
Abstract
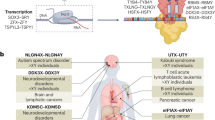
Sex chromosomes are a fundamental aspect of sex-biased biology, but the extent to which homologous X–Y gene pairs (‘the gametologs’) contribute to sex-biased phenotypes remains hotly debated. Although these genes tend to exhibit large sex differences in expression throughout the body (XX females can express both X members, and XY males can express one X and one Y member), there is conflicting evidence regarding the degree of functional divergence between the X and Y members. Here we develop and apply co-expression fingerprint analysis to characterize functional divergence between the X and Y members of 17 gametolog gene pairs across >40 human tissues. Gametolog pairs exhibit functional divergence between the sexes that is driven by divergence between the X versus Y members (assayed in males), and this within-pair divergence is greatest among pairs with evolutionarily distant X and Y members. These patterns reflect that X versus Y gametologs show coordinated patterns of asymmetric coupling with large sets of autosomal genes, which are enriched for functional pathways and gene sets implicated in sex-biased biology and disease. Our findings suggest that the X versus Y gametologs have diverged in function and prioritize specific gametolog pairs for future targeted experimental studies.
This is a preview of subscription content, access via your institution
Access options
Access Nature and 54 other Nature Portfolio journals
Get Nature+, our best-value online-access subscription
$32.99 / 30 days
cancel any time
Subscribe to this journal
Receive 12 digital issues and online access to articles
$119.00 per year
only $9.92 per issue
Buy this article
- Purchase on SpringerLink
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout





Similar content being viewed by others

Data availability
The data used for the analyses described in this manuscript were obtained from the GTEx Project (dbGaP accession no. phs000424/GRU).
Code availability
All code used to produce the results in this manuscript is available via GitHub at github.com/ardecasien/gametologs.
References
Natri, H., Garcia, A. R., Buetow, K. H., Trumble, B. C. & Wilson, M. A. The pregnancy pickle: evolved immune compensation due to pregnancy underlies sex differences in human diseases. Trends Genet. 35, 478–488 (2019).
Merikangas, A. K. & Almasy, L. Using the tools of genetic epidemiology to understand sex differences in neuropsychiatric disorders. Genes Brain Behav. 19, e12660 (2020).
Mazure, C. M. & Swendsen, J. Sex differences in Alzheimer’s disease and other dementias. Lancet Neurol. 15, 451–452 (2016).
Bae, Y. J. et al. Reference intervals of nine steroid hormones over the life-span analyzed by LC–MS/MS: effect of age, gender, puberty, and oral contraceptives. J. Steroid Biochem. Mol. Biol. 193, 105409 (2019).
Arnold, A. P. The end of gonad-centric sex determination in mammals. Trends Genet. 28, 55–61 (2012).
Cortez, D. et al. Origins and functional evolution of Y chromosomes across mammals. Nature 508, 488–493 (2014).
Ross, M. T. et al. The DNA sequence of the human X chromosome. Nature 434, 325–337 (2005).
Wilson, M. A. & Makova, K. D. Genomic analyses of sex chromosome evolution. Ann. Rev. Genomics Hum. Genet. https://doi.org/10.1146/annurev-genom-082908-150105 (2009).
Lahn, B. T. & Page, D. C. Four evolutionary strata on the human X chromosome. Science 286, 964–967 (1999).
Wilson, M. A. & Makova, K. D. Evolution and survival on eutherian sex chromosomes. PLoS Genet. https://doi.org/10.1371/journal.pgen.1000568 (2009).
Sayres, M. A. W., Wilson Sayres, M. A. & Makova, K. D. Gene survival and death on the human Y chromosome. Mol. Biol. Evol. https://doi.org/10.1093/molbev/mss267 (2013).
Slavney, A., Arbiza, L., Clark, A. G. & Keinan, A. Strong constraint on human genes escaping X-inactivation is modulated by their expression level and breadth in both sexes. Mol. Biol. Evol. 33, 384–393 (2016).
Naqvi, S., Bellott, D. W., Lin, K. S. & Page, D. C. Conserved microRNA targeting reveals preexisting gene dosage sensitivities that shaped amniote sex chromosome evolution. Genome Res. 28, 474–483 (2018).
Bellott, D. W. et al. Mammalian Y chromosomes retain widely expressed dosage-sensitive regulators. Nature 508, 494–499 (2014).
GTEx Consortium et al. Landscape of X chromosome inactivation across human tissues. Nature 550, 244–248 (2017).
Balaton, B. P., Cotton, A. M. & Brown, C. J. Derivation of consensus inactivation status for X-linked genes from genome-wide studies. Biol. Sex. Differ. 6, 1–11 (2015).
Skaletsky, H. et al. The male-specific region of the human Y chromosome is a mosaic of discrete sequence classes. Nature 423, 825–837 (2003).
Godfrey, A. K. et al. Quantitative analysis of Y-chromosome gene expression across 36 human tissues. Genome Res. 30, 860–873 (2020).
Raznahan, A. et al. Sex-chromosome dosage effects on gene expression in humans. Proc. Natl Acad. Sci. USA 115, 7398–7403 (2018).
Liu, S. et al. Aneuploidy effects on human gene expression across three cell types. Proc. Natl Acad. Sci. USA 120, e2218478120 (2023).
San Roman, A. K. et al. The human Y and inactive X chromosomes similarly modulate autosomal gene expression. Cell Genomics 4, 100462 (2024).
Roldan, E. R. & Gomendio, M. The Y chromosome as a battle ground for sexual selection. Trends Ecol. Evol. 14, 58–62 (1999).
Nurk, S. et al. The complete sequence of a human genome. Science 376, 44–53 (2022).
Rhie, A. et al. The complete sequence of a human Y chromosome. Nature 621, 344–354 (2023).
Shen, H. et al. Sexually dimorphic RNA helicases DDX3X and DDX3Y differentially regulate RNA metabolism through phase separation. Mol. Cell 82, 2588–2603.e9 (2022).
Gažová, I., Lengeling, A. & Summers, K. M. Lysine demethylases KDM6A and UTY: the X and Y of histone demethylation. Mol. Genet. Metab. 127, 31–44 (2019).
Johansson, M. M. et al. Spatial sexual dimorphism of X and Y homolog gene expression in the human central nervous system during early male development. Biol. Sex Differ. 7, 5 (2016).
Martínez-Pacheco, M. et al. Expression evolution of ancestral XY gametologs across all major groups of placental mammals. Genome Biol. Evol. 12, 2015–2028 (2020).
Venkataramanan, S., Gadek, M., Calviello, L., Wilkins, K. & Floor, S. N. DDX3X and DDX3Y are redundant in protein synthesis. RNA 27, 1577–1588 (2021).
Walport, L. J. et al. Human UTY (KDM6C) is a male-specific Nϵ-methyl lysyl demethylase. J. Biol. Chem. 289, 18302–18313 (2014).
Gozdecka, M. et al. UTX-mediated enhancer and chromatin remodeling suppresses myeloid leukemogenesis through noncatalytic inverse regulation of ETS and GATA programs. Nat. Genet. 50, 883–894 (2018).
Lan, F. et al. A histone H3 lysine 27 demethylase regulates animal posterior development. Nature 449, 689–694 (2007).
Nguyen, T. A. et al. A cluster of autism-associated variants on X-linked NLGN4X functionally resemble NLGN4Y. Neuron 106, 759–768.e7 (2020).
Wang, Z., Sun, L. & Paterson, A. D. Major sex differences in allele frequencies for X chromosomal variants in both the 1000 Genomes Project and gnomAD. PLoS Genet. 18, e1010231 (2022).
Lucotte, E. A., Laurent, R., Heyer, E., Ségurel, L. & Toupance, B. Detection of allelic frequency differences between the sexes in humans: a signature of sexually antagonistic selection. Genome Biol. Evol. 8, 1489–1500 (2016).
Ciesielski, T. H., Bartlett, J., Iyengar, S. K. & Williams, S. M. Hemizygosity can reveal variant pathogenicity on the X-chromosome. Hum. Genet. 142, 11–19 (2023).
Jolly, L. A. et al. Missense variant contribution to USP9X-female syndrome. npj Genom. Med 5, 53 (2020).
Turner, T. N. et al. Sex-based analysis of de novo variants in neurodevelopmental disorders. Am. J. Hum. Genet. 105, 1274–1285 (2019).
Laumonnier, F. et al. X-linked mental retardation and autism are associated with a mutation in the NLGN4 gene, a member of the neuroligin family. Am. J. Hum. Genet. 74, 552–557 (2004).
Allocco, D. J., Kohane, I. S. & Butte, A. J. Quantifying the relationship between co-expression, co-regulation and gene function. BMC Bioinformatics 5, 1–10 (2004).
Lea, A. et al. Genetic and environmental perturbations lead to regulatory decoherence. eLife 8, e40538 (2019).
Oliver, S. Guilt-by-association goes global. Nature 403, 601–603 (2000).
Li, L. et al. Joint embedding of biological networks for cross-species functional alignment. Bioinformatics 39, btad529 (2023).
Lu, Y., Feng, Z., Zhang, S. & Wang, Y. Annotating regulatory elements by heterogeneous network embedding. Bioinformatics 38, 2899–2911 (2022).
GTEx Consortium. The GTEx Consortium atlas of genetic regulatory effects across human tissues. Science 369, 1318–1330 (2020).
Hughes, J. F. et al. Strict evolutionary conservation followed rapid gene loss on human and rhesus Y chromosomes. Nature 483, 82–86 (2012).
Veerappa, A. M., Padakannaya, P. & Ramachandra, N. B. Copy number variation-based polymorphism in a new pseudoautosomal region 3 (PAR3) of a human X-chromosome-transposed region (XTR) in the Y chromosome. Funct. Integr. Genomics 13, 285–293 (2013).
Cotter, D. J., Brotman, S. M. & Wilson Sayres, M. A. Genetic diversity on the human X chromosome does not support a strict pseudoautosomal boundary. Genetics 203, 485–492 (2016).
Trombetta, B., Sellitto, D., Scozzari, R. & Cruciani, F. Inter- and intraspecies phylogenetic analyses reveal extensive X–Y gene conversion in the evolution of gametologous sequences of human sex chromosomes. Mol. Biol. Evol. 31, 2108–2123 (2014).
Oliva, M. et al. The impact of sex on gene expression across human tissues. Science 369, eaba3066 (2020).
Lopes-Ramos, C. M. et al. Sex differences in gene expression and regulatory networks across 29 human tissues. Cell Rep. 31, 107795 (2020).
Rodríguez-Montes, L. et al. Sex-biased gene expression across mammalian organ development and evolution. Science 382, eadf1046 (2023).
Langfelder, P. & Horvath, S. WGCNA: an R package for weighted correlation network analysis. BMC Bioinformatics 9, 1–13 (2008).
Hartman, R. J. G., Mokry, M., Pasterkamp, G. & den Ruijter, H. M. Sex-dependent gene co-expression in the human body. Sci. Rep. 11, 18758 (2021).
Cai, J. J., Borenstein, E. & Petrov, D. A. Broker genes in human disease. Genome Biol. Evol. 2, 815–825 (2010).
Piñero, J., Berenstein, A., Gonzalez-Perez, A., Chernomoretz, A. & Furlong, L. I. Uncovering disease mechanisms through network biology in the era of next generation sequencing. Sci. Rep. 6, 24570 (2016).
Xu, J. & Li, Y. Discovering disease-genes by topological features in human protein–protein interaction network. Bioinformatics 22, 2800–2805 (2006).
Liao, B.-Y. & Weng, M.-P. Unraveling the association between mRNA expressions and mutant phenotypes in a genome-wide assessment of mice. Proc. Natl Acad. Sci. USA 112, 4707–4712 (2015).
Fass, S. B. et al. Relationship between sex biases in gene expression and sex biases in autism and Alzheimer’s disease. Biol. Sex Differ. 15, 47 (2024).
May, T., Adesina, I., McGillivray, J. & Rinehart, N. J. Sex differences in neurodevelopmental disorders. Curr. Opin. Neurol. 32, 622–626 (2019).
Satterstrom, F. K. et al. Large-scale exome sequencing study implicates both developmental and functional changes in the neurobiology of autism. Cell 180, 568–584.e23 (2020).
Iossifov, I. et al. The contribution of de novo coding mutations to autism spectrum disorder. Nature 515, 216–221 (2014).
De Rubeis, S. et al. Synaptic, transcriptional and chromatin genes disrupted in autism. Nature 515, 209–215 (2014).
Wilfert, A. B. et al. Recent ultra-rare inherited variants implicate new autism candidate risk genes. Nat. Genet. 53, 1125–1134 (2021).
Zhou, X. et al. Integrating de novo and inherited variants in 42,607 autism cases identifies mutations in new moderate-risk genes. Nat. Genet. 54, 1305–1319 (2022).
Köglsberger, S. et al. Gender-specific expression of ubiquitin-specific peptidase 9 modulates tau expression and phosphorylation: possible implications for tauopathies. Mol. Neurobiol. 54, 7979–7993 (2017).
Riera-Escamilla, A. et al. Large-scale analyses of the X chromosome in 2,354 infertile men discover recurrently affected genes associated with spermatogenic failure. Am. J. Hum. Genet. 109, 1458–1471 (2022).
Walsh, M. J. M., Wallace, G. L., Gallegos, S. M. & Braden, B. B. Brain-based sex differences in autism spectrum disorder across the lifespan: a systematic review of structural MRI, fMRI, and DTI findings. Neuroimage Clin. 31, 102719 (2021).
DeCasien, A. R., Guma, E., Liu, S. & Raznahan, A. Sex differences in the human brain: a roadmap for more careful analysis and interpretation of a biological reality. Biol. Sex. Differ. 13, 43 (2022).
Wyckoff, G. J., Li, J. & Wu, C.-I. Molecular evolution of functional genes on the mammalian Y chromosome. Mol. Biol. Evol. 19, 1633–1636 (2002).
Gerrard, D. T. & Filatov, D. A. Positive and negative selection on mammalian Y chromosomes. Mol. Biol. Evol. 22, 1423–1432 (2005).
Zhou, Y. et al. Eighty million years of rapid evolution of the primate Y chromosome. Nat. Ecol. Evol. 7, 1114–1130 (2023).
San Roman, A. K. et al. The human inactive X chromosome modulates expression of the active X chromosome. Cell Genom. 3, 100259 (2023).
Bray, N. L., Pimentel, H., Melsted, P. & Pachter, L. Near-optimal probabilistic RNA-seq quantification. Nat. Biotechnol. 34, 525–527 (2016).
Webster, T. H. et al. Identifying, understanding, and correcting technical artifacts on the sex chromosomes in next-generation sequencing data. Gigascience 8, giz074 (2019).
Soneson, C., Love, M. I. & Robinson, M. D. Differential analyses for RNA-seq: transcript-level estimates improve gene-level inferences. F1000Res. 4, 1521 (2015).
Robinson, M. D. & Oshlack, A. A scaling normalization method for differential expression analysis of RNA-seq data. Genome Biol. 11, R25 (2010).
Smyth, G. K. in Bioinformatics and Computational Biology Solutions Using R and Bioconductor (eds Gentleman, R. et al.) 397–420 (Springer, 2021).
Wang, Y., Hicks, S. C. & Hansen, K. D. Addressing the mean-correlation relationship in co-expression analysis. PLoS Comput. Biol. https://doi.org/10.1371/journal.pcbi.1009954 (2022).
Giurgiu, M. et al. CORUM: the comprehensive resource of mammalian protein complexes—2019. Nucleic Acids Res. 47, D559–D563 (2018).
Benjamini, Y. & Hochberg, Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J. R. Stat. Soc. Ser. B 57, 289–300 (1995).
Suzuki, R. & Shimodaira, H. Pvclust: an R package for assessing the uncertainty in hierarchical clustering. Bioinformatics 22, 1540–1542 (2006).
Fisher, R. A. in Breakthroughs in Statistics: Methodology and Distribution (eds Kotz, S. & Johnson, N. L.) 66–70 (Springer, 1992).
Alfons, A., Anderegg, N., Aragon, T. et al. DescTools: tools for descriptive statistics. R package version 0.99.45 (2022).
Pandey, R. S., Wilson Sayres, M. A. & Azad, R. K. Detecting evolutionary strata on the human x chromosome in the absence of gametologous y-linked sequences. Genome Biol. Evol. 5, 1863–1871 (2013).
Durinck, S. et al. BioMart and Bioconductor: a powerful link between biological databases and microarray data analysis. Bioinformatics 21, 3439–3440 (2005).
McInnes, L., Healy, J. & Melville, J. UMAP: Uniform Manifold Approximation and Projection. J. Open Source Softw. 3, 861 (2018).
Krijthe, J. H. Rtsne: T-distributed stochastic neighbor embedding using Barnes–Hut implementation version 0.13. GitHub https://github.com/jkrijthe/Rtsne (2023).
Hoffman, G. E. & Schadt, E. E. variancePartition: interpreting drivers of variation in complex gene expression studies. BMC Bioinformatics 17, 1–13 (2016).
Bhuva, D. D., Cursons, J., Smyth, G. K. & Davis, M. J. Differential co-expression-based detection of conditional relationships in transcriptional data: comparative analysis and application to breast cancer. Genome Biol. 20, 1–21 (2019).
Charrad, M., Ghazzali, N., Boiteau, V. & Niknafs, A. NbClust: an R package for determining the relevant number of clusters in a data set. J. Stat. Softw. 61, 1–36 (2014).
Wu, T. et al. clusterProfiler 4.0: A universal enrichment tool for interpreting omics data. Innovation 2, 100141 (2021).
Urbut, S. M., Wang, G., Carbonetto, P. & Stephens, M. Flexible statistical methods for estimating and testing effects in genomic studies with multiple conditions. Nat. Genet. 51, 187–195 (2019).
Stephens, M. False discovery rates: a new deal. Biostatistics 18, 275–294 (2017).
Bovy, J., Hogg, D. W. & Roweis, S. T. Extreme deconvolution: Inferring complete distribution functions from noisy, heterogeneous and incomplete observations. Ann. Appl. Stat. 5, 1657–1677 (2011).
Acknowledgements
We thank N. Snyder-Mackler for feedback on earlier versions of this manuscript and L. Lacbawan for helping us create Fig. 1. The GTEx Project was supported by the Common Fund of the Office of the Director of the National Institutes of Health, and by National Cancer Institute (NCI), National Human Genome Research Institute (NHGRI), National Heart, Lung, and Blood Institute (NHLBI), National Institute on Drug Abuse (NIDA), National Institute of Mental Health (NIMH) and National Institute of Neurological Disorders and Stroke (NINDS). The data used for the analyses described in this manuscript were obtained from dbGaP (accession no. phs000424/GRU). This research was supported (in part) by the Intramural Research Program of the NIMH (1ZIAMH002949-09).
Author information
Authors and Affiliations
Contributions
A.R. and A.R.D. conceived and designed the study. A.R. oversaw the study. A.T. facilitated access to the data. A.R.D. and K.T. analysed the data. S.L. provided analytical support. A.R.D. and A.R. led the writing of the manuscript, and all authors provided valuable feedback and advice during its preparation.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Ecology & Evolution thanks the anonymous reviewers for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data
Extended Data Fig. 1 Methods diagram.
a. Bottom portion of Fig. 1. b. Methods for estimating the significance of asymmetric coupling (overall and per pair) using the CLIP method. c. Method for calculating overall asymmetric coupling.
Extended Data Fig. 2 Mean adjusted expression ratios for each gametolog pair in each tissue.
Comparisons are shown between MX versus FXX (a), MXY versus FXX (b), and MX versus MY (c). Note these results are consistent with those presented by Godfrey and colleagues18.
Extended Data Fig. 3 Clustering of tissues according to functional divergence values.
Hierarchical clustering dendrograms of tissues for between-sex CFD (aCFDXXF–XYM) (a), within-pair absolute CFD (aCFDXM–YM) (b), and within-pair signed CFD (sCFDXM–YM) (c). Red values are AU (Approximately Unbiased) p-values and green values are BP (Bootstrap Probability) values (two-sided) (Methods).
Extended Data Fig. 4 Comparisons between CFD measures calculated using all genes or autosomal genes only.
a. Figure 2c using autosomal genes only. Mean (± standard error) between-sex functional divergence measures per tissue (averaged across gametolog pairs; N = 11-14 pairs, see Supplementary Table 2). Open squares indicate tissues where gametologs exhibit higher mean normalized aCFDXXF–XYM values compared to a null distribution (two-sided; padj < 0.05; Methods). Filled circles indicate tissues where normalized aCFDXXF–XYM values are significantly higher than normalized aCFDXXF–XM values (paired t-tests: padj < 0.05, Methods). Filled squares indicate tissues where gametologs exhibit higher mean normalized aCFDXXF–XM values compared to a null distribution (padj < 0.05, Methods). b-f. Top: aCFDXXF–XYM (b), sCFDXXF–XYM (c), aCFDXXF–XM (d), aCFDXM–YM (e) or sCFDXM–YM (f) per pair and tissue using autosomal genes only (y-axis) or all genes (x-axis). Bottom: histograms of the difference between aCFDXXF–XYM (b), sCFDXXF–XYM (c), aCFDXXF–XM (d), aCFDXM–YM (e) or sCFDXM–YM (f) values estimated using all genes or autosomal genes only (> 0 = higher values when using all genes).
Extended Data Fig. 5 Gametolog X-Y functional divergence predicts between-sex functional divergence.
a. Figure 2e within each tissue. Regression lines are provided for each tissue (black lines, confidence intervals are shaded) b. Between-sex signed co-expression fingerprint divergence (sCFDXXF–XYM) modeled as a function of within-pair signed co-expression fingerprint divergence in males (sCFDXM–YM). Regression lines are provided for each tissue (see legend) and across all data points (black line, confidence interval is shaded). Pearson’s correlation value and associated p-value are shown for the latter.
Extended Data Fig. 6 Regulatory and structural similarity measures.
a. Correlations across regulatory structural dissimilarity measures. Darker blue = more positive correlation; darker red = more negative correlation. b. Structural dissimilarity measures grouped by evolutionary strata (see Fig. 3b or Supplementary Table 5 for N pairs per strata). Data and colors match those in Fig. 3b. Boxplots depict medians (horizonal lines), interquartile ranges [IQRs] (boxes), 1.5*IQRs (whiskers), and outliers (points). c. Within-pair functional divergence aCFDXM–YM (across all pairs and tissues) grouped by evolutionary strata (see Fig. 3b or Supplementary Table 5 for N pairs per strata; see Supplementary Table 2 for N tissues per pair). Boxplots depict medians (horizonal lines), interquartile ranges [IQRs] (boxes), 1.5*IQRs (whiskers), and outliers (points). d. Within-pair functional divergence aCFDXM–YM (across all tissues) grouped by evolutionary strata and gametolog pair (see legend) (see Supplementary Table 2 for N tissues per pair). Boxplots depict medians (horizonal lines), interquartile ranges [IQRs] (boxes), 1.5*IQRs (whiskers), and outliers (points).
Extended Data Fig. 7 Relationships between X-Y gemetolog expression/co-expression and functional divergence.
a. X–Y co-expression values in the current study versus those estimated by Godfrey and colleagues18. We recomputed X–Y co-expression values to be comparable to our estimates of functional divergence (aCFDXXF–XYM and sCFDXXF–XYM). Differences between X–Y co-expression values across studies are the result of methodological differences, as our study: i) used a newer version of the GTEx data (v8 versus v7); ii) removed age effects when calculating adjusted expression levels (Methods); and iii) normalized co-expression values by expression level using spatial quantile normalization (Methods). Regression lines are provided for each tissue (see legend) and across all data points (black line, confidence interval is shaded). These values are highly similar (ρ = 0.617, p < 2.2e-16; linear model: slope = 0.418; p < 2.2e-16) across pair–tissue combinations. Spearman’s rank order correlation and the corresponding p-value are provided. b. aCFDXXF–XYM versus X–Y member co-expression. Regression lines are provided for each tissue (see legend) and across all data points (black line, confidence interval is shaded). Absolute within-pair divergence (aCFDXM–YM) necessarily exhibits a strong negative correlation with the level of co-expression between the X and Y members themselves (within each pair) (ρ = -0.768, p < 2.2e-16). Spearman’s rank order correlation and the corresponding p-value are provided. c. sCFDXXF–XYM versus X–Y member co-expression. Regression lines are provided for each tissue (see legend) and across all data points (black line, confidence interval is shaded). Note that relative to aCFDXXF–XYM (Extended Data Fig. 7b), this relationship is relatively muted for signed divergence (sCFDXM–YM) (ρ = -0.172, p < 0.001). Spearman’s rank order correlation and the corresponding p-value are provided. d. aCFDXM–YM versus the median Y/X expression ratio. Regression lines are provided for each tissue (see legend) and across all data points (black line, confidence interval is shaded). Spearman’s rank order correlation and the corresponding p-value are provided. e. sCFDXM–YM versus the median Y/X expression ratio. Regression lines are provided for each tissue (see legend) and across all data points (black line, confidence interval is shaded). Spearman’s rank order correlation and the corresponding p-value are provided. f. Differences in X–Y expression variance versus differences in X–Y mean expression. Regression lines are provided for each tissue (see legend) and across all data points (black line, confidence interval is shaded). Spearman’s rank order correlation and the corresponding p-value are provided. g. Extended Data Fig. 7d excluding TMSB4X/Y. Regression lines are provided for each tissue (see legend) and across all data points (black line, confidence interval is shaded). Spearman’s rank order correlation and the corresponding p-value are provided. h. Extended Data Fig. 7e excluding TMSB4X/Y. Regression lines are provided for each tissue (see legend) and across all data points (black line, confidence interval is shaded). Spearman’s rank order correlation and the corresponding p-value are provided. i. Extended Data Fig. 7f excluding TMSB4X/Y. Regression lines are provided for each tissue (see legend) and across all data points (black line, confidence interval is shaded). j. Y/X expression ratios (log2) across all pair–tissue combinations. Purple = Y > X expression. Red = X > Y expression.
Extended Data Fig. 8 Distributions of asymmetric coupling.
a. Figure 4d excluding tissue-specific genes. Color indicates direction of coupling (see Fig. 4b legend). Note that although the modal number of tissues in which genes showed significant X- or Y-biased coupling was one (Fig. 4d), the current plot suggests that this pattern is driven by genes with tissue-specific expression. b. Count of genes with significant asymmetric coupling (CLIP padj < 0.05) to a given gametolog pair in a maximum of N tissues. Color indicates direction of coupling (see Fig. 4b legend). c. Extended Data Figure 8b excluding tissue-specific genes. Color indicates direction of coupling (see Fig. 4b legend). d. Count of genes with significant asymmetric coupling (CLIP padj < 0.05) with a maximum of N pairs within a given tissue. Color indicates direction of coupling (see Fig. 4b legend). Note that, while the modal number of gametolog pairs that genes were asymmetrically coupled to (across all tissues) was eight (Fig. 4d), the modal number of pairs within a given tissue was only two (shown here). e. Number of genes with significant (CLIP padj < 0.05) asymmetric coupling for each pair–tissue using the subsamples (N = 66 males, Methods; Supplementary Tables 9,10) versus the entire sample (ρ = 0.598, p < 2.2e-16). Regression lines with confidence intervals are shown. f. Number of genes with significant (CLIP padj < 0.05) asymmetric coupling for each pair using the subsamples (N = 66 males, Methods; Supplementary Tables 9,10) versus the entire sample (ρ = 0.981, p = 1.124e-10). Regression lines with confidence intervals are shown. g. Number of genes with Methods (CLIP padj < 0.05) asymmetric coupling for each tissue using the subsamples (N = 66 males, Methods; Supplementary Tables 9,10) versus the entire sample (ρ = 0.428, p = 0.005). Regression lines with confidence intervals are shown. h. Boxplots of the proportion of pair-tissue combinations in which genes are asymmetrically coupled (CLIP padj < 0.05). ANOVA p = 1.08e-05; Tukey’s HSD padj = 5.93e-05 for X > autosomal. Boxplots depict medians (horizonal lines), interquartile ranges [IQRs] (boxes), 1.5*IQRs (whiskers), and outliers (points). i. Boxplots of asymmetric coupling values among autosomal, X chromosome, and Y chromosome genes with significant asymmetric coupling (CLIP padj < 0.05). Values are shown for each gametolog pair (see Supplementary Table 8 for N genes x tissues per pair). Colored dots on the x-axis indicate significance of comparisons (see bottom legend, Supplementary Table 11; Tukey’s HSD results are only shown for pairs in which ANOVA pad < 0.05). Boxplots depict medians (horizonal lines), interquartile ranges [IQRs] (boxes), 1.5*IQRs (whiskers), and outliers (points). j. Boxplots of asymmetric coupling values among autosomal, X chromosome, and Y chromosome genes with significant asymmetric coupling (CLIP padj < 0.05). Values are shown for each tissue (see Supplementary Table 8 for N genes x pairs per tissue). Colored dots on the x-axis indicate significance of comparisons (see bottom legend, Supplementary Table 11; Tukey’s HSD results are only shown for tissues in which ANOVA padj < 0.05). Non-gametolog Y chromosome genes show strong Y-biased coupling in the testes (for example, HSFY1/2) and stomach (for example, DAZ1/2/4). Boxplots depict medians (horizonal lines), interquartile ranges [IQRs] (boxes), 1.5*IQRs (whiskers), and outliers (points).
Extended Data Fig. 9 Delineating the polarity and biological patterning of X-Y gametolog functional divergence.
a. PCA of asymmetric coupling values across all pairs–tissues (represented by each point, see legend). b. tSNE of asymmetric coupling values across all pairs–tissues (represented by each point, see legend). c. UMAP of asymmetric coupling values across tall pairs–tissues (represented by each point, see legend). d. PCA of asymmetric coupling values across all pairs–tissues (represented by each point, see legend). Similar tissues have been collapsed (Methods). e. tSNE of asymmetric coupling values across all pairs–tissues (represented by each point, see legend). Similar tissues have been collapsed (Methods). f. UMAP of asymmetric coupling values across all pairs–tissues (represented by each point, see legend). Similar tissues have been collapsed (Methods). g. Mean asymmetric coupling values with N = 15 gametolog pairs (excluding AMELX/Y and RPS4X/Y2, which are not expressed) for genes in each cluster (averaged across tissues). N = 8 clusters were derived from N = 11,498 genes whose asymmetric coupling values were predicted by gametolog pair only (see Fig. 4g) (clusters with < 20 genes were removed). Clusters and pairs are ordered according to hierarchical clustering. Asterisks indicate padj < 0.05 (versus null, two-sided; Methods). h. Extended Data Fig. 9g, but with mean sCFDXM–YM values shown for each tissue. i. Extended Data Fig. 9g, but with pairs ranked within each cluster according to their mean sCFDXM–YM values. j. Top enriched biological processes and associated −log10(p-values) for each cluster. Top 2 categories are shown for each cluster. k. Top enriched cellular compartments and associated −log10(p-values) for each cluster. Top 2 categories are shown for each cluster. l. Similar to Extended Data Fig. 9g, but for genes whose expression is predicted by tissue only (see Supplementary Table 13, Column L). Mean asymmetric coupling values in each tissue for genes in each cluster. N = 3 clusters were derived from N = 40 genes whose asymmetric coupling values were predicted by tissue only (Fig. 4g). Clusters and pairs are ordered according to hierarchical clustering. Asterisks indicate padj < 0.05 (versus null, two-sided; Methods). m. Extended Data Fig. 9l, but with mean sCFDXM–YM values shown for each pair. n. Extended Data Fig. 9l, but with tissues ranked within each cluster according to their mean sCFDXM–YM values. o. Extended Data Fig. 9j, but for genes whose expression is predicted by tissue only (see Supplementary Table 13, Column L). Top enriched biological processes and associated −log10(p-values) for each cluster. Top 4 categories are shown for each cluster. p. Extended Data Fig. 9k, but for genes whose expression is predicted by tissue only (see Supplementary Table 13, Column L). Top enriched cellular compartments and associated −log10(p-values) for each cluster. Top 4 categories are shown for each cluster. q. Similar to Extended Data Fig. 9g, but for genes whose expression is predicted by both pair and tissue (see Supplementary Table 13, Column M). Mean asymmetric coupling values for each pair (top) or tissue (bottom) for genes in each cluster. N = 5 clusters were derived from N = 1,141 genes whose asymmetric coupling values were predicted by pair and tissue (Fig. 4g). Clusters and pairs are ordered according to hierarchical clustering. Asterisks indicate padj < 0.05 (versus null, two-sided; Methods). r. Extended Data Fig. 9q, but with mean sCFDXM–YM values shown for each tissue (top) or pair (bottom). s. Similar to Extended Data Fig. 9j, but for genes whose expression is predicted by pair and tissue (see Supplementary Table 13, Column M). Top enriched biological processes and associated −log10(p-values) for each cluster. Top 2 categories are shown for each cluster. t. Similar to Extended Data Fig. 9k, but for genes whose expression is predicted by pair and tissue (see Supplementary Table 13, Column M). Top enriched cellular compartments and associated −log10(p-values) for each cluster. Top 2 categories are shown for each cluster.
Extended Data Fig. 10 X-Y functional divergence predicts sex differences in expression and co-expression.
a. Figure 5a with tissue specific regression lines. b. Enrichment results between overall asymmetric coupling and sex-biased co-expression (Methods). OR = odds ratio. c. Mean scaled co-expression values between all pairs of X-biased genes and Y-biased genes in each tissue in males (green) versus females (yellow) (tissue-specific results in Supplementary Table 23). d. Data used for Fig. 5d. All pair-level asymmetric coupling values for N = 24 ASD risk genes with significant overall asymmetric coupling (CLIP padj<0.05) in BA24 are shown (see right column for direction of overall asymmetric coupling; ASD risk genes are separated by functional category). Only significant pair-level values (CLIP padj<0.05) are included in Fig. 5d, and in this figure they are outlined in black.
Supplementary information
Supplementary Information
Supplementary Notes and Discussion.
Rights and permissions
About this article

Cite this article
DeCasien, A.R., Tsai, K., Liu, S. et al. Evolutionary divergence between homologous X–Y chromosome genes shapes sex-biased biology. Nat Ecol Evol 9, 448–463 (2025). https://doi.org/10.1038/s41559-024-02627-x
Received:
Accepted:
Published:
Issue date:
DOI: https://doi.org/10.1038/s41559-024-02627-x
This article is cited by
-
A cross-species analysis of neuroanatomical covariance sex differences in humans and mice
Biology of Sex Differences (2025)
