be dodecameric Mpx-V3 particles, which comprised 29.15% (calculated from 2D class averages) of total particles from the Ns-TEM images.
Western blotting was performed against anti-NSP-10 sera. The purified refolded Mpx-V3 (5 µg) was resolved on 12% SDS-PAGE along with a pre-stained marker and transferred onto a polyvinylidene fluoride (PVDF) membrane. The membrane was blocked in 3% BSA at 4 °C overnight. The membrane was probed with anti-E8L (1:1000) (#abx376565), anti-H3L (1:500) (#abx376562, Abbexa Ltd.) monoclonal antibodies, and anti-NSP-10 mouse polyclonal antibody (1:500) at 37 °C for 2 h. Next, the blot was probed with anti-mouse HRP-conjugated secondary antibody (1:2000), and the membrane was washed with PBST twice and a final wash with PBS. The signal was developed using an ECL reagent (Bio-Rad Laboratories, USA).
Ethics statement, mice immunization, and intranasal virus challenge
The guidelines by the Committee for the Control and Supervision of Experiments on Animals (New Delhi) were followed faithfully throughout the study. The protocol of the present study was approved by the Institutional Animal Ethics Committee (IAEC) of Translational Health Science and Technology Institute, Faridabad, India (IAEC approval no: IAEC/THSTI/226 for VACV and IAEC/THSTI/ 343 for MPXV). All of the animals were housed in climate- and photoperiod-controlled rooms, supplemented with conventional rodent pellets, and had free access to water. The temperature and humidity were kept at 23 ± 2 °C and 60%, respectively. The study adhered to the ARRIVE guidelines. The study involved female BALB/c mice in five distinct animal experiments were conducted to evaluate the cellular and humoral immune responses, as well as the protective efficacy, of the Mpx-V3 immunogen formulated with two different adjuvants. The investigator was not blinded to the treatment groups; however, the immunization, challenge, and surgical teams remained blinded throughout the procedures. These procedures included the intramuscular administration of the test agents, the collection of blood and spleen samples from mice, and the challenge conducted via intranasal (IN) and scarification methods. For blood collection, mice were first anaesthetized using Ketamine/Xylazine solution (50 mg/kg Ketamine and 5 mg/kg Xylazine) prepared in PBS and injected 0.5 mL via IP route in the lower abdominal area and then 200 μL blood was collected from un-conscious mice via lateral tail vein (on study day 0, day 14 post-prime, day 14 post-boost) from all animals under sedation. The serum was separated by centrifugation at 3000 rpm for 20 min, and it was stored at −80 °C until needed. In all challenge studies, mice were anaesthetized with vapor isoflurane, and then an intranasal infection of the virus was given. For spleen, lung, and trachea collection, mice were euthanized with CO2, and every effort was made to minimize suffering.
In experiment 1, the immunogenicity of Mpx-V3 immunogen was evaluated; for this, female BALB/c mice (20–25 g) of 6–8 weeks of age were used for immunization with immunogen Mpx-V3 prepared with AddaVaxTM or Alhydrogel. Mice were grouped into five groups having six animals in each group and not having a collective weight difference of ±2 g. The five groups were Mpx-V3+Alhydrolgel, Alhydrogel, Mpx-V3+AddaVaxTM, AddaVaxTM, and naïve control. Mice were immunized following a one prime–one boost regimen, and 14 days after every immunization, the blood sample was collected for serum harvesting and later estimated for antibody titers, virus neutralization potential, and cytokine levels in the serum. In the second experiment, 6–8 weeks old female BALB/c mice were similarly immunized with Mpx-V3 antigens with AddaVaxTM or Alhydrogel (n = 3 per group), and 2 weeks after boost, mice were euthanized for the collection of splenic T cells for studying the T cell-mediated immune response.
For the third animal experiment dealing with the VACV challenge study, the same immunization protocol was repeated in 40 (n = 8/group) mice divided into five groups (Fig. 4a). Three weeks after the boost dose, mice were intranasally challenged with 50 μL of 1 × 107 PFU/mL of VACV. Mice were regularly monitored for the development of any clinical signs of VACV infection, weight loss, mortality, and changes in rectal temperature. On the 5th dpi, two mice from each group were euthanized, and organs (lungs, trachea, liver, and spleen) were harvested after extensive perfusion with PBS for determination of the viral titers and histopathological analysis.
The fourth animal experiment involved the assessment of the protective efficacy of Mpx-V3 antigens against the VACV via the tail scarification technique. Thirty-one female BALB/c mice (n = 7 in adjuvanted Mpx-V3 groups, n = 6 in adjuvant control groups, and n = 5 in naïve control group, total groups—5) aged 6–7 weeks (weighed around 18–22 g) were vaccinated by the tail scarification technique92,93. Briefly, the mice from all the groups were anaesthetized and placed on a flat surface and a droplet of 10 μL of virus inoculum (1 × 107 PFU/mL) was deposited on the dorsal base of the tail (1 cm from the base of the tail) then an upper layer of the tail was scratched with a needle (25 G × 0.5 in.) through the droplet 20–25 times along 1 cm towards the tip of the tail. Following the tail scarification, the mice were observed for 21 days for the development of tail lesions and scabs. Apart from the skin lesions and scabs, the mice were also observed for the appearance of any significant clinical signs and morbidity.
In the fifth animal experiment, the protective efficacy of Mpx-V3 was evaluated upon homologous challenge with the MPXV. We immunized 6–8-week-old mice (n = 7 per group, a total of five groups) with 25 μg of Mpx-V3 along with AddaVaxTM or Alhydrogel. In a prime-boost strategy, mice were immunized via intramuscular immunizations (Fig. 6a). 21 days after the booster dose, mice were shifted to THSTI’s ABSL3 facility and challenged via an intranasal route with 50 μL of 1 × 106 PFUs/mL of MPXV (clade IIb A.2.1, GISAID ID: EPI_ISL_159123322), and animals were monitored closely for 14 dpi. The blood sample was collected 2 weeks post every immunization, and 6th day post-viral challenge, lungs and trachea were harvested to determine the viral load and histopathology.
Th1/Th2 response and antibody titers estimation
Th1/Th2 cytokine levels were measured in the serum collected from mice immunized with Mpx-V3 with adjuvants (AddaVaxTM or Alhydrogel) after the booster dose (14 days) using a mouse Th1/Th2 CBA kit (#560485, BD Biosciences, USA). The standard dilutions of cytokines and test samples were incubated with cytokine capture beads and PE-detection reagent at room temperature for 3 h in the dark conditions. After the incubation, beads were washed twice with the wash buffer and resuspended in the wash buffer for data acquisition using BD FACS Canto II Flow Cytometer (BD Biosciences, CA, USA)94. Instrument setting for sample acquisition was done prior to the experiment as per the manufacturer’s instructions. The data analysis was performed using the software FCAP Array, and graphs were plotted on GraphPad Prism.
For antibody titers, indirect ELISA was used. Mpx-V3 and NSP-10 protein in carbonate buffer was coated in an ELISA plate at a concentration of 1 µg/mL overnight. The plates were blocked with 5% skimmed milk for 1 h at room temperature, and after incubation plates were washed with 0.05% PBS-Tween-20 wash buffer and probed with test serum samples (starting with 1:50 and triple dilution for later wells) as primary antibodies. After incubation, plates were washed thrice with PBS-T-20 wash buffer and probed with anti-mouse HRP-tagged secondary antibody (1:2000), followed by 1 h incubation. For IgG isotyping, in another round of experiments, secondary antibodies against IgG1(#115-035-205), IgG2a (#115-035-206), IgG2b (#115-035-207), and IgG3 (#115-035-209) were used as secondary antibodies. After incubation, the plates were washed and developed with tetramethylbenzidine (TMB) (#002023, Sigma-Aldrich, India) substrate, and the reaction was allowed to develop for 5-min and then stopped with 1 N sulfuric acid. The absorbance of plates was recorded at 450 nm on an ELISA reader (Bio-Rad). The Th1 to Th2 ratio was calculated by dividing the IgG1 endpoint titers by the IgG2a endpoint titers. The endpoint titers were determined for IgG and each subtype, which is the reciprocal of the highest test serum dilution giving a reading above the cut-off. In each assay, the sera from PBS-treated mice were considered as a negative control to calculate the cut-off value. The cut-off value was calculated by multiplying the average OD value of negative control samples + 3 times the SD of the OD value of negative control samples. The test result was considered positive when the signal-to-cut-off ratio was ≥1.
Measurement of splenic T cell response against the administration of the Mpx-V3 antigen
Briefly, the single-cell suspensions of splenocytes from the Mpx-V3 immunized or naïve control mice after the 3 weeks of the booster dose were prepared in RPMI-1640 medium and collected by centrifugation for 5 min at 300 × g. The splenocytes from each group were seeded in the 96-well plates and stimulated with E8L (abx376565) and H3L (abx376562) proteins for 3–4 h in a CO2 incubator. After incubation, the splenocytes were washed with FACS buffer and incubated with the fluorescently labeled extracellular marker (anti-mouse CD4; BD PharmigenTM, 561832 and anti-mouse CD8a; PharmigenTM, 557668 antibodies) on ice for 30 min. Once the extracellular staining was done, splenocytes were fixed and permeabilized for the intracellular staining with anti-mouse IL-2 (PharmigenTM; 560538) and IFN-y (PharmigenTM; 561479) antibodies. Finally, the splenocytes were washed twice with FACS buffer and fixed in 1% paraformaldehyde. The samples were acquired in a FACSCantoTM II (BD Biosciences) flow cytometry system, and the analysis was performed using FlowJo software (BD Biosciences, Version 9.0).
Histopathology
Histopathology assessment was done for two of the challenge studies. For the histopathology of both the challenge studies (IN challenge with VACV and MPXV), the organs were collected after extensive perfusion with 1× PBS, fixed in 10% formaldehyde and the samples were further processed at the Institute of Liver and Biliary Sciences, New Delhi, India for mounting, sectioning, and H&E staining. The histopathological scoring of the organs harvested from the Mpx-V3 immunized and non-immunized mice was assessed for each sample section by a trained pathologist at The Institute of Liver and Biliary Sciences (ILBS), Delhi, India. Similarly, for the MPXV challenge study, the samples were processed and scored at the THSTI histopathology facility.
Statistical analysis
Results are presented as mean ± SD, and differences between two or multiple groups were analyzed for statistical significance by Student’s t-test, or one-way ANOVA followed by Dunnett’s test or Tukey’s post-test. It was followed by post hoc tests inbuilt in GraphPad Prism 9 (GraphPad Software Inc., San Diego, CA, USA) to test for normal distribution of the samples by the Shapiro-Wilk normality test. Statistical analysis was performed where *p < 0.05, **p < 0.01, ***p < 0.001, and ****p < 0.0001 were considered significant.
Data availability
All data associated with this study are present in the paper or the supplementary figures and will be provided by the corresponding author upon request.
References
Bankar, N. J. et al. Emerging and re-emerging viral infections: An Indian perspective. Cureus 14, e30062 (2022).
Besombes, C. et al. Seasonal patterns of Mpox Index Cases, Africa, 1970-2021. Emerg. Infect. Dis. 30, 1017–1021 (2024).
Begum, J. P. S. et al. Emergence of monkeypox: a worldwide public health crisis. Hum. Cell 36, 877–893 (2023).
2022 Mpox Outbreak Global Map. Mpox, Poxvirus, CDC. https://www.cdc.gov/poxvirus/mpox/response/2022/world-map.html.
Kibungu, E. M. et al. Clade I-associated mpox cases associated with sexual contact, the Democratic Republic of the Congo. Emerg. Infect. Dis. 30, 172–176 (2024).
Nachega, J. B. et al. The surge of mpox in Africa: a call for action. Lancet Glob. Health https://doi.org/10.1016/S2214-109X(24)00187-6 (2024).
Moss, B. Poxvirus DNA replication. Cold Spring Harb. Perspect. Biol. 5, a010199 (2013).
Fields virology. WorldCat.org. https://search.worldcat.org/title/Fields-virology/oclc/71812790.
Shchelkunov, S. N. et al. Human monkeypox and smallpox viruses: genomic comparison. FEBS Lett. 509, 66–70 (2001).
Sammartino, J. C. et al. Characterization of immune response against monkeypox virus in cohorts of infected patients, historic and newly vaccinated subjects. J. Med. Virol. 95, e28778 (2023).
Harrison, C. Monkeypox response relies on three vaccine suppliers. Nat. Biotechnol. 40, 1306–1307 (2022).
Chiuppesi, F. et al. Synthetic modified vaccinia Ankara vaccines confer cross-reactive and protective immunity against mpox virus. Commun. Med. 4, 19 (2024).
Freyn, A. W. et al. An mpox virus mRNA-lipid nanoparticle vaccine confers protection against lethal orthopoxviral challenge. Sci. Transl. Med. 15, eadg3540 (2023).
Jacob-Dolan, C. et al. Comparison of the immunogenicity and protective efficacy of ACAM2000, MVA, and vectored subunit vaccines for Mpox in rhesus macaques. Sci. Transl. Med. 16, eadl4317 (2024).
Alakunle, E. et al. A comprehensive review of monkeypox virus and mpox characteristics. Front. Cell. Infect. Microbiol. 14, 1360586 (2024).
Monkeypox: experts give virus variants new names. https://www.who.int/news/item/12-08-2022-monkeypox--experts-give-virus-variants-new-names.
Yu, X., Shi, H. & Cheng, G. Mpox virus: its molecular evolution and potential impact on viral epidemiology. Viruses 15, 995 (2023).
Lu, J. et al. Mpox (formerly monkeypox): pathogenesis, prevention, and treatment. Signal Transduct. Target Ther. 8, 458 (2023).
Garcia-Atutxa, I., Mondragon-Teran, P., Huerta-Saquero, A. & Villanueva-Flores, F. Advancements in monkeypox vaccines development: a critical review of emerging technologies. Front. Immunol. 15, (2024).
Wang, Y., Yang, K. & Zhou, H. Immunogenic proteins and potential delivery platforms for mpox virus vaccine development: a rapid review. Int. J. Biol. Macromol. 245, 125515 (2023).
Hirao, L. A. et al. Multivalent smallpox DNA vaccine delivered by intradermal electroporation drives protective immunity in nonhuman primates against lethal monkeypox challenge. J. Infect. Dis. 203, 95–102 (2011).
Meseda, C. A. et al. Enhanced immunogenicity and protective effect conferred by vaccination with combinations of modified vaccinia virus Ankara and licensed smallpox vaccine Dryvax in a mouse model. Virology 339, 164–175 (2005).
Hooper, J. W. et al. Molecular smallpox vaccine delivered by alphavirus replicons elicits protective immunity in mice and non-human primates. Vaccine 28, 494 (2009).
Hooper, J. W., Custer, D. M. & Thompson, E. Four-gene-combination DNA vaccine protects mice against a lethal vaccinia virus challenge and elicits appropriate antibody responses in nonhuman primates. Virology 306, 181 (2003).
Kasai, N., Sakamoto, A. & Uchida, M. A combined modality for anaplastic large-cell carcinoma of the thyroid. Auris Nasus Larynx 12, S72–S74 (1985).
Sang, Y. et al. Monkeypox virus quadrivalent mRNA vaccine induces immune response and protects against vaccinia virus. Signal Transduct. Target Ther. 8, 172 (2023).
Chu, Q. et al. Non-replicating vaccinia virus NTV as an effective next-generation smallpox and monkeypox vaccine: evidence from mouse and rhesus monkey models. Emerg. Microbes Infect. 12, 2278900 (2023).
Jacobs, B. L. et al. Vaccinia virus vaccines: past, present and future. Antivir. Res. 84, 1–13 (2009).
Moss, B. Smallpox vaccines: targets of protective immunity. Immunol. Rev. 239, 8–26 (2011).
Su, D. et al. Dodecamer structure of severe acute respiratory syndrome coronavirus nonstructural protein nsp10. J. Virol. 80, 7902–7908 (2006).
Nguyen, B. & Tolia, N. H. Protein-based antigen presentation platforms for nanoparticle vaccines. npj Vaccines 6, 70 (2021).
Krachmarova, E., Ivanov, I. & Nacheva, G. Nucleic acids in inclusion bodies obtained from E. coli cells expressing human interferon-gamma. Microb. Cell Factories 19, 1–9 (2020).
Singh, S. M. & Panda, A. K. Solubilization and refolding of bacterial inclusion body proteins. J. Biosci. Bioeng. 99, 303–310 (2005).
Maachupalli-Reddy, J., Kelley, B. D. & De Bernardez Clark, E. Effect of inclusion body contaminants on the oxidative renaturation of hen egg white lysozyme. Biotechnol. Prog. 13, 144–150 (1997).
Castellanos-Serra, L. R. et al. Expression and folding of an interleukin-2-proinsulin fusion protein and its conversion into insulin by a single step enzymatic removal of the C-peptide and the N-terminal fused sequence. FEBS Lett. 378, 171–176 (1996).
Marston, F. A. The purification of eukaryotic polypeptides synthesized in Escherichia coli. Biochem J. 240, 1–12 (1986).
Kane, J. F. & Hartley, D. L. Formation of recombinant protein inclusion bodies in Escherichia coli. Trends Biotechnol. 6, 95–101 (1988).
Schein, C. H. Production of soluble recombinant proteins in bacteria. Nat. Biotechnol. 7, 1141–1149 (1989).
De Marco, A. et al. Bacterial inclusion bodies are industrially exploitable amyloids. FEMS Microbiol. Rev. 43, 53–72 (2019).
Ganjave, S. D., Dodia, H., Sunder, A. V., Madhu, S. & Wangikar, P. P. High cell density cultivation of E. coli in shake flasks for the production of recombinant proteins. Biotechnol. Rep. 33, e00694 (2022).
Kangwa, M. et al. High-level fed-batch fermentative expression of an engineered Staphylococcal protein A based ligand in E. coli: purification and characterization. AMB Express 5, 1–10 (2015).
Honary, S. & Zahir, F. Effect of zeta potential on the properties of nano-drug delivery systems—a review (Part 1). Trop. J. Pharm. Res. 12, 255–264 (2013).
Akbarian, M. & Chen, S. H. Instability challenges and stabilization strategies of pharmaceutical proteins. Pharmaceutics 14, 2533 (2022).
Németh, Z. et al. Quality by design-driven zeta potential optimisation study of liposomes with charge imparting membrane additives. Pharmaceutics 14, 1798 (2022).
Obata, T. et al. Effect of storage temperature on the dispersibility of commercially available 0.1% fluorometholone ophthalmic suspension. PLoS ONE 17, e0277311 (2022).
Lee, J. et al. Combined effects of zeta-potential and temperature of nanopores on diffusioosmotic ion transport. Anal. Chem. 93, 14169–14177 (2021).
Szabó, T. et al. The effect of lipopolysaccharides from Salmonella enterica on the size, density, and compressibility of phospholipid vesicles. Biomimetics (Basel) 10, 55 (2025).
Lakowicz, J. R. Principles of Fluorescence Spectroscopy 1–954. https://doi.org/10.1007/978-0-387-46312-4/COVER (2006).
Sharon, M. et al. Tail scarification with Vaccinia virus Lister as a model for evaluation of smallpox vaccine potency in mice. Vaccine 25, 7743–7753 (2007).
Falendysz, E. A., Lopera, J. G., Rocke, T. E. & Osorio, J. E. Monkeypox virus in animals: current knowledge of viral transmission and pathogenesis in wild animal reservoirs and captive animal models. Viruses 15, 905 (2023).
Bouvet, M. et al. Coronavirus Nsp10, a critical co-factor for activation of multiple replicative enzymes. J. Biol. Chem. 289, 25783–25796 (2014).
Sawicki, S. G. et al. Functional and genetic analysis of coronavirus replicase-transcriptase proteins. PLoS Pathog. 1, 0310–0322 (2005).
Carter, D. C., Wright, B., Jerome, W. G., Rose, J. P. & Wilson, E. A unique protein self-assembling nanoparticle with significant advantages in vaccine development and production. J. Nanomater. 2020, 4297937 (2020).
Wang, X., Gu, Z., Sheng, S., Song, R. & Jin, R. The current state and progress of Mpox vaccine research. China CDC Wkly 6, 118–125 (2024).
Shen-Gunther, J., Cai, H. & Wang, Y. Genomic and antigenic differences between monkeypox virus and vaccinia vaccines: insights and implications for vaccinology. Int. J. Mol. Sci. 26, 1428 (2025).
Kupritz, J., Pahwa, S. & Pallikkuth, S. Serosurvey of immunity to monkeypox (Mpox) virus antigens in people living with HIV in South Florida. Pathogens 12, 1355 (2023).
Valax, P. & Georgiou, G. Molecular characterization of beta-lactamase inclusion bodies produced in Escherichia coli. 1. Composition. Biotechnol. Prog. 9, 539–547 (1993).
Rudolph, R., Lilie, H. & Schwarz, E. In vitro folding of inclusion body proteins on an industrial scale. Biotechnology 5, 110–123 (2008).
Rudolph, R. & Lilie, H. In vitro folding of inclusion body proteins. FASEB J. 10, 49–56 (1996).
Walsh, G. Biopharmaceutical benchmarks 2018. Nat. Biotechnol. 36, 1136–1145 (2018).
Buscajoni, L., Martinetz, M. C., Berkemeyer, M. & Brocard, C. Refolding in the modern biopharmaceutical industry. Biotechnol. Adv. 61, 108050 (2022).
Williams, D. C., Van Frank, R. M., Muth, W. L. & Burnett, J. P. Cytoplasmic inclusion bodies in Escherichia coli producing biosynthetic human insulin proteins. Science 215, 687–689 (1982).
Mehta, D., Chirmade, T., Tungekar, A. A., Gani, K. & Bhambure, R. Cloning and expression of antibody fragment (Fab) I: effect of expression construct and induction strategies on light and heavy chain gene expression. Biochem. Eng. J. 176, 108189 (2021).
Chen, Y. C., Lin, W. T., Wu, J. W. & Liu, H. S. Efficient lysozyme refolding at a high final concentration and a low dilution factor. Process Biochem. 47, 1883–1888 (2012).
Kang-Pettinger, T. et al. Identification, binding, and structural characterization of single domain anti-PD-L1 antibodies inhibitory of immune regulatory proteins PD-1 and CD80. J. Biol. Chem. 299, 102769 (2023).
Zhansaya, A. et al. Recombinant expression and purification of extracellular domain of the programmed cell death protein receptor. Rep. Biochem. Mol. Biol. 8, 347 (2020).
Valente, C. A., Monteiro, G. A., Cabral, J. M. S., Fevereiro, M. & Prazeres, D. M. F. Optimization of the primary recovery of human interferon alpha2b from Escherichia coli inclusion bodies. Protein Expr. Purif. 45, 226–234 (2006).
Mousavi, S. B. & Davarpanah, S. J. Solvent extraction of recombinant interferon alpha-2b from inclusion bodies and efficient refolding at high protein concentrations. Protein Expr. Purif. 197, 106110 (2022).
Riggs, A. D. Bacterial production of human insulin. Diabetes Care 4, 64–68 (1981).
Gani, K., Bhambure, R., Deulgaonkar, P., Mehta, D. & Kamble, M. Understanding unfolding and refolding of the antibody fragment (Fab). I. In-vitro study. Biochem. Eng. J. 164, 107764 (2020).
Rathore, A. S., Agarwal, H., Sharma, A. K., Pathak, M. & Muthukumar, S. Continuous processing for production of biopharmaceuticals. Prep. Biochem.Biotechnol. 45, 836–849 (2015).
Oberg, K., Chrunyk, B. A., Wetzel, R. & Fink, A. L. Nativelike secondary structure in interleukin-1 beta inclusion bodies by attenuated total reflectance FTIR. Biochemistry 33, 2628–2634 (1994).
García-Fruitós, E. et al. Aggregation as bacterial inclusion bodies does not imply inactivation of enzymes and fluorescent proteins. Microb. Cell Factories4, 1–6 (2005).
Ventura, S. & Villaverde, A. Protein quality in bacterial inclusion bodies. Trends Biotechnol. 24, 179–185 (2006).
Lamm, R. et al. Detailed small-scale characterization and scale-up of active YFP inclusion body production with Escherichia coli induced by a tetrameric coiled coil domain. J. Biosci. Bioeng. 129, 730–740 (2020).
Chen, H. et al. Purification of inclusion bodies using PEG precipitation under denaturing conditions to produce recombinant therapeutic proteins from Escherichia coli. Appl. Microbiol. Biotechnol. 101, 5267–5278 (2017).
Panda, A. K., Khan, R. H., Appa, Rao, K. B. C. & Totey, S. M. Kinetics of inclusion body production in batch and high cell density fed-batch culture of Escherichia coli expressing ovine growth hormone. J. Biotechnol. 75, 161–172 (1999).
Yousaf, M., Ismail, S., Ullah, A. & Bibi, S. Immuno-informatics profiling of monkeypox virus cell surface binding protein for designing a next generation multi-valent peptide-based vaccine. Front. Immunol. 13, 1035924 (2022).
Bhattacharya, M., Chatterjee, S., Nag, S., Dhama, K. & Chakraborty, C. Designing, characterization, and immune stimulation of a novel multi-epitopic peptide-based potential vaccine candidate against monkeypox virus through screening its whole genome encoded proteins: an immunoinformatics approach. Travel Med. Infect. Dis. 50, 102481 (2022).
Waqas, M. et al. Immunoinformatics design of multivalent epitope vaccine against monkeypox virus and its variants using membrane-bound, enveloped, and extracellular proteins as targets. Front. Immunol. 14, 1091941 (2023).
Zhao, Q. et al. Characterization of virus-like particles in GARDASIL® by cryo transmission electron microscopy. Hum. Vaccines Immunother. 10, 734–739 (2014).
Jerajani, K. et al. Analytical and preformulation characterization studies of human papillomavirus virus-like particles to enable quadrivalent multi-dose vaccine formulation development. J. Pharm. Sci. 111, 2983–2997 (2022).
Zeng, J. et al. Mpox multi-antigen mRNA vaccine candidates by a simplified manufacturing strategy afford efficient protection against lethal orthopoxvirus challenge. Emerg. Microbes Infect. 12, 2204151 (2023).
Zuiani, A. et al. A multivalent mRNA monkeypox virus vaccine (BNT166) protects mice and macaques from orthopoxvirus disease. Cell 187, 1363–1373.e12 (2024).
Grigoryan, L. et al. Adjuvanting a subunit SARS-CoV-2 vaccine with clinically relevant adjuvants induces durable protection in mice. npj Vaccines 7, 55 (2022).
Yadav, N. et al. Comparative immunogenicity analysis of intradermal versus intramuscular administration of SARS-CoV-2 RBD epitope peptide-based immunogen in vivo. Microbes Infect. 23, 104843 (2021).
Kumar, V., Kumar, R., Jain, V. K. & Nagpal, S. Preparation and characterization of nanocurcumin based hybrid virosomes as a drug delivery vehicle with enhanced anticancerous activity and reduced toxicity. Sci. Rep. 11, 1–14 (2021).
Kumar, V., Wadhwa, R., Kumar, N. & Maurya, P. K. A comparative study of chemically synthesized and Camellia sinensis leaf extract-mediated silver nanoparticles. 3 Biotech 9, 7 (2019).
Kimanius, D., Dong, L., Sharov, G., Nakane, T. & Scheres, S. H. W. New tools for automated cryo-EM single-particle analysis in RELION-4.0. Biochem. J. 478, 4169–4185 (2021).
Jumper, J. et al. Highly accurate protein structure prediction with AlphaFold. Nature 596, 583–589 (2021).
Pettersen, E. F. et al. UCSF Chimera—a visualization system for exploratory research and analysis. J. Comput. Chem. 25, 1605–1612 (2004).
Mota, B. E. F. et al. Adverse events post smallpox-vaccination: insights from tail scarification infection in mice with Vaccinia virus. PLoS ONE 6, e18924 (2011).
Bray, M. et al. Cidofovir protects mice against lethal aerosol or intranasal cowpox virus challenge. J. Infect. Dis. 181, 10–19 (2000).
Medeiros, N. I. & Gomes, J. A. S. Cytometric bead array (CBA) for measuring cytokine levels in Chagas disease patients. Methods Mol. Biol. 1955, 309–314 (2019).
Acknowledgements
We sincerely thank Prof Ganesan Karthikeyan, Executive Director, iBRIC-THSTI, for the suggestions, edits, and critical inputs. We thank Dr Pramod Kumar Garg, former Director, for his full support and valuable input and guidance. We sincerely thank DBT Secretary Dr Rajesh S. Gokhale and Dr Jyoti Logani for their continuous support and encouragement. We thank ICMR Director General Dr Rajiv Bahl for his timely support in assisting and obtaining the Mpox strain. We thank Dr Jayanta Bhattacharya, Dean, BRIC-THSTI, for the support and guidance. We thank Dr Naveen Kumar, Director, ICMR-National Institute of Virology (NIV), and Dr Pragya D. Yadav, Scientist-F, ICMR-NIV, for providing the MPXV (clade IIb A.2.1, GISAID ID:EPI_ISL_159123322) strain. The following reagent was obtained through BEI Resources, NIAID, NIH: Monkeypox Virus, WRAIR 7-61, NR-27. The following reagent was obtained through BEI Resources, NIAID, NIH: Vaccinia Virus, Western Reserve (Mouse adapted), NR-56. We thank our IBSC chair Dr Krishnamohan Atmakuri, and the committee, RCGM committee, IAEC, and ABSL3 committee for their timely approvals and support in carrying out the study. We thank Mr M. V Santo, Head Administration, iBRIC-THSTI, for continuous support and encouragement. We thank our industry partner Panacea Biotec Ltd, India technical team, Mr Harshet Jain, and Dr Amulya Panda, for continuous support and technical guidance. We thank Mr Amit Kumar and Mr Aman Kumar from our lab for helping in animal experimentation and obtaining the reagents, respectively. We thank Dr Amit Pandey, Dr B. N. Panda, and Mr Prabhanjan Kumar Dwivedi from THSTI-EAF, BSL3, ABSL3, Histopathology facility, and staff for providing support and timely help for immunization experiments and processing histopathology samples. We thank Dr Mahima Gupta for assisting with the pilot-scale fermentation in the Vaccine Design and Development Center (VDDC), THSTI. We thank Mr Vijay Kumar of the Regional Centre for Biotechnology (RCB) for assisting with DLS and CD experiments. Part of the work was carried out at the Advanced Technology Platform Centre (ATPC) of RCB. We thank the TEM facility at the Indian Institute of Science, Bangalore, for time and support. The following reagent was obtained through BEI Resources, NIAID, NIH: Vector pET-28a(+) Containing the SARS-Related Coronavirus 2, Wuhan-Hu-1 Non-Structural Protein-10 Gene, NR-53502. The works were supported by Translational Health Science and Technology Institute (THSTI) core funding and the INDCEPI project, the Translational Research Project (TRP) funded by the Biotechnology Research and Innovation Council (BRIC) of the Department of Biotechnology, Ministry of Science and Technology, Government of India. Ns-TEM work at IISc was supported through intramural funding support to V.M.P. from the Indian Institute of Science, Bangalore. A.C. is supported via the Prime Minister Research Fellowship (PMRF), Govt. of India.
Author information
Authors and Affiliations
Contributions
S.S. and S.A. designed the experiments, analyzed and interpreted the data, and S.S. wrote the manuscript, and all authors edited. S.A. designed the antigens. R.A., P.V., V.K., R.K., S.M., G.K., A.S., Ak.C, V.M., S.Si., P.Y., K.T., Z.A.R. performed the experiment. Z.A.R., V.K., and A.A. performed the T cell assays, analyzed the data. R.A. and A.S. expressed and purified the immunogen and carried out the biochemical analysis. P.V. performed and supervised the animal experiments and analysis of humoral responses. V.K. performed the biophysical characterization along with R.A. and performed and supervised the tail scarification experiment along with P.V. Ns-TEM imaging and analyses were carried out by An.C. and V.M.P. R.K. established the VACV in vitro assays with K.T. and in vivo, animal model along with P.V. and V.K. G.K. performed the bioinformatic analysis and in vitro assays. A.S. and V.M. assisted R.A. in batch purifications of antigens and other in vitro biochemical assays. V.K. and Ak.C. performed Mpox challenge studies. S.Si., assisted in the in vivo experiments for animal handling during immunization and blood collection at different time points. DY performed the small-scale fermenter batch production of Mpx-V3, and S.K.S. performed all the standard superdex column experiments. Ab.Ch., P.P., S.K.A. from Panacea Biotech helped in biophysical repetition, pilot-scale fermentation inputs. All authors have read and agreed to the published version of the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The Translational Health Science and Technology Institute (THSTI) has filed patent PCT International Application No. PCT/IB2022/059713||202111046243|| Multiepitope Self-Assembled Nanoparticle vaccine platform (msn-vaccine platform) and uses thereof, and listed S.S., S.A., R.K., and A.A., and technology has been transferred to Panacea Biotec Ltd, India for development of DP and DS. Indian Patent Provisional Application No.: 202411072889 filed on September 26, 2024, Title: an engineered polypeptide complex for generating protective immune responses against monkeypox (MPXV). All other authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Ahuja, R., Vishwakarma, P., Kumar, V. et al. Next-gen novel nanocage-based multivalent vaccine candidate to tackle the rising menace of Mpox. npj Vaccines 10, 117 (2025). https://doi.org/10.1038/s41541-025-01174-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41541-025-01174-1
