
Abstract
Among all enzymatic metallocofactors, those found in nitrogenases, the P and L or M clusters, stand out for their intricate structures. They are assembled by proteins of the Nif gene cluster from Fe2S2 rhombs—the smallest building blocks in FeS cluster chemistry—through a sequence of reactions constructing a Fe8S8 precursor. To advance our understanding of how enzymes selectively build such elaborate inorganic molecules, here we parallel the biosynthetic pathway by reporting the rational stepwise assembly of [Fe8S8]m+ (m = 2, 4, 6) clusters from [Fe2S2]2+ rhombs within an extensive cyclic synthetic network. A [Fe8S8]4+ cluster of unique topology is identified, for which we coin the term ‘interlocked’ double cubane. As a molecular analogue of the NifB K cluster, a proposed precursor to both the P and L or M clusters, its preparation and the characterization of all related intermediates, offers fundamental insights into the molecular mechanisms governing the assembly of both biogenic and synthetic FeS clusters.

Similar content being viewed by others


Main
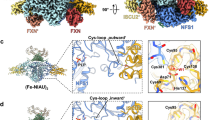
Iron–sulfur (FeS) clusters function as enzymatic metallocofactors in many critical metabolic processes1,2. While most of them are bi-, tri- or tetranuclear, larger assemblies are rare and have only been structurally characterized within two families: as μ2-sulfido-bridged double-cubane-type clusters in the aptly named double-cubane cluster protein3,4 and, more prominently, as the octanuclear P, L and M clusters found in nitrogenases and nitrogenase-like enzymes5,6,7,8. The biosyntheses of these clusters, particularly nitrogenases, are intricate and proceed through distinct yet mechanistically related pathways. Even though the P and M clusters mature separately from one another, both are assembled by proteins encoded in the Nif gene cluster through the intermediacy of a Fe8S8 precursor7,8,9,10; in a first step, NifS and NifU mobilize Fe and S to produce Fe2S2 rhombs, which are reductively combined to Fe4S4 cubanes7,8,11,12,13 (Figs. 1a and 2a). Towards the M cluster, two of the Fe4S4 cubanes are recruited by NifB, site differentiated14 (Figs. 1b and 2b), merged (Fig. 2b) and fused to form the Fe8S8 precursor. NifB itself contains three Fe4S4 clusters: one is a radical SAM cluster, while the other two, referred to as the K1 and K2 clusters, constitute the building blocks for the M cluster. A recent crystallographic structure of the enzyme revealed that the K1 and K2 clusters may be coordinatively unsaturated or bound to each other (Figs. 1c and 2e), forming a double cubane15. Based on an alternative refinement of the original diffraction data, another arrangement of the clusters was also proposed: rather than existing as two discrete Fe4S4 units, the K1 and K2 clusters may adopt a configuration in which they form a single Fe8S8 complex, simply termed the K cluster16. Its FeS topology is similar to that of the Fe8S7 PNcluster, but is distinguished by an eighth μ2(S2−) ligand (Figs. 1d and 2e and Supplementary Fig. 97). During subsequent stages, several enzymes work together with NifB to functionalize this K cluster with a carbide and a ninth μ2(S2−) ligand, yielding the Fe8S9C L cluster. The latter is then transferred to NifEN, wherein a hetero-metal (Mo or V) and a homocitrate ligand are installed, enabling the formation of the final MFe7S9C M cluster7,8,17,18,19,20 (Fig. 2c). By contrast, although it also requires many extra proteins, the Fe8S8 precursor to the P cluster, which is potentially K-cluster-like, matures directly within NifDK via the loss of one of its S atoms7,8,21,22 (Fig. 2d).

a, Crystal structure of the NifU-like protein, Aquifex aeolicus IscU with a bound [2Fe–2S] cluster (Protein Data Bank (PDB) ID 2Z7E)13. A close-up view of the metallocofactor is shown as well. b, Crystal structure of Methanotrix thermoacetophila NifB with bound radical SAM and K1 clusters (PDB 6Y1X)14. A close-up view of the K1 cluster is shown alongside. c,d, Crystal structure refinements of Methanobacterium thermoautotrophicum NifB with a full complement of FeS cofactors, as proposed by ref. 15 (c; PDB 7JMB) and ref. 16 (d; PDB 7BI7). An alternative view of the [8Fe–8S] K cluster (d) is presented in Supplementary Fig. 97. For each solution of the structure, a close-up view of the K1 and K2 clusters (c) and the K cluster (d) are provided.

a, Conversion of Fe and S into Fe2S2 clusters in NifU and NifS and their reductive fusion to Fe4S4 clusters. Here the arbitrary ligand, L, is usually a cysteinate or histidine residue, but may also refer to any other amino acid able and known to bind to Fe4S4 cofactors, for example, aspartate. b, Site differentiation and spatial merging of Fe4S4 clusters to form the K1 and K2 clusters in NifB. c, Last steps of M-cluster maturation from the Fe8S8 K-cluster intermediate, encompassing installation of the additional carbide and sulfide ligands, as well as the Mo ion. d, A hypothetically simple route from a K- (or K-like-) cluster precursor to the PNcluster via S(µ2) removal. e, Structures of the NifB K cluster proposed in refs. 16 and 15.
Parallel to the structural and biosynthetic characterizations of these metallocofactors (vide supra)7,8,23,24,25, chemists also pursued their synthetic replication26,27,28,29. Notable examples include (1) the fusion of catechol- or tris(pyrazole)borate- and phosphine- or chloride-supported MFe3S4 (M=V, Mo, W) heterocubanes30,31 to form edge-bridged double-heterocubanes32,33,34,35, which rearrange to M2Fe6S7 clusters on treatment with hydrosulfide or hydroselenide36,37,38; (2) P-cluster models of the type Fe8S(7-x)Ox (x = 0, 1)39,40,41,42,43,44, which are accessible by self-assembly, but whose mechanism of formation remains elusive40,41,44; and (3) all-Fe, ligand-, edge- and face-bridged double or multicubanes, which were stabilized by phosphines, N-heterocyclic carbenes or tripodal chelating tris-thiolates as supporting ligands45,46,47,48,49. However, although successful at replicating several of the nitrogenases’ metallocofactors’ core structures, these synthetic results remain disconnected from the biosynthetic pathways, because they do not capture the stepwise and coordinated assembly of simple FeS building blocks into complex clusters.
Departing from traditional approaches, we thus describe here a rational, cyclic and reversible chemical network of FeS cluster interconversions, enabling the stepwise assembly of synthetic [Fe2S2]2+ rhombs—the simplest FeS building blocks—to [Fe8S8]m+ (m = 2, 4, 6) clusters. Among these, the [Fe8S8]4+ cluster exhibits the same ‘core’ FeS topology as that proposed for the K cluster in ref. 16 (Figs. 1d and 2e), which we herein call an ‘interlocked’ double cubane (ildc). Its ultimate and penultimate precursors are themselves Fe8S8 clusters, namely, a ligand-bridged [Fe8S8]6+ or an edge-bridged [Fe8S8]2+ double cubane, respectively. Both resemble the structure that was proposed in ref. 15 for the K1 and K2 clusters (Figs. 1c and 2e). On the basis of the characterization of these clusters’ electronic, vibrational and 57Fe nuclear spectroscopic signatures, it is suggested that they could aid the identification of such intermediates in NifB and NifDK, respectively, while also providing fundamental insight into the factors governing their formation and interconversion.
Results
Rationalizing oxidation-state-dependent cluster conversions
The initial goal of our synthetic strategy was to achieve site differentiation, as this was proposed to play a key role in inducing cluster conversion14,50. In enzymes, Fe4S4-site differentiation occurs as the regiospecific substitution of a cysteine ligand, typically by histidine51, driven by an asymmetric cofactor binding pocket. Efforts to replicate this behaviour have been explored using rigid multidentate thiolate ligands49,52 or bulky N-heterocyclic carbenes53. However, these strategies are not transposable to [Fe4S4(RS)4]n− clusters bearing simple monodentate thiolate ligands due to the latter’s high symmetry, which prevents precise control over the stoichiometry of ligand substitutions. Leveraging our group’s expertise with the Kn[Fe4S4(DmpS)4] (n = 0–4; where Dmp is 2,6-dimesitylphenyl) model system54,55,56, and specifically the evaluation of the oxidation-state dependence of the Fe–S(μ3) and Fe–S(thiolate) bond covalency using X-ray absorption spectroscopy, we rationalized an original strategy to promote ligand exchange, rooted in the cluster’s electronic structure (Fig. 3a,b)54; the covalent character of a chemical bond is evaluated using the α2-parameter, which is often referred to as ‘covalency’57. However, α2 quantifies the ligand character in unoccupied (or half-occupied) metal 3d orbitals, which are covalently mixed with the ligand orbitals (here, S 3p), forming a ψ*(Fe–L) frontier molecular orbital. The α2-parameter, which may adopt any value between 0% and 100%, hence carries information about the localization of the frontier molecular orbital or, reciprocally, the polarization of the Fe–L bond. As such, and counterintuitively, the covalent character of a bond is maximal when it is not polarized, that is, when α2 is ~50%. For this work, it is important to realize that cubanes in the [Fe4S4]3+/[Fe4S4]4+ oxidation states are characterized by exceptionally large values of α2 for their Fe–S(thiolate) bonds (>50%). This means that the singly occupied ψ*(Fe–SR) molecular orbitals are predominantly localized on (or polarized towards) S, and therefore the binding of a less covalent ligand to Fe can trigger homolytic cleavage of the Fe–S(thiolate) bond. As the bonding electrons (that is, those in the corresponding ψ(Fe–SR) orbitals) are predominantly localized on Fe, this process reduces the Fe4S4 core by one electron, while the thiolate is formally oxidized to a radical species (Fig. 3a). At the same time, because of the core’s reduction, the remaining Fe–S(thiolate) bonds will be again more ‘covalent’ than before (α2 closer to 50%), and dimerization of the thiolate radical renders the reaction irreversible. This ensures that only a specific number of thiolate ligands are exchanged per cluster, allowing control of the substitution stoichiometry. Such mechanisms are disfavoured in cubanes in the [Fe4S4]0/[Fe4S4]1+ oxidation states, which exhibit low Fe–S(thiolate) bond covalencies (<50%). In contrast to their oxidized congeners, the fact that their singly occupied ψ*(Fe–L/SR) molecular orbitals are predominantly localized on Fe and their ψ(Fe–L/SR) orbitals remain on the ligand or thiolate makes them prone to (heterolytic) ligand exchange mechanisms (Fig. 3b).

a, Mechanism for oxidatively triggered covalency-driven (reductive) site differentiation of [Fe4S4(RS)4]n− clusters by an arbitrary ligand, L, via homolytic bond cleavage. More specifically, L refers to a (μ1-bound) neutral or anionic dative ligand, which is less covalent than a thiolate and may be weakly π acidic. The overall cluster negative charge (n) increases by 1 if L is anionic and remains the same if L is neutral. By contrast, the core reduction level, (4 − n), decreases by 1, with each ligand substitution. b, Electronic origin of redox-neutral ligand loss from a reduced Fe4S4 complex via heterolytic bond cleavage. Here the overall negative cluster charge (n) decreases by 1 if L is anionic, and remains the same if L is neutral. Both behaviours (those shown in a and b, respectively) are ascribed to specific [Fe4S4](4−n)+ oxidation states based on our study of the corresponding Fe–Lt (where Lt is a terminal ligand) bond covalencies (α2)54. c, Synthetic pathway from a [Fe2S2]2+ complex via canonical and site-differentiated [Fe4S4](4−n)+ (n = 1, 2, 3) complexes to edge-bridged and ligand-bridged [Fe8S8]2+/[Fe8S8]6+ double cubanes, as well as an interlocked [Fe8S8]4+ cluster, topologically paralleling the proposed K cluster’s architecture. Equivalents (eq.) of reagents are given with respect to the educt cluster. In cases, where reductive elimination of disulfide occurs, this is indicated and the reaction was balanced accordingly. In all clusters, R denotes the 2,6-dimesitylphenyl residue. Below each structure, the corresponding sum formula and the overall core oxidation state/substitution pattern/cluster topology are indicated. Refer to the Supplementary Information for the exact synthetic conditions of each step.
Constructing and traversing the synthetic cycle
The considerations outlined above were successfully implemented to construct the synthetic cycle shown in Fig. 3c, wherein repeated site differentiation and redox chemistry enabled FeS cluster conversion. Detailed procedures, additional synthetic explorations, as well as supporting discussions of the basic characterization data for all shown compounds are compiled in the Supplementary Information. Figure 3c highlights the central role of the canonical [Fe4S4]2+ complex, K2[Fe4S4(DmpS)4], which constitutes the junction, connecting the [2Fe–2S]-to-[4Fe–4S], [4Fe–4S]-site-differentiating, as well as [4Fe–4S]-to-[8Fe–8S] cluster interconversion cycles (highlighted in grey, green and brown boxes, respectively).
As anticipated from the Fe–S(thiolate) bond covalency considerations (Fig. 3a,b), [4Fe–4S]-site-differentiation is enabled by cluster oxidation: the redox congeners of K2[Fe4S4(DmpS)4] in the [Fe4S4]3+ and [Fe4S4]4+ states readily undergo homolytic Fe–S(thiolate) bond cleavage to exchange their DmpS−ligands for the less covalent 1,2,4,5-tetramethylimidazole (Im*) ligand. Thereby, the cluster core is reduced by one electron per additional Im*, until the [Fe4S4]2+ state is reached. This enabled the syntheses of K[Fe4S4(DmpS)3(Im*)] (ref. 58), [Fe4S4(DmpS)3(Im*)] and [Fe4S4(DmpS)2(Im*)2], whereby the last two are interconvertible depending on the stoichiometry of Im*, owing to the [Fe4S4]3+ redox level of [Fe4S4(DmpS)3(Im*)]. Conversely, because the [Fe4S4]2+–(DmpS−) bond is more covalent than the [Fe4S4]2+–(Im*) bond, Im* ligands are easily substituted from [Fe4S4(DmpS)3(Im*)]− or [Fe4S4(DmpS)2(Im*)2] by stoichiometric DmpSK, re-forming K2[Fe4S4(DmpS)4].
The [2Fe–2S]-to-[4Fe–4S] cluster conversion chemistry occurs on reaching the all-ferric oxidation state59: [Fe4S4(DmpS)4] readily coordinates four pyridine (py) molecules, resulting in cubane scission, to form 2 equivalents of [Fe2S2(DmpS)2(py)2]. Pyridine ligands can be removed reversibly using B(C6F5)3, leading to the fusion of two [Fe2S2]2+ rhombs and re-forming [Fe4S4(DmpS)4], while the reduction of [Fe2S2(DmpS)2(py)2] reconstitutes K2[Fe4S4(DmpS)4] through the fusion of two transient [Fe2S2]1+ synthons. Notably, the fusion of a [Fe2S2]2+ with a [Fe2S2]1+ cluster, yielding a [Fe4S4]3+ complex, was not observed.
In contrast to the [2Fe–2S]-to-[4Fe–4S] and [4Fe–4S]-site-differentiation cycles, [4Fe–4S]-to-[8Fe–8S] conversion chemistry is initiated reductively: addition of one electron and one K+ ion to K[Fe4S4(DmpS)3(Im*)] yields the edge-bridged [Fe8S8]2+ double cubane K4[Fe8S8(DmpS)6] (ebdc; Fig. 4a) via fusion of two site-differentiated [Fe4S4]1+ cubanes. This result is counterintuitive, considering that the Im*-substitution of one of the thiolate ligands shifts the cluster’s redox potential anodically, which, in turn, suggests an overall better stabilization of lower oxidation states (Supplementary Fig. 68). However, ligand-field considerations offer a different perspective regarding the cluster’s reactivity: reduction affords less covalent Fe4S4–ligand bonds55, enabling the facile heterolytic dissociation of the least covalent one (arguably that to Im*) and generating a coordinatively unsaturated Fe centre. The self-coordination of this intermediate via two µ4(S2−) ligands, affording ebdc, results in a more stable ligand field, in which net covalency is maximized (that is, α2 approaching 50%), compared with the one in which Im* ligation is maintained. ebdc can be reversibly oxidized by up to four electrons; its fully oxidized congener, however, does not possess an edge-bridged architecture. Instead, the four-electron oxidation is accompanied by what we propose to be a sliding motion of the two Fe4S4 subclusters, until they symmetrically face each other. Concomitantly, two of the µ1(DmpS−) ligands convert to µ2(DmpS−) ligands, yielding the ligand-bridged [Fe8S8]6+ double cubane [Fe8S8(DmpS)6] (lbdc; Fig. 4b,d). Although lbdc is formally ‘homoleptic’, the inequivalent DmpS− ligands (bridging and terminal) render it site differentiated. We thus found that the primary coordination sphere is destabilized enough to induce spontaneous cluster conversion: metastable lbdc slowly converts to the ‘interlocked’ [Fe8S8]4+ double cubane, [Fe8S8(DmpS)4] (ildc; Fig. 4c), probably through a transition state initiated by a tilting motion of lbdc’s two ligand-bridged Fe4S4 subclusters, whereby the µ2(DmpS−) ligands act as hinges. Simultaneously, two µ3(S2−) ligands rearrange to one µ2(S2−) and one µ6(S2−) ligand, while two of the formerly µ1(DmpS−) ligands undergo reductive elimination (Fig. 4d). This mechanistic scenario is supported by the fact that stoichiometric amounts of (DmpS)2 were observed through 1H NMR (Supplementary Figs. 54–56) in situ during the transformation of lbdc to ildc. Given the precedents of covalency-driven homolytic Fe–S(thiolate) bond cleavage at [Fe4S4]3+/[Fe4S4]4+ clusters in this work, it seems reasonable to assume that the reductive elimination of (DmpS)2 from lbdc—formally composed of two [Fe4S4]3+ clusters—occurs homolytically as well. Although ildc appears coordinatively saturated and electrochemically robust (Supplementary Fig. 72), it readily converts back to Fe4S4 complexes in presence of competing ligands: 4 equivalents of Im* or DmpSK ‘unlock’ and cleave the double cubane, yielding 2 equivalents of [Fe4S4(DmpS)2(Im*)2] or K2[Fe4S4(DmpS)4], respectively, both of which are interconvertible themselves. As such, these transformations close the [4Fe–4S]-to-[8Fe–8S] cycle and integrate it into the network of [2Fe–2S]-to-[4Fe–4S]-to-[8Fe–8S] conversions (Fig. 3c).

a–c, Solid-state molecular structures of ebdc (a), lbdc (b) and ildc (c) in crystals of K4[Fe8S8(DmpS)6]·3.5(C7H8), [Fe8S8(DmpS)6]·3(C7H8) and [Fe8S8(DmpS)4], respectively. Displacement ellipsoids are shown at the 50% probability level for ebdc and ildc, and at 30% for lbdc, and are only displayed for the Fe, S and K atoms. Cocrystallized solvent molecules and hydrogen atoms have been omitted for clarity. d, Proposed mechanism for the interconversions of the three Fe8S8 clusters (ebdc, lbdc and ildc). Structures marked by a double dagger are postulated transition states.
Beyond these results, our synthetic explorations lead to the discovery of a variety of additional reactivity patterns, yielding unique FeS architectures and contributing to our fundamental understanding of this type of cluster conversion chemistry (Supplementary Figs. 2–5 and 81–91). Two species are particularly noteworthy: the edge-bridged [Fe12S12]o triple cubane, K6[Fe12S12(DmpS)6] (ebtc; Supplementary Figs. 4 and 89), the preparation of which underscores the validity of our synthetic strategy (Fig. 3b), and [Fe24S24(DmpS)10] (Supplementary Figs. 3 and 88), an extremely large structurally characterized molecular FeS cluster.
Electronic and vibrational signatures of the [8Fe–8S] clusters
We proposed that the change in the topology of the Fe8S8 clusters would be reflected in their spectroscopic properties. To this end, the spectroscopic signatures of ebdc, lbdc and ildc were compared by ultraviolet-visible light (UV-vis) electronic absorption, 57Fe nuclear resonance vibrational spectroscopy (NRVS) and 57Fe Mössbauer spectroscopy (Fig. 5). While ebdc and lbdc exhibit UV-vis electronic absorption spectra with features at similar energies as their [Fe4S4]1+/[Fe4S4]3+ redox congeners, ildc is characterized by electronic transitions that are atypical for an FeS cluster possessing equal numbers of FeII and FeIII ions, and thus, an average oxidation state of Fe2.5; the intense peak at 467 nm falls within the range of the maxima observed for the more oxidized, Fe2.75-containing clusters55 and the shoulder at 631 nm is equally unusual (Fig. 5a). Similarly, the 57Fe partial vibrational density of states (PVDOS) spectra of ebdc and lbdc appear close to those of the canonical [Fe4S4]1+ and [Fe4S4]3+ complexes, respectively60,61,62, whereas ildc shows distinct vibrational bands, which deviate from the modes observed for its [Fe4S4]2+ counterpart (Fig. 5b). The most notable differences are evident in the region of the spectrum associated with FeS bending and twisting (or ‘breathing’) motions, around 80–220 cm−1. This shows that, while ebdc and lbdc are formally Fe8S8 clusters, their vibrational (and electronic) behaviour closely resembles that of cubanes because they retain intact Fe4S4 building blocks. Conversely, the FeS skeleton of ildc is topologically rearranged, rendering its properties distinct from those of Fe4S4 complexes. The most intense mode in its 57Fe PVDOS spectrum occurs at approximately 80 cm−1. A simulation of the normal modes using density functional theory (DFT) revealed a vibrational profile similar to experiment (Supplementary Fig. 111), wherein the most intense mode occurs around 100 cm−1; the latter identifying as ‘asymmetric twisting’ of the two Fe4S3 subunits. A mode at roughly 200 cm−1 is of further interest, because FeS cubanes usually display minimal intensity in this region61. Similar vibrations have, however, been observed for the M cluster, which possesses a µ6(C4−) ligand and an associated cluster breathing mode around 180–190 cm−1 (ref. 63). Inspection of the calculated modes around 200 cm−1 confirmed that the cluster topology of ildc, possessing a µ6(S2−) ligand (S1), indeed induces breathing modes at comparable energies (around 204 cm−1), despite the substantially different mass of its central μ6 ligand. This similar energy results from the fact that, in this particular mode, the central µ6 atom is not significantly displaced compared with its surrounding Fe and S atoms. A very different situation was observed for the mode appearing at 599 cm−1 for the M cluster63, which involves significant motion of the central atom. For ildc, simulation suggests that it appears at around 260 cm−1, 295–305 cm−1 and 370 cm−1 (corresponding to the three respective dimensions of µ6(S2−) motion). As for the cubanes, the PVDOS around 400 cm−1 is primarily derived from Fe–S(thiolate) stretches 60,61,62. Notably, however, the displacement of the other distinct sulfide ligand, namely, the µ2(S2−) sulfide (S2), occurs at a rather high energy of 420 cm−1 (marked by a black arrow in Fig. 5b). This mode is neither found in cubanes, nor in double cubanes such as lbdc or ebdc (Fig. 5b), and it should also not occur in Fe8S7 PN-type clusters, which lack µ2(S2−) ligands. We thus anticipate that this vibrational feature could serve as a useful signature to identify such a K cluster in the more complex biological environments.

a, UV-vis electronic absorption spectra of 1 × 10−4 M toluene solutions of lbdc (magenta), ildc (yellow) and ebdc (blue), are shown as bold solid lines. For comparison, the spectra of the canonical and site-differentiated Fe4S4 complexes in the relevant oxidation states are also shown: they include K3[Fe4S4(DmpS)4] (dotted blue), K2[Fe4S4(DmpS)4] (dotted yellow), K[Fe4S4(DmpS)3(Im*)] (dashed yellow) and [Fe4S4(DmpS)2(Im*)2] (dotted-dashed yellow), K[Fe4S4(DmpS)4] (dotted magenta) and [Fe4S4(DmpS)3(Im*)] (dashed magenta). b, 57Fe NRVS PVDOS spectra of lbdc, ildc and ebdc (solid coloured lines). For comparison, the spectra of the corresponding canonical cubanes in their respective oxidation states are shown alongside, as dotted coloured lines. All spectra were recorded on powdered 57Fe enriched (>95%) samples between 30 K and 50 K. c–e, The 80 K Mössbauer spectra (vertical bars) recorded on powder samples of lbdc (c), ildc (d) and ebdc (e). No external magnetic field was applied in d and e, whereas a 0.06 T external magnetic field was applied along the γ ray direction in c. Simulations are overlaid as thick grey solid lines and components are displayed above the spectra as coloured thin solid lines. The nuclear parameters are the following: doublet 1 (violet) δ = 0.48 mm s−1, ∆EQ = 0.81 mm s−1; doublet 2 (magenta) δ = 0.31 mm s−1, ∆EQ = 0.92 mm s−1; common linewidth ΓFWHM = 0.39 mm s−1 (c); doublet 1 (brown) δ = 0.34 mm s−1, ∆EQ = 0.89 mm s−1; doublet 2 (yellow) δ = 0.68 mm s−1, ∆EQ = 2.57 mm s−1; common linewidth ΓFWHM = 0.43 mm s−1 (d) and doublet 1 (light blue): δ = 0.51 mm s−1, ∆EQ = 0.89 mm s−1, ΓFWHM = 0.66/0.78 mm s−1; doublet 2 (dark blue): δ = 0.55 mm s−1, ∆EQ = 1.56 mm s−1, ΓFWHM = 0.57/0.61 mm s−1 (e). FWHM, full-width at half-maximum.
Valence topologies of the [8Fe–8S] clusters
To further understand the asystematic electronic and vibrational properties of ildc, compared with ebdc and lbdc, the valence topology of the three Fe8S8 clusters was rationalized by 57Fe Mössbauer spectroscopy (Fig. 5c–e): lbdc and ebdc exhibit zero-field 80 K powder spectra with two main absorption lines (Fig. 5c,e), while ildc shows four (Fig. 5d). Regardless of the simulation model (refer to the Supplementary Information), the average isomer shift of the spectra, δavg, coincides well with the clusters’ average Fe oxidation states55. However, while the simulations of the spectra of ebdc and lbdc suggest delocalized Fe valences, typical for Fe4S4 complexes (individual isomer shifts, δi, are 0.45 < δi < 0.58 mm s−1 for ebdc and 0.31 < δi < 0.48 mm s−1 for lbdc), the unique simulation of ildc’s spectrum presents discrete doublets at δi = 0.34 and 0.68 mm s−1 (Fig. 5d). These values are substantiated by the analysis performed on the 5.7 K spectra recorded with 0.06 to 7 T magnetic fields applied along the γ ray direction (Supplementary Fig. 102a–d) and that performed on the 293 K zero-field spectrum (Supplementary Fig. 101 and Supplementary Table 11), evidencing a diamagnetic ground state with localized ferric and ferrous sites, even at room temperature. Therefore, we establish a Robin–Day class I (valence-trapped) mixed-valence state in ildc, for which a bond-valence sum analysis of the crystallographic structure suggests that [Fe3,Fe4] are in the +II and Fe1 and Fe2 in the +III oxidation states (Supplementary Table 10). Although there are conflicting interpretations of Mössbauer spectra in the literature40,44, similar valence-trapped ground states were suggested for the μ6(S2−)-ligand containing [Fe8S7]4+ clusters (possessing six FeII and two FeIII atoms)64. However, to date, no direct confirmation of this phenomenon by variable-temperature variable-field Mössbauer spectroscopy has been provided, and the origin and nature of the valence-trapped state has not been rationalized. The fact that the [Fe4S4]2+ subcluster motif in ildc (possessing two FeII and two FeIII atoms) has localized FeII and FeIII sites is particularly intriguing, because it stands in contrast with the fully delocalized [Fe4S4]2+ (two FeII and two FeIII) cluster core of ‘neat’ cubanes and the (at least partially) delocalized cuboidal [Fe4S3C]0-motif of the M cluster (likewise possessing two FeII and two FeIII atoms)65,66,67.
This apparent discrepancy prompted us to investigate possible electronic origins of the trapped valences in ildc by complementing our spectroscopic investigations with broken-symmetry DFT calculations. The qualitative molecular orbital diagram of the energetically most favoured broken-symmetry solution is summarized in Fig. 6a, alongside its spin-density isosurface plot (Fig. 6b). Among all converged wavefunctions, this one produced 57Fe Mössbauer isomer shifts and PVDOS in best agreement with experiment (Fig. 6a, Supplementary Table 12 and Supplementary Fig. 111) and is thus in clear support of the valence-trapped configuration. Although they are not real molecular properties (observables), the populations of localized orbitals were then evaluated to assess and visualize electronic delocalization: while this method predicts perfectly symmetric double-exchange (or spin-dependent delocalization, SDD) on the Fe2S2 subclusters of [Fe4S4(DmpS)4]2− (Fig. 6d–f), the comparable orbitals of ildc are significantly desymmetrized towards the itinerant electrons residing on [Fe3,Fe4] (Fig. 6c). This model (Fig. 6a) thus constitutes an appropriate electronic basis for the valence-trapped state of ildc.

a, Qualitative molecular orbital diagram for the electronic structure of ildc calculated with DFT. The α spins are drawn in green and β spins in purple. Local minority spins pertinent to SDD are drawn in grey and the corresponding molecular orbital is circled in grey. The spin and valence topologies of the complex are schematically drawn in separate (symmetric) halves of the molecule. For completion, the former is inverted, while the latter is mirrored over the symmetry plane indicated as a grey line. The experimental 57Fe Mössbauer isomer shift values associated with each Fe site are shown alongside, with the respective theoretical DFT-derived isomer shift value given in brackets. b, Calculated spin-density isosurface plot (isodensity cut-off 0.015) for ildc (left). c, Isosurface plot (isodensity cut-off 0.04) of one of the four equivalent orbitals relevant to the evaluation of SDD, which are highlighted in a grey circle in a. The corresponding atomic populations (φ) are φ(Fe1) = 16% and φ([Fe3,Fe4]) = 77%. d, Comparative qualitative molecular orbital diagrams for [Fe4S4(DmpS)4]2− and its associated measured (and calculated) 57Fe Mössbauer isomer shifts determined at equivalent temperature (refer to Supplementary Fig. 103 for the spectra and simulations), and at the equivalent level of theory. The Fe2S2 subclusters across which SDD occurs are highlighted in grey. e,f, Comparative spin-density isosurface plot (e) and one of the two equivalent orbitals relevant to SDD for [Fe4S4(DmpS)4]2− (f). The corresponding atomic populations (φ) are φ(Fe1) = 47% and φ(Fe2) = 48%.
From a structural perspective, the geometric parameters of the delocalized [Fe4S4]2+ core of [Fe4S4(DmpS)4]2−, the trapped [Fe4S4]2+ subcluster of ildc and the (at least partially) delocalized [Fe4S3C]0 motif of the M cluster were considered (Supplementary Fig. 110): while the average Fe–S(μ3) bonds in both ildc (2.283(2) Å) and the M cluster (2.274 Å)68 are similar to those of canonical [Fe4S4]2+ clusters (2.285 Å)55,56, the Fe–L(μ6) bonds to the central ligand are significantly longer in ildc (2.436(2) Å; L=S) and significantly shorter in the M cluster (1.990 Å; L=C)68. Therefore, whereas the M cluster’s ‘compacted’ [Fe2S(μ3)C(μ6)]1− subclusters appear to sustain SDD between their Fe atoms, probably due to the short Fe–C bonds and high charge-to-bridged-atom ratio of the μ6(C4−) ligand, ildc’s ‘expanded’ [Fe2S(μ3)S(μ6)]1+ subclusters do not, offering a simple geometric argument for the observed effect.
Discussion
The sequence of reactions required to traverse the entire network of FeS cluster conversions shown in Fig. 3c offers valuable insights into potential mechanisms at stake in the biogenesis of the nitrogenase P-, L- and M-cluster cofactors. All three octa-Fe clusters (ebdc, lbdc and ildc) can serve as molecular models for the Fe8S8 K cluster, the structure of which remains a topic of discussion (vide supra) and, together, may represent molecular snapshots of the [4Fe–4S]-to-[8Fe–8S] fusion occurring in NifB7,8. We foresee that our analysis of the clusters’ essential spectroscopic fingerprints (Fig. 5) will aid in confirming, respectively disproving their involvement in the corresponding cofactor maturation pathways. For this, particularly the trapped valences and the identification of the characteristic Fe–S(μ2) stretching vibration in ildc should represent useful spectroscopic features. However, we point out that among the three Fe8S8 complexes, ildc is distinct and exhibits identical FeS topology to that proposed recently in ref. 16. Our results show that such a structure can be systematically assembled through stepwise synthesis, beginning from an [Fe2S2]2+ complex as the simplest FeS cluster building block. Depending on whether they occur in a concerted or stepwise manner, this involves approximately nine fundamental chemical steps:
-
(1)
reduction of two [Fe2S2]2+ clusters to [Fe2S2]1+ clusters;
-
(2)
fusion of two [Fe2S2]1+ clusters to form a canonical [Fe4S4]2+ cluster;
-
(3)
one-electron oxidation to a [Fe4S4]3+ cluster;
-
(4)
reductive site differentiation of the canonical cluster by a labile noncanonical ligand, regenerating a [Fe4S4]2+ cluster;
-
(5)
reduction, leading to the loss of the labile noncanonical ligand, yielding a coordinatively unsaturated [Fe4S4]1+ cubane;
-
(6)
fusion of two coordinatively unsaturated cubanes to form an edge-bridged double cubane;
-
(7)
oxidation of the edge-bridged double cubane;
-
(8)
rearrangement of the cluster architecture into a ligand-bridged double cubane through a sliding motion and
-
(9)
interlocking of the two Fe4S4 subclusters by mobilization of µ3(S2−) ligands and a tilting motion along the hinge provided by the two bridging ligands, concomitant with reductive elimination of disulfide.
These cluster conversions are exclusively driven by alternating redox- and site-differentiation reactions, which repeatedly destabilize the FeS cluster’s primary coordination sphere and sequentially force it to relax into a new, more stable structure, while also increasing its size. In a broader context, we thus emphasize that two key factors appear to govern FeS cluster conversion chemistry: (1) the FeS cluster’s ligands, and (2) its oxidation state. In enzymes, these are controlled by the residues accessible in the cofactor binding pocket and the local electrochemical potential. We also emphasize that the cluster oxidation state and the associated ligand covalencies control whether and how Fe4S4 site differentiation occurs; polarized Fe–S(thiolate) bonds are prone to cleave either homolytically if α2 is >50%, or heterolytically if α2 is <50% (with respect to the ligand). In between, highly covalent bonds (α2 ≈ 50%) are stable, rendering [Fe4S4]2+ complexes the thermodynamic sink of FeS cluster conversion chemistry. This was exactly reflected in the reactivity observed in this work, where we demonstrated homolytic Fe–ligand bond cleavage for [Fe4S4]3+ and [Fe4S4]4+ complexes versus heterolytic bond cleavage in [Fe4S4]1+ and [Fe4S4]0, as well as that observed in previous studies, where we reported the aggregation of [Fe4S4]0 clusters via (heterolytic) loss of DmpS− ligands56. Given that the reduction potential of the DmpS− ligand (roughly −1.2 to −1.4 V versus normal hydrogen electrode; Supplementary Fig. 59) closely matches that of cysteinate (−1.38 to −1.45 V versus normal hydrogen electrode)69, it appears plausible that similar considerations could govern the reactivity of natural Fe4S4 cofactors.
Furthermore, our results indicate that ildc, or a topologically similar cluster, could represent a key intermediate at which the biosynthetic pathways of the P and M clusters diverge. Considering the uncertainties of knowing whether the K1 and K2 clusters fuse to an interlocked topology before or after the C-atom transfer from SAM7,8,17,18, and whether the eighth S of the P cluster is lost before, during or following cluster interlocking21,22, it is plausible that an interlocked topology of fused Fe4S4 synthons serves as a precursor in both biosynthetic pathways, aligning with the fact that they are thought to be evolutionarily related9,10.
Conclusion
Altogether, this work introduced an original approach to control FeS cluster conversion chemistry through alternating redox- and site-differentiation reactions. On this basis, we replicated the initial steps of M-cluster maturation, starting from a Fe2S2 rhomb, and ultimately arriving at a model for the Fe8S8 K cluster, within a coherent synthetic cycle. The isolation and characterization of all (meta)stable intermediate products allowed rationalizing the conditions driving each of the (electro)chemical steps and provides a framework to better understand and identify similar species during the biogenesis of the P and M clusters.
Methods
Experiments were carried out under a dry, oxygen-free Ar atmosphere using Schlenk-line and glove-box techniques. All solvents and reagents were rigorously dried and deoxygenated before use. Compounds were experimentally characterized by various techniques, including single-crystal X-ray diffraction, 1H/13C NMR, UV-vis electronic absorption, cyclic voltammetry, 57Fe NRVS, 57Fe Mössbauer and elemental analysis. Furthermore, ildc and [Fe4S4(DmpS)4]2− were theoretically probed using DFT calculations. Further details are available in the Supplementary Information with this article.
Data availability
The Supplementary Information (in PDF format) contains all synthetic procedures, experimental details, characterization data, computational methods and results, as well as additional supporting discussions relevant to this work. All unprocessed raw data for nonstandard characterization techniques (NRVS and 57Fe Mössbauer spectra) that were used to obtain the graphs shown in Figs. 5a–e and/or the Supplementary Information are provided by the authors from the ETH Zürich Research Collection at https://doi.org/10.3929/ethz-b-000739805. All molecular structures derived from single-crystal X-ray diffraction analyses are deposited as crystallographic information files in the Cambridge Structural Database under the accession numbers 2389114 ([Fe2S2(DmpS)2(py)2]), 2389123 (ebdc), 2389116 (lbdc), 2389115 (ildc, unit cell 2), 2389110 (ildc, unit cell 1), 2389111 ([Fe4S4(DmpS)2(Im)2]·K[Fe4S4(DmpS)3(Im)]), 2389112 ([Fe4S4(DmpS)3(Im)]·0.5[Fe(Im)6]), 2389122 ([Fe10S10(DmpS)6(Im)2]), 2389124 ([Fe4S4(DmpS)4]·0.5[Fe(MeCN)6]), 2389127 ([Fe4S4(DmpS)3(THF)3]), 2389125 (DmpSSSDmp), 2389120 (Im*·B(C6F5)3), 2389119 ([Fe24S24(DmpS)10]), 2389109 (K6[Fe12S12(DmpS)6]), 2389126 ([Fe4S4(DmpS)3(Im*)]), 2389118 (K[Fe4S4(DmpS)3(Im*)]), 2389117 ([18-C-6]K[Fe4S4(DmpS)3(Im*)]), 2389113 ([Fe4S4(DmpS)2(Im*)2]), 2389121 ([Fe8S7(DmpS)3Cl2]) and 2389108 ([Fe(DmpS)Cl]2). Copies of the data can be obtained free of charge at https://www.ccdc.cam.ac.uk/structures/. All data were analysed using standard software plugins whenever appropriate, as described in the Supplementary Information. The DFT-optimized XYZ coordinates of ildc are available on Supplementary Data 1 and animations of the theoretical normal modes are provided in Supplementary Video 1.
Code availability
All data were analysed using standard software plugins whenever appropriate, as described in the Supplementary Information.
References
Boncella, A. E. et al. The expanding utility of iron-sulfur clusters: their functional roles in biology, synthetic small molecules, maquettes and artificial proteins, biomimetic materials, and therapeutic strategies. Coord. Chem. Rev. 453, 214229 (2022).
Beinert, H., Holm, R. H., & Münck, E. Iron-sulfur clusters: nature’s modular, multipurpose structures. Science 277, 653–659 (1997).
Jeoung, J.-H. & Dobbek, H. ATP-dependent substrate reduction at an [Fe8S9] double-cubane cluster. Proc. Natl Acad. Sci. USA 115, 2994–2999 (2018).
Jeoung, J.-H., Nicklisch, S. & Dobbek, H. Structural basis for coupled ATP-driven electron transfer in the double-cubane cluster protein. Proc. Natl Acad. Sci. USA 119, e2203576119 (2022).
Einsle, O. & Rees, D. C. Structural enzymology of nitrogenase enzymes. Chem. Rev. 120, 4969–5004 (2020).
Trncik, C., Detemple, F. & Einsle, O. Iron-only Fe-nitrogenase underscores common catalytic principles in biological nitrogen fixation. Nat. Catal. 6, 415–424 (2023).
Ribbe, M. W., Hu, Y., Hodgson, K. O. & Hedman, B. Biosynthesis of nitrogenase metalloclusters. Chem. Rev. 114, 4063–4080 (2014).
Burén, S., Jiménez-Vicente, E., Echavarri-Erasun, C. & Rubio, L. M. Biosynthesis of nitrogenase cofactors. Chem. Rev. 120, 4921–4968 (2020).
Brigle, K. E., Weiss, M. C., Newton, W. E. & Dean, D. R. Products of the iron-molybdenum cofactor-specific biosynthetic genes, nifE and nifN, are structurally homologous to the products of the nitrogenase molybdenum-iron protein genes, nifD and nifK. J. Bacteriol. 169, 1547–1553 (1987).
Fani, R., Gallo, R. & Liò, P. Molecular evolution of nitrogen fixation: the evolutionary history of the nifD, nifK, nifE, and nifN genes. J. Mol. Evol. 51, 1–11 (2000).
Zheng, L., White, R. H., Cash, V. L., Jack, R. F. & Dean, D. R. Cysteine desulfurase activity indicates a role for NIFS in metallocluster biosynthesis. Proc. Natl Acad. Sci. USA 90, 2754–2758 (1993).
Yuvaniyama, P., Agar, J. N., Cash, V. L., Johnson, M. K. & Dean, D. R. NifS-directed assembly of a transient [2Fe-2S] cluster within the NifU protein. Proc. Natl Acad. Sci. USA 97, 599–604 (2000).
Shimomura, Y., Wada, K., Fukuyama, K. & Takahashi, Y. The asymmetric trimeric architecture of [2Fe–2S] IscU: implications for its scaffolding during iron–sulfur cluster biosynthesis. J. Mol. Biol. 383, 133–143 (2008).
Fajardo, A. S. et al. Structural insights into the mechanism of the radical SAM Carbide synthase NifB, a key nitrogenase cofactor maturating enzyme. J. Am. Chem. Soc. 142, 11006–11012 (2020).
Kang, W. et al. X-ray crystallographic analysis of NifB with a full complement of clusters: structural insights into the radical SAM-dependent carbide insertion during nitrogenase cofactor assembly. Angew. Chem. Int. Ed. 60, 2364–2370 (2021).
Jenner, L. P., Cherrier, M. V., Amara, P., Rubio, L. M. & Nicolet, Y. An unexpected P-cluster like intermediate en route to the nitrogenase FeMo-co. Chem. Sci. 12, 5269–5274 (2021).
Hu, Y. & Ribbe, M. W. Biosynthesis of nitrogenase FeMoco. Coord. Chem. Rev. 255, 1218–1224 (2011).
Wiig, J. A., Hu, Y. & Ribbe, M. W. Refining the pathway of carbide insertion into the nitrogenase M-cluster. Nat. Commun. 6, 8034 (2015).
Tanifuji, K. et al. Tracing the incorporation of the ‘ninth sulfur’ into the nitrogenase cofactor precursor with selenite and tellurite. Nat. Chem. 13, 1228–1234 (2021).
Hu, Y. et al. Nitrogenase Fe protein: a molybdate/homocitrate insertase. Proc. Natl Acad. Sci. USA 103, 17125–17130 (2006).
Hu, Y., Fay, A. W., Lee, C. C. & Ribbe, M. W. P-cluster maturation on nitrogenase MoFe protein. Proc. Natl Acad. Sci. USA 104, 10424–10429 (2007).
Lee, C. C. et al. Stepwise formation of P-cluster in nitrogenase MoFe protein. Proc. Natl Acad. Sci. USA 106, 18474–18478 (2009).
Burns, R. C., Holsten, R. D. & Hardy, R. W. F. Isolation by crystallization of the MoFe protein of Azotobacter nitrogenase. Biochem. Biophys. Res. Commun. 39, 90–99 (1970).
Mortenson, L. E., Morris, J. A. & Jeng, D. Y. Purification, metal composition and properties of molybdo-ferredoxin and azoferredoxin, two of the components of the nitrogen-fixing system of Clostridium pasteurianum. Biochim. Biophys. Acta 141, 516–522 (1967).
Kirn, J. & Rees, D. C. Crystallographic structure and functional implications of the nitrogenase molybdenum–iron protein from Azotobacter vinelandii. Nature 360, 553–560 (1992).
Holm, R. H. & Lo, W. Structural conversions of synthetic and protein-bound iron–sulfur clusters. Chem. Rev. 116, 13685–13713 (2016).
Lee, S. C., Lo, W. & Holm, R. H. Developments in the biomimetic chemistry of cubane-type and higher nuclearity iron–sulfur clusters. Chem. Rev. 114, 3579–3600 (2014).
Venkateswara Rao, P. & Holm, R. H. Synthetic analogues of the active sites of iron–sulfur proteins. Chem. Rev. 104, 527–560 (2004).
Ohki, Y. & Tatsumi, K. New synthetic routes to metal-sulfur clusters relevant to the nitrogenase metallo-clusters. Z. Anorg. Allg. Chem. 639, 1340–1349 (2013).
Fomitchev, D. V., McLauchlan, C. C. & Holm, R. H. Heterometal cubane-type MFe3S4 clusters (M=Mo, V) trigonally symmetrized with hydrotris(pyrazolyl)borate(1−) and tris(pyrazolyl)methanesulfonate(1−) capping ligands. Inorg. Chem. 41, 958–966 (2002).
Coucouvanis, D. et al. Synthesis, characterization, and reactivity of new clusters that contain the [MFe3S4]0 core, M = molybdenum, tungsten. A weakly perturbed [MFe3S4]0 unit structurally and electronically analogous to the reduced three-ion centers in ferredoxins. J. Am. Chem. Soc. 114, 2472–2482 (1992).
Demadis, K. D., Campana, C. F. & Coucouvanis, D. Synthesis and structural characterization of the new Mo2Fe6S8(PR3)6(Cl4-cat)2 clusters. double cubanes containing two edge-linked [MoFe3S4]2+ reduced cores. J. Am. Chem. Soc. 117, 7832–7833 (1995).
Hauser, C., Bill, E. & Holm, R. H. Single- and double-cubane clusters in the multiple oxidation states [VFe3S4]3+,2+,1+. Inorg. Chem. 41, 1615–1624 (2002).
Berlinguette, C. P., Miyaji, T., Zhang, Y. & Holm, R. H. Precursors to clusters with the topology of the PN cluster of nitrogenase: edge-bridged double cubane clusters [(Tp)2Mo2Fe6S8L4]z: synthesis, structures, and electron transfer series. Inorg. Chem. 45, 1997–2007 (2006).
Scott, T. A. & Holm, R. H. VFe3S4 single and double cubane clusters: synthesis, structures, and dependence of redox potentials and electron distribution on ligation and heterometal. Inorg. Chem. 47, 3426–3432 (2008).
Berlinguette, C. P. & Holm, R. H. Edge-bridged Mo2Fe6S8 to PN-type Mo2Fe6S9 cluster conversion: structural fate of the attacking sulfide/selenide nucleophile. J. Am. Chem. Soc. 128, 11993–12000 (2006).
Zhang, Y. & Holm, R. H. Synthesis of a molecular Mo2Fe6S9 cluster with the topology of the PN cluster of nitrogenase by rearrangement of an edge-bridged Mo2Fe6S8 double cubane. J. Am. Chem. Soc. 125, 3910–3920 (2003).
Osterloh, F., Achim, C. & Holm, R. H. Molybdenum–iron–sulfur clusters of nuclearities eight and sixteen, including a topological analogue of the P-cluster of nitrogenase. Inorg. Chem. 40, 224–232 (2001).
Ohta, S., Ohki, Y., Hashimoto, T., Cramer, R. E. & Tatsumi, K. A nitrogenase cluster model [Fe8S6O] with an oxygen unsymmetrically bridging two proto-Fe4S3 cubes: relevancy to the substrate binding mode of the FeMo cofactor. Inorg. Chem. 51, 11217–11219 (2012).
Moula, G. et al. Synthesis of a nitrogenase PN-cluster model with [Fe8S7(μ-Sthiolate)2] core from the all-ferric [Fe4S4(Sthiolate)4] cubane synthon. Angew. Chem. Int. Ed. 60, 15792–15797 (2021).
Ohki, Y., Tanifuji, K., Yamada, N., Cramer, R. E. & Tatsumi, K. Formation of a nitrogenase P-cluster [Fe8S7] core via reductive fusion of two all-ferric [Fe4S4] clusters. Chem. Asian J. 7, 2222–2224 (2012).
Ohki, Y., Sunada, Y., Honda, M., Katada, M. & Tatsumi, K. Synthesis of the P-cluster inorganic core of nitrogenases. J. Am. Chem. Soc. 125, 4052–4053 (2003).
Ohki, Y., Ikagawa, Y. & Tatsumi, K. Synthesis of new [8Fe-7S] clusters: a topological link between the core structures of P-cluster, FeMo-co, and FeFe-co of nitrogenases. J. Am. Chem. Soc. 129, 10457–10465 (2007).
Ohki, Y. et al. Synthesis, structures, and electronic properties of [8Fe-7S] cluster complexes modeling the nitrogenase P-cluster. J. Am. Chem. Soc. 131, 13168–13178 (2009).
Goh, C., Segal, B. M., Huang, J., Long, J. R. & Holm, R. H. Polycubane clusters: synthesis of [Fe4S4(PR3)4]1+,0 (R = But, Cy, Pri) and [Fe4S4]0 core aggregation upon loss of phosphine. J. Am. Chem. Soc. 118, 11844–11853 (1996).
Brown, A. C. & Suess, D. L. M. An iron–sulfur cluster with a highly pyramidalized three-coordinate iron center and a negligible affinity for dinitrogen. J. Am. Chem. Soc. 145, 20088–20096 (2023).
Deng, L. & Holm, R. H. Stabilization of fully reduced iron−sulfur clusters by carbene ligation: the [FenSn]0 oxidation levels (n = 4, 8). J. Am. Chem. Soc. 130, 9878–9886 (2008).
Rao, P. V., Bhaduri, S., Jiang, J., Hong, D. & Holm, R. H. On [Fe4S4]2+−(μ2-SR)−MII bridge formation in the synthesis of an A-cluster analogue of carbon monoxide dehydrogenase/acetylcoenzyme A synthase. J. Am. Chem. Soc. 127, 1933–1945 (2005).
Terada, T. et al. Tridentate thiolate ligands: application to the synthesis of the site-differentiated [4Fe-4S] cluster having a hydrosulfide ligand at the unique iron center. Chem. Asian J. 7, 920–929 (2012).
Rettberg, L. A. et al. Identity and function of an essential nitrogen ligand of the nitrogenase cofactor biosynthesis protein NifB. Nat. Commun. 11, 1757 (2020).
Bak, D. W. & Elliott, S. J. Alternative FeS cluster ligands: tuning redox potentials and chemistry. Curr. Opin. Chem. Biol. 19, 50–58 (2014).
Stack, T. D. P. & Holm, R. H. Subsite-differentiated analogs of biological [4Fe-4S]2. clusters: synthesis, solution and solid-state structures, and subsite-specific reactions. J. Am. Chem. Soc. 110, 2484–2494 (1988).
Bostelaar, T. M., Brown, A. C., Sridharan, A. & Suess, D. L. M. A general method for metallocluster site-differentiation. Nat. Synth. 2, 740–748 (2023).
Grunwald, L., Abbott, D. F. & Mougel, V. Gauging iron–sulfur cubane reactivity from covalency: trends with oxidation state. JACS Au 4, 1315–1322 (2024).
Grunwald, L. et al. A complete biomimetic iron-sulfur cubane redox series. Proc. Natl Acad. Sci. USA 119, e2122677119 (2022).
Grunwald, L., Inoue, M., Carril, P. C., Wörle, M. & Mougel, V. Gated electron transfers at synthetic iron-sulfur cubanes. Chem 10, 365–387 (2024).
Glaser, T., Hedman, B., Hodgson, K. O. & Solomon, E. I. Ligand edge X-ray absorption spectroscopy: a direct probe of ligand–metal covalency. Acc. Chem. Res. 33, 859–868 (2000).
Ohki, Y. et al. Synthetic analogues of [Fe4S4(Cys)3(His)] in hydrogenases and [Fe4S4(Cys)4] in HiPIP derived from all-ferric [Fe4S4{N(SiMe3)2}4]. Proc. Natl Acad. Sci. USA 108, 12635–12640 (2011).
Tanifuji, K., Tajima, S., Ohki, Y. & Tatsumi, K. Interconversion between [Fe4S4] and [Fe2S2] clusters bearing amide ligands. Inorg. Chem. 55, 4512–4518 (2016).
Mitra, D. et al. Dynamics of the [4Fe-4S] cluster in Pyrococcus furiosus D14C ferredoxin via nuclear resonance vibrational and resonance Raman spectroscopies, force field simulations, and density functional theory calculations. Biochemistry 50, 5220–5235 (2011).
Xiao, Y. et al. Dynamics of an [Fe4S4(SPh)4]2− cluster explored via IR, Raman, and nuclear resonance vibrational spectroscopy (NRVS)-analysis using 36S substitution, DFT calculations, and empirical force fields. Dalton Trans. 18, 2192–2201 (2006).
Mitra, D. et al. Cluster vibrations and structure in nitrogenase Fe protein at three oxidation levels via combined NRVS, EXAFS, and DFT analyses. J. Am. Chem. Soc. 135, 2530–2543 (2013).
Xiao, Y. et al. How nitrogenase shakes—initial information about P-cluster and FeMo-cofactor normal modes from nuclear resonance vibrational spectroscopy (NRVS). J. Am. Chem. Soc. 128, 7608–7612 (2006).
Shoji, M. et al. Theory of chemical bonds in metalloenzymes V: hybrid-DFT studies of the inorganic [8Fe–7S] core. Int. J. Quantum Chem. 106, 3288–3302 (2006).
Yoo, S. J., Angove, H. C., Papaefthymiou, V., Burgess, B. K. & Münck, E. Mössbauer study of the MoFe protein of nitrogenase from Azotobacter vinelandii using selective 57Fe enrichment of the M-centers. J. Am. Chem. Soc. 122, 4926–4936 (2000).
Bjornsson, R., Neese, F. & DeBeer, S. Revisiting the Mössbauer isomer shifts of the FeMoco cluster of nitrogenase and the cofactor charge. Inorg. Chem. 56, 1470–1477 (2017).
Badding, E. D., Srisantitham, S., Lukoyanov, D. A., Hoffman, B. M. & Suess, D. L. M. Connecting the geometric and electronic structures of the nitrogenase iron–molybdenum cofactor through site-selective 57Fe labelling. Nat. Chem. 15, 658–665 (2023).
Spatzal, T. et al. Evidence for interstitial carbon in nitrogenase FeMo cofactor. Science 334, 940–940 (2011).
Zhu, Q., Costentin, C., Stubbe, J. & Nocera, D. G. Disulfide radical anion as a super-reductant in biology and photoredox chemistry. Chem. Sci. 14, 6876–6881 (2023).
Acknowledgements
F. Campell, M. Mössner, V. Bandmann, N. Herren, J.-M. Rojas and P. Gärtner are acknowledged for synthesizing [FeCp2]+ salts as part of ETH Zürich’s third semester inorganic and organic chemistry laboratory practical course (AOCP II). L.G. thanks Dr. P. L. Türtscher for insightful discussions. We are grateful to N. Nagasawa and K. Tamasaku for their assistance with 57Fe NRVS measurements, conducted at SPring-8 BL19LXU (RIKEN proposal nos. 20230009 and 20240030). We also thank T. Grunwald for her design of the table of contents artwork. L.G. and V.M. thank the European Research Council under the European Union’s Horizon 2020 research and innovation programme (grant agreement no. 853064). L.G. also thanks the Swiss Scholarship of the Chemical Industry (SSCI) for funding. Research by S.P.C. was supported by grant nos. NIH GM-65440 and NSF MCB-2149122. G.B., P.D. and M.C. thank the French National Agency for Research (Labex ARCANE, grant nos. CBH-EUR-GS, ANR-17-EURE-0003 and ANR-23-CE07-0043) for funding.
Funding
Open access funding provided by Swiss Federal Institute of Technology Zurich.
Author information
Authors and Affiliations
Contributions
L.G. conducted and developed the synthetic work with support from M.L.W. and H.S. L.G. conducted all computational analyses. L.G. and G.B. analysed data and visualized it. M.W., M.C., P.D., H.W., Y.Y., S.P.C. and G.B. contributed to spectroscopic data collection and analysis. L.G. and V.M. wrote the article with support from all authors. L.G. and V.M. designed and conceptualized the research. V.M. supervised the project.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Chemistry thanks the anonymous reviewers for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Supplementary Information
Supplementary Methods (synthetic procedures, instrumentation, computational details), Discussion, Notes 1–6, Figs. 1–112, Tables 1–12 and Refs. 1–74.
Supplementary Data 1
XYZ format coordinates for the broken-symmetry DFT-optimized geometry of ildc.
Supplementary Video 1
GIF animations of broken-symmetry DFT-calculated normal modes of ildc.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Grunwald, L., Weber, M.L., Seng, H. et al. Stepwise and reversible assembly of [2Fe–2S] rhombs to [8Fe–8S] clusters and their topological interconversions. Nat. Chem. 17, 1586–1595 (2025). https://doi.org/10.1038/s41557-025-01895-9
Received:
Accepted:
Published:
Version of record:
Issue date:
DOI: https://doi.org/10.1038/s41557-025-01895-9
