
Abstract
Aquatic bacterial rhodopsin proton pumps harvest light energy for photoheterotrophic growth and are known to contain hydroxylated carotenoids that expand the wavelengths of light utilized, but these have not been characterized in marine archaea. Here, by combining a marine chromophore extract with purified archaeal rhodopsins identified in marine metagenomes, we show light energy transfer from diverse hydroxylated carotenoids to heimdallarchaeial rhodopsins (HeimdallRs) from uncultured marine planktonic members of ‘Candidatus Kariarchaeaceae’ (‘Candidatus Asgardarchaeota’). These light-harvesting antennas absorb in the blue-light range and transfer energy to the green-light-absorbing retinal chromophore within HeimdallRs, enabling the use of light that is otherwise unavailable to the rhodopsin. Furthermore, we show elevated proton pumping by the antennas in HeimdallRs under white-light illumination, which better simulates the light conditions encountered by these archaea in their natural habitats. Our results indicate that light-harvesting antennas in microbial rhodopsins exist in families beyond xanthorhodopsins and proteorhodopsins and are present in both marine bacteria and archaea.
Similar content being viewed by others
Main
Proteorhodopsins and xanthorhodopsins are microbial light-driven proton pumps identified in widespread and abundant bacterial, archaeal and some eukaryotic groups in marine and freshwater environments1. It has been predicted that ~50% of microbes in the ocean’s photic zone possess these rhodopsins2 and use them to harvest light energy to sustain a photoheterotrophic lifestyle3,4.
The phenomenon of energy transfer from light-harvesting ketolated (containing a keto group) carotenoid antennas to the retinal chromophore in two xanthorhodopsins was first demonstrated two decades ago5,6. The recent discovery that many proteorhodopsins and xanthorhodopsins can bind hydroxylated (containing a hydroxyl group) carotenoid antennas7 suggested that such light-harvesting complexes have a substantial global impact on rhodopsin-mediated phototrophy in aquatic environments. These antennas transfer the absorbed energy in the violet- and blue-light range (~420–480 nm) to the green-absorbing retinal chromophore (~550 nm)5,6,7, therefore enabling use of light that is otherwise not available to them. The binding is facilitated by a lateral opening in the rhodopsin (known as the fenestration), exposing the retinal β-ionone ring to one of the rings of the cyclic carotenoid antenna6,7,8 and therefore enabling energy transfer. Currently, light energy transfer from hydroxylated carotenoids (lutein, zeaxanthin and nostoxanthin) has been reported only for bacterial proteorhodopsins and xanthorhodopsins7,9.
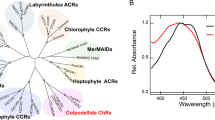
Several marine group II and III archaea (MGII/III, ‘Candidatus Poseidoniia’)10 contain both proteorhodopsins and proton-pumping ACB (Archaea clade B) rhodopsins11,12,13,14 (Fig. 1a and Supplementary Fig. 1). While proteorhodopsins from ‘Ca. Poseidoniia’ are not fenestrated due to bulky residues at position G156 in transmembrane helix 5 (TM5), most ACB rhodopsins possess a fenestration in proximity to the retinal ring facilitated by a leucine residue unusual for this position (Fig. 1a,b). In addition to rhodopsins from ‘Ca. Poseidoniia’, a proton-pumping rhodopsin was recently reported for ‘Candidatus Heimdallarchaeia’ (‘Candidatus Asgardarchaeota’)15,16. This protein (referred to here as HeimdallR1) has a glycine at position 156 and is predicted to have a fenestration exposing the retinal ring similarly to Salinibacter xanthorhodopsin8 and Kin4B8-xanthorhodopsin7. These observations suggest that some of these archaeal proton pumps might utilize carotenoid antennas, despite the evolutionary distance separating them from the more common bacterial proteorhodopsin and xanthorhodopsin families and the presence of non-canonical motifs in transmembrane helix 3 (TM3) (Fig. 1a). The appearance of proton-pumping rhodopsins with a potential for antenna binding in ‘Ca. Heimdallarchaeia’ is of particular interest as members of this Asgard group are the closest archaeal relatives of eukaryotes17,18 and nothing is known about photoheterotrophy among them. In this study, we searched for rhodopsin–carotenoid complexes originating in marine archaea.

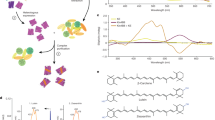
a, NeighborNet network of proteorhodopsins (PRs), xanthorhodopsins (XRs), Archaea clade B (ACB) rhodopsins, HeimdallRs and related rhodopsin families. The three clades of outward proton pumps appearing in marine archaea are highlighted: Archaea clade A proteorhodopsins (ACA) among marine group II and marine group III (Thermoplasmatota: ‘Ca. Poseidoniia’), the ACB family among marine group II and the family of HeimdallRs among the ‘Ca. Kariarchaeaceae’ (‘Ca. Asgardarchaeota’: ‘Ca. Heimdallarchaeia’). Indicated are (from inside out): activity, transmembrane helix 3 motif, presence and type of fenestration, alias and type of carotenoid antenna (-OH for xanthophylls with hydroxyl at carbon C3 and =O for xanthophylls with keto group at carbon C4). Abbreviated family names are as follows: ESR, E. sibiricum rhodopsin; MACR, marine actinobacterial clade rhodopsins; P1, clade P1 (actinobacterial DSE and related DTE rhodopsins); P4, clade P4; Proteo-SR, proteorhodopsin-related sensory rhodopsins; TwRs, twin-peaked rhodopsins. b, Structural comparison of fenestrated rhodopsins: S. ruber xanthorhodopsin with salinixanthin (PDB: 3DDL)8, Kin4B8-xanthorhodopsin with zeaxanthin (PDB: 7YTB)7, ACB-G35 rhodopsin (AlphaFold 3 model50) and HeimdallR1 (PDB: 9JTQ, this study) versus a non-fenestrated rhodopsin, bacteriorhodopsin (BR; PDB: 1IW6)85. The structures are aligned on the basis of the retinal β-ionone ring position, and the fenestration zone is highlighted. For clarity, salinixanthin and zeaxanthin are not shown in the enlarged fenestration zone of S. ruber xanthorhodopsin and Kin4B8-xanthorhodopsin structures, respectively. c, Absorbance spectra of ACB-G35 rhodopsin and HeimdallR1 before (purple) and after (brown) incubation, and wash, with a marine chromophore extract. d, Absorbance changes of HeimdallR1 before (purple) and after (orange) incubation, and wash, with pure lutein, diatoxanthin, fucoxanthin and β-carotene. The carotenoid structures are shown at the bottom.
Results
Asgard rhodopsin HeimdallR1 binds environmental carotenoids
Two representative fenestrated archaeal rhodopsins, ACB-G35 rhodopsin and HeimdallR1 (Fig. 1b) were incubated with a marine (Mediterranean Sea) chromophore extract containing a natural mix of carotenoids (Methods). After purifying the proteins from the chromophore extract mixture, a change in absorbance was observed for HeimdallR1 but not for the ACB rhodopsin (Fig. 1c), suggesting binding of specific chromophores to HeimdallR1. High-performance liquid chromatography diode array detector (HPLC-DAD) analysis of the complexes showed that the enriched chromophores consisted mainly of cyclic hydroxylated (lutein, diatoxanthin and fucoxanthin) and non-hydroxylated (β-carotene) carotenoids (Extended Data Fig. 1; see carotenoid structures in Fig. 1d). When switching to pure carotenoids, the non-symmetric hydroxylated carotenoids lutein, diatoxanthin and fucoxanthin showed binding to HeimdallR1, while β-carotene did not (Fig. 1d). Interestingly, this carotenoid specificity of HeimdallR1 is similar but not identical to that of the bacterial Kin4B8-xanthorhodopsin which does not bind fucoxanthin (Extended Data Fig. 2). This indicates a unique binding preference of HeimdallR1 towards fucoxanthin and further implies that carotenoid binding in fenestrated rhodopsins is a complex trait.
Distribution and diversity of heimdallarchaeial rhodopsins
HeimdallR1 from the Red Sea has thus far remained an isolated monotypic branch15,16. By searching multiple databases for similar sequences, we found three groups of closely related proteins: HeimdallR1-like, HeimdallR2-like and HeimdallR3, in marine and estuarine locations (Fig. 2a, Extended Data Fig. 3 and Supplementary Fig. 2). The HeimdallR1- and HeimdallR2-like subclades were closest to each other with pairwise identities between 81% and 86%. The third subclade comprised a single protein, HeimdallR3, obtained via co-assembly of metagenomic data from the Groves Creek Marsh (Georgia, USA), which showed an identity of 63–67% to HeimdallR1/R2.

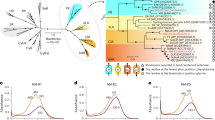
a, Global distribution of heimdallarchaeial rhodopsins. Samples in which HeimdallRs were detected are indicated with squares, with their size proportional to their percentage among all regular microbial rhodopsins. Samples lacking HeimdallRs (but containing other archaeal proton pumps) are indicated with crosses. b, Top: phylogenetic relationships between members of the family ‘Ca. Kariarchaeaceae’ based on concatenated alignment of 153 markers present in at least 60% of the genomes. The position of the root is indicated with a triangle. Rapid bootstrap support values are indicated for branches with support >90. The scaffold Kari_Gs0128817 was placed on the phylogenetic tree and its position is highlighted in red and supplied with the corresponding likelihood weight ratio. Genomes with HeimdallR genes are indicated with stars coloured by the corresponding rhodopsin subclade (Extended Data Fig. 3). Histograms reflect the incidence of phylogenetic markers in the final alignment for each genome, and pink squares mark the presence of the two genes used for phylogenetic placement of Kari_Gs0128817. Bottom: genomic context of the HeimdallR genes from the three clades. Genes used to place Kari_Gs0128817 on the tree are highlighted.
The two widespread subclades were found in metagenome-assembled genomes (MAGs) belonging to the same archaeal species ‘Candidatus Kariarchaeum pelagium’19 despite the sequence divergence exceeding expected intraspecific variation20. To clarify the origin of this divergence, we recruited metagenomic contigs from diverse locations to the core pan-genome of ‘Ca. K. pelagium’ (Extended Data Fig. 4). While genetic variation at most of the loci in the genome was within the limits expected for a single prokaryotic species, the heimdallR gene and genes in its immediate vicinity showed an unusually high variation without any indication that the corresponding region might be duplicated in the genome (Extended Data Fig. 4b,c). This strongly suggests that the appearance of the HeimdallR1-like and HeimdallR2-like alleles in ‘Ca. K. pelagium’ is a result of introgression from a related unsampled species. Incidentally, phylogenetic relationships between the ‘Ca. K. pelagium’ MAGs were inconsistent with the HeimdallR1/R2 divide (Fig. 2b), which might be a result of binning artefacts or ongoing intraspecific homologous recombination.
Together, HeimdallRs can be characterized as a minor yet widespread family of proton pumps: comprising up to 0.77% among genes for regular microbial rhodopsins (7.2% of marine archaeal rhodopsin pumps) and up to 0.95% of their transcripts (21% of archaeal rhodopsin pumps) in the open-ocean sunlit stations from Tara Oceans21 (Fig. 2a and Supplementary Fig. 2). HeimdallRs and ‘Ca. K. pelagium’ appear at shallow depths in both open-ocean and coastal locations (including estuaries and salt marshes) between parallels 40° N and 24° S with peak abundances in the Indian Ocean and the Red Sea, along the North-West Atlantic coast and the East Pacific coast (Fig. 2a and Extended Data Fig. 4a).
Sequence alignment and structure predictions showed that all HeimdallRs have fenestrations as in HeimdallR1, and heterologous expression of HeimdallR2 and HeimdallR3 indeed showed that they can also bind lutein, diatoxanthin and fucoxanthin (Extended Data Fig. 5). This indicates that all pelagic ‘Ca. Kariarchaeum’ possess rhodopsins capable of binding xanthophylls. Nevertheless, gene content analysis of the ‘Ca. K. pelagium’ pan-genome revealed only genes for biosynthesis of linear carotenoids (Extended Data Fig. 3). This indicates that both the retinal and the xanthophylls must be obtained by these archaea from the environment.
Gene content analysis of the other ‘Ca. Kariarchaeaceae’ yielded no genes for proton-pumping rhodopsins, consistent with their deep-sea and subsurface habitats. Phylogenetic relationships within the family and their ties to other heimdallarchaeia point to a singular origin of the pelagic and putatively photoheterotrophic lifestyle of ‘Ca. Kariarchaeum’ derived from an ancestral dark habitat (Fig. 2b).
Energy transfer from different carotenoids to HeimdallRs
Xanthophyll binding to different members of the HeimdallR family indicates that this ability is physiologically relevant and is supported by selection. We thus tested whether the carotenoids function as light-harvesting antennas in HeimdallRs. Fluorescence analysis of HeimdallR1–carotenoid complexes demonstrated that energy transfer takes place from lutein, diatoxanthin or fucoxanthin to the retinal chromophore (Fig. 3a). Fluorescence measurements revealed energy transfer from these xanthophylls also to the retinal in the closely related HeimdallR2, while for HeimdallR3, a clear fluorescence signal could be observed only with lutein (Extended Data Fig. 5). Analogously, laser-flash photolysis revealed an increase in transient absorption signal for HeimdallR1 bound to lutein or fucoxanthin when excited at the violet–blue (425–490 nm) region, indicating the enhancement of the retinal isomerization quantum yield (Fig. 3b). Notably, the transient absorption signal of HeimdallR1 was enhanced 1.6-fold compared with that of Kin4B8-xanthorhodopsin with lutein, indicating more efficient energy transfer in the former7.

a, Fluorescence excitation spectra of HeimdallR1 upon incubation with (orange) or without (purple) lutein (top), diatoxanthin (middle) or fucoxanthin (bottom); emission was recorded at 720 nm. b, The ratios of transient absorption (TA) change in HeimdallR1 with and without lutein (top) and with and without fucoxanthin (bottom) at different excitation wavelengths (425, 450, 465, 480, 545 and 590 nm for lutein and 440, 465, 580, 490, 530, 540, 550 and 600 nm for fucoxanthin) (bars coloured according to the colour of excitation light). The absorption spectra of HeimdallR1 without (purple line) and with (orange line for lutein and coral for fucoxanthin) xanthophylls are overlaid. The red dashed lines indicate no difference between with and without xanthophyll. Tables with the quantum yield (QY) percentages and the pictures of the purified proteins are shown next to the corresponding results. c, Light-minus-dark difference FTIR spectra at 77 K upon illumination of HeimdallR1 with (red) or without (black) fucoxanthin, HeimdallR1 with (red) or without (black) lutein, HeimdallR1 with lutein in H2O (red) or D2O (blue), and Kin4B8-xanthorhodopsin with (green) or without (black) lutein. Hydrated films of lipid-reconstituted protein with H2O are illuminated at 540 nm light (solid lines), which forms the red-shifted K intermediate. Each peak originates from a hydrogen out-of-plane (HOOP) vibration of the retinal chromophore, which shifts upon xanthophyll binding to HeimdallR1, but not to Kin4B8-xanthorhodopsin. One division of the y axis corresponds to 0.0006 absorbance units. LUT, lutein; DIATO, diatoxanthin; FUCO, fucoxanthin.
The influence of the xanthophyll binding on the retinal isomer composition in HeimdallR1 was investigated using HPLC of retinal oximes produced by hydrolysing the retinal Schiff base (RSB) with hydroxylamine. In the dark-adapted (DA) form, the all-trans configuration (Fig. 4a) was dominant. The binding of lutein and fucoxanthin further increased the all-trans form. The retinal photoisomerized to the 13-cis form, which was reflected in transient absorption changes, representing red-shifted (K and O) and blue-shifted (M) photointermediates (Fig. 4b–d) in addition to L, N and HeimdallR1ʹ intermediates with maximum absorption wavelengths (λamax) close to the initial state (Fig. 4e). A sharp peak at 471 and 480 nm was observed when lutein or fucoxanthin, respectively, were bound to HeimdallR1 (Fig. 4c,e), suggesting that xanthophyll structures are influenced by a large conformational change of the protein moiety (Fig. 4f).

a, Chromatogram of HPLC analyses (left) and the compositions of the retinal isomers (right) (n = 3, mean ± s.d.) under the dark (DA, blue), light (green) and light-adapted (LA, orange) conditions, where AT, 13C, 11C, syn and anti indicate all-trans, 13-cis, 11-cis, syn and anti configurations, respectively. b, Two-dimensional plot of transient absorption change. The labels of positive peaks indicate the absorption increase derived from the absorptions of K, M, O intermediates, and β-band. The negative peaks labelled in HeimdallR1 represent the bleaching of the initial state. c, Transient absorption spectra at different time points of HeimdallR1 without (top), with lutein (middle) and with fucoxanthin (bottom). * indicates the absorption change of xanthophylls in c and d. d, Time course of the transient absorption change of HeimdallR1 without (top), with lutein (middle) and with fucoxanthin (bottom). e, Absorption spectra of photointermediates of HeimdallR1 without (left), with lutein (middle) and with fucoxanthin (right). * indicates the peaks derived from the absorption change of xanthophylls. f, Photocycle models of HeimdallR1 without (top), with lutein (middle) and with fucoxanthin (bottom).
To investigate how the retinal binding pocket is affected by xanthophyll binding, we purified HeimdallR1 without carotenoids, as well as with lutein or fucoxanthin (Extended Data Fig. 6a). Binding of both xanthophylls elicited an apparent blue shift in the λamax, which was not observed with bacterial rhodopsin proton pumps6,7. To determine whether this shift reflects a real change in the absorption spectra of the retinal, we subtracted the absorption spectra of the isolated xanthophylls from the spectra of HeimdallR1 complexed with xanthophylls (blue and green spectra in Extended Data Fig. 6a). This revealed that the retinal λamax of HeimdallR1 bound to lutein (540 nm) was shorter than that of HeimdallR1 alone (551 nm), whereas the retinal λamax of HeimdallR1 complexed with fucoxanthin (556 nm) was not significantly different from the unbound protein. Meanwhile, a small λamax shift was observed on the acidic side of HeimdallR1 alone, whereas HeimdallR1 with lutein or fucoxanthin exhibited larger red shifts (20 nm and 11 nm, respectively, Extended Data Fig. 6b). This effect is caused by the protonation of the counterion in the third transmembrane helix (TM3) as known for many microbial rhodopsins. The retinal counterion of HeimdallR1, D81, is fully protonated at pH <4.0 irrespective of xanthophyll binding. Hence, the difference in the λamax shift is due to the difference in the degree of counterion protonation at neutral pH, and D81 of HeimdallR1 binding lutein is more deprotonated than that of HeimdallR1 alone at pH 7.0 (Extended Data Fig. 6c,d). Since the deprotonated counterion can act as the proton acceptor to receive H+ from the RSB22, the higher deprotonation suggests that lutein binding would enhance proton pumping, which aligns with the large M formation for HeimdallR1 binding lutein (Fig. 4b–e).
To gain further structural insights into the xanthophyll binding, we measured light-induced differences in Fourier transform infrared (FTIR) spectra of HeimdallR1 with or without lutein or fucoxanthin at 77 K. Extended Data Fig. 7a,b shows UV–visible spectra and formation of the K intermediate at 77 K, respectively. While light-induced difference FTIR spectra looked similar for HeimdallR1 with or without xanthophylls (Extended Data Fig. 7c), we observed two xanthophyll-dependent frequency shifts. One was seen in the C=C stretching frequency of the retinal chromophore (Extended Data Fig. 7d), which is, however, not due to structural reasons, as C=C stretch reflects λamax (Extended Data Fig. 7b). Indeed, the spectra of the C-C stretch region (Extended Data Fig. 5e) and amide-I (Extended Data Fig. 7h) were coincident, indicating that xanthophyll binding has no effect on chromophore structure and peptide backbone. The second shift was observed for a hydrogen out-of-plane (HOOP) band of the retinal chromophore in the K intermediate of HeimdallR1, while no such shift was observed for Kin4B8-xanthorhodopsin (Fig. 3c and Extended Data Fig. 7f). As HOOP bands appear by chromophore distortions, we conclude that binding of lutein or fucoxanthin to HeimdallR1 influences the retinal distortion in the K intermediate. Interestingly, this peak was broader for HeimdallR1 binding fucoxanthin, indicating larger structural heterogeneity. As the HOOP band is downshifted by an H/D exchange (Fig. 3c and Extended Data Fig. 7g), the chromophore distortion is located near the Schiff base. Therefore, xanthophyll binding to HeimdallR1 alters the structure of the retinal moiety furthest from the fenestration. This is in clear contrast to Kin4B8, which binds xanthophylls without generating structural changes upon retinal photoisomerization7.
Enhanced proton pumping in HeimdallR1 containing carotenoids
The influence of carotenoid binding on the efficiency of proton pumping of HeimdallR1 was investigated by expressing the protein in Escherichia coli spheroplasts. When illuminated with white light (400–700 nm), the spheroplasts demonstrated an enhanced light-dependent outward proton flux upon the addition of lutein or fucoxanthin but not of diatoxanthin (Fig. 5a top). Introduction of a fenestration-blocking mutation, G141F (xanthorhodopsin position Gly156), abolished carotenoid binding (Extended Data Fig. 2c). Under the same illumination conditions and in the presence of lutein, diatoxanthin or fucoxanthin, the pumping activity of the non-fenestrated HeimdallR-G141F was reduced significantly (Fig. 5a bottom). This decline is probably caused by non-specific adsorption of the carotenoids to the spheroplasts, resulting in masking of light. This implies that under our experimental settings, the observed proton flux in wild-type HeimdallR1 with all three xanthophyll antennas is in effect underestimated. Importantly, the enhancement of the pumping activity in HeimdallR1 when bound to lutein was maintained over a range of decreasing light intensities (2,000, 860, 590 and 515 µmol m−2 s−1, Fig. 5b). This suggests that in the photic zone, under open-ocean water conditions23, xanthophyll antennas enhance rhodopsin performance from the very surface to tens of metres deep.

a, Light-driven proton-pumping rates in HeimdallR1 and HeimdallR1-G141F E. coli spheroplasts with and without lutein, diatoxanthin or fucoxanthin (depicted in orange, mustard and red, respectively) under white-light illumination (400–700 nm, at 860 µmol m−2 s−1). Each dot represents the proton-pumping rate of HeimdallR1 and HeimdallR1-G141F with a xanthophyll over without the xanthophyll. A total of 26 ratios were used. b, Light-driven proton-pumping rates in HeimdallR1 (depicted in orange circles) and HeimdallR1-G141F (depicted in grey circles) E. coli spheroplasts with and without lutein under various white-light intensities (400–700 nm, at 2,000, 860, 590 and 515 µmol m−2 s−1). Each dot represents the proton-pumping rate of HeimdallR1 and HeimdallR1-G141F with lutein over without lutein. A total of 37 ratios were used. In both panels, boxplots represent the lower quartile, median and the upper quartile, and the whiskers depict 1.5× the interquartile range. The red dashed lines correspond to no differences in proton-pumping activity relative to HeimdallR1 or HeimdallR1-G141F without a xanthophyll. Highly significant effects were found only for the factor of fenestration (wild type vs G141F mutation) in both cases (Supplementary Table 1).
Structure of HeimdallR1
We succeeded in purifying fucoxanthin-bound HeimdallR1 (Extended Data Fig. 8a) and determined the crystal structure of HeimdallR1 at 2.0-Å resolution (Extended Data Fig. 8b and Supplementary Table 2). HeimdallR1 appeared as a monomer in the crystal packing (Extended Data Fig. 8c), leaving its physiological oligomeric structure unknown. The protein has the canonical architecture with seven transmembrane helices (TM1–7) and an all-trans retinal (ATR) (Fig. 6a,b and Extended Data Fig. 8d) with an overall high similarity to microbial rhodopsins. HeimdallR1 shows a noteworthy deformation in TM6: a π-bulge followed by a 310-helix segment, also known for green-absorbing proteorhodopsin24 and Exiguobacterium sibiricum rhodopsin25 but absent in xanthorhodopsins, NQ chloride and sodium pumps and related families (Extended Data Fig. 8e), which makes it a prominent structural hallmark of proteorhodopsin-like families. HeimdallR1 diverges from the other members of the proteorhodopsin–xanthorhodopsin superclade most conspicuously in the structure of the loop portion (Extended Data Fig. 8f,g): intracellular loop 3 (ICL3) forms a short membrane-extending α-helix and tightly interacts with TM6 (Fig. 6b and Extended Data Fig. 8h). While a similar helix in ICL3 is also present in HeimdallR2, HeimdallR3 appears to lack it (Extended Data Fig. 8i). Another salient structural detail of HeimdallR1 is an unusually extensive cavity at the cytoplasmic side that directly connects the putative proton donor residue K92 with the bulk solvent, which distinguishes it from E. sibiricum rhodopsin with which it shares the non-canonical cationic donor25,26.

a,b, Overall structure of HeimdallR1 with the retinal chromophore, viewed from the membrane plane (a) and the intracellular side (b). c,d, Fenestrations in HeimdallR1 (c) and Kin4B8-xanthorhodopsin (PDB ID: 8I2Z) (d). e, Conservation of the residues surrounding the fenestrations in HeimdallRs and Kin4B8-xanthorhodopsin. Glycine (G) and tyrosine (Y) are coloured orange and cyan, respectively, while polar [threonine (T), serine (S)] and hydrophobic [valine (V), isoleucine (I), leucine (L), alanine (A)] residues are coloured green and blue, respectively. f–h, HeimdallR1 structure, energy minimized using the hybrid QM/MM method (f), and the docking of lutein (g) and fucoxanthin (h) along the outer surface of TM6. Black dashed lines indicate hydrogen bonds. i, Zeaxanthin-bound Kin4B8-xanthorhodopsin (PDB ID: 8I2Z). See Extended Data Fig. 9 for zoom-in on the fenestration area.
As predicted, the structure of HeimdallR1 features a fenestration in the vicinity of the retinal ring owing to two glycine residues, G141 and G145 (Fig. 6c). The residues in the bottom of the fenestration are hydrophilic (Fig. 6d,e) as in Kin4B8-xanthorhodopsin, thus favourable for carotenoid binding. However, the resolved crystal structure lacks fucoxanthin that was dissociated under the conditions used for crystallization (Extended Data Fig. 8c). We thus explored the binding of xanthophylls (lutein and fucoxanthin) to HeimdallR1 using hybrid quantum mechanics/molecular mechanics (QM/MM) simulations27 (Fig. 6f–i).
This approach revealed that the carotenoids can form an intricate hydrogen bonding network with water molecules, T142 and Y206 via the hydroxyl groups present on the rings. We observed that lutein can bind via both rings, preferably through the β-ionone ring (C5 atom of the ring directed towards retinal) where the hydroxyl group forms hydrogen bonds with WAT331 and T142, apart from the C-H…π interaction of a methyl group with F149 (Fig. 6g and Extended Data Fig. 9b). The ε-ring at the other end is stabilized mainly by C-H…π interaction of methyl with F171 on ICL3 (Supplementary Fig. 3). Fucoxanthin can fit in the fenestration through the keto end and bind to the protein similar to lutein through hydrogen bonding with WAT331 and T142, and π-interaction with F149 (Fig. 6h and Extended Data Fig. 9c). Fucoxanthin binding in the fenestration via the allene end would not be possible due to steric clashes between its acetate moiety and the protein.
Calculated excitation energies revealed a blue-shifted absorption spectrum upon binding of lutein or fucoxanthin to HeimdallR1 (Supplementary Table 3 and Fig. 4). Natural transition orbital analysis showed mainly local excitations on the retinal and lutein/fucoxanthin, but also charge transfer from retinal to the respective carotenoid (Extended Data Fig. 10). Electron densities are largely localized on the retinal and the extended π-network of the carotenoid backbone with no significant contribution from the rings. This indicates that the rings of the carotenoids are important for binding with the rhodopsin but not for energy transfer.
When comparing the calculated structures of HemidallR1 with Kin4B8-xanthorhodopsin, the xanthophylls lie similarly transversely against the outer surface of TM6 (Fig. 6i and Extended Data Fig. 9d). However, the angles between the polyene chain and the protein are very different in HeimdallR1 and Kin4B8 due to the helix in ICL3 and the π-bulge in TM6. Moreover, the orientation of the ring is different in Kin4B8 such that its hydroxyl interacts with S208. Most importantly, T142 and F149, which are critical for fucoxanthin and lutein binding in the QM/MM model, are not conserved in Kin4B8 (Fig. 6e). The amino-acid difference in the fenestration may explain the unique binding and energy transfer of fucoxanthin to HeimdallR1.
Discussion
Two different spectral tuning strategies are currently known in oceanic microbial rhodopsins. The first is determined mainly by changes in a single amino acid at position 105 (leucine28,29 and methionine30 in green-absorbing proteorhodopsins, and glutamine28,29 in blue-absorbing proteorhodopsins) and enables proteorhodopsins to absorb light according to depth, as blue light penetrates deeper in clear oceanic waters23. The second strategy is the use of carotenoid antennas absorbing blue light for energy transfer to xanthorhodopsins5,6,7, green-absorbing proteorhodopsins7 and HeimdallRs (this study), which otherwise absorb in green. Our present findings, illustrating the advantageous nature of rhodopsin–xanthophyll complexes under white-light illumination (Fig. 5b), suggest that the second strategy follows the solar conditions encountered by marine microbes within aquatic ecosystems (that is, multichromatic light availability at most depths in the photic zone23), therefore allowing these microbes to utilize available light more efficiently.
Asgard archaea possess three unrelated families of rhodopsins15: heliorhodopsins and schizorhodopsins found in ‘Candidatus Lokiarchaeia’, ‘Candidatus Thorarchaeia’ and ‘Ca. Heimdallarchaeia’, and the outward proton pumps HeimdallRs, found in ‘Ca. Heimdallarchaeia’. On the basis of these observations, it was suggested that during their evolutionary history, ‘Ca. Asgardarchaeota’ were present in sunlit habitats15. This work demonstrates that a genuine planktonic lifestyle in the photic zone evolved only in the ‘Ca. Kariarchaeaceae’, a change correlated with the acquisition of HeimdallRs (Fig. 2a). Moreover, a comparison with the widespread photoheterotrophic ‘Ca. Poseidoniia’ demonstrates that the lifestyle transition in the ‘Ca. Heimdallarchaeia’ must be relatively recent, as the pelagic and putatively photoheterotrophic forms in the latter are restricted to a single genus-level clade (‘Ca. Kariarchaeum’) and the diversity of HeimdallRs is shallow compared with both types of proton-pumping rhodopsins of ‘Ca. Poseidoniia’ (Fig. 1a). The acquisition of HeimdallRs by ‘Ca. Kariarchaeum’ further spotlights the metabolic diversity among the ‘Ca. Heimdallarchaeia’19,31, although given its late date, it contributes little to the understanding of the metabolism of the first eukaryotic common ancestor that probably had a heimdallarchaeial origin17,18.
Notwithstanding the parallels between ‘Ca. Poseidoniia’ and ‘Ca. Kariarchaeum’, the three groups of proton-pumping rhodopsins appearing in the planktonic Archaea: proteorhodopsins (Archaea clade A rhodopsins, or ACA) and Archaea clade B (ACB) rhodopsins in ‘Ca. Poseidoniia’11,12,13,14 and heimdallarchaeia rhodopsins (HeimdallRs) in ‘Ca. Kariarchaeaceae’, diverge in their ability to form two-chromophore complexes. None of the ‘Ca. Poseidoniia’ proton pumps appear to be capable of binding carotenoid antennas. Thus, archaeal proteorhodopsins are not fenestrated and while some ACB rhodopsins, such as ACB-G35, do feature a fenestration, it is dissimilar to the fenestration in bacterial xanthorhodopsins and proteorhodopsins and is consequently incapable of mediating carotenoid binding as shown by our experiments (Fig. 1c). All HeimdallRs, on the other hand, possess the canonical fenestration similar to that of the carotenoid-binding proteorhodopsins and xanthorhodopsins. In line with this, we discovered that these rhodopsins from ‘Ca. Kariarchaeum’, thus far uniquely among microbial rhodopsins, can utilize such dissimilar xanthophyll antennas as lutein and fucoxanthin. These pelagic Asgard archaea do not appear to possess known genes necessary for retinal or xanthophyll synthesis and it is thus possible that this lack of specificity might reflect a spectrum of antennas recruited by them from the environment. Understanding of the exact mechanism by which the tripartite opsin–retinal–xanthophyll complex is assembled will require isolation of ‘Ca. Kariarchaeum’ in culture.
Methods
Proteins chosen for expression
Three representatives from the HeimdallR family were selected on the basis of protein sequence dissimilarity: HeimdallR1 (GenBank accession number MBS85746.1) from MAG RS678 (Archaea, ‘Ca. Asgardarchaeota’, ‘Ca. Heimdallarchaeia’, order UBA460, ‘Ca. Kariarchaeaceae’, ‘Ca. K. pelagium’) from the Red Sea (Tara Oceans)32, HeimdallR2 (GenBank accession number NDB54273.1) from MAG WBC_A_4_184 (‘Ca. K. pelagium’) from the estuary of the Apalachicola River, Florida33, and HeimdallR3 from ‘Ca. Kariarchaeum’ scaffold Gs0128817_NODE_76 obtained from metagenomic data from Groves Creek Marsh, Skidaway Island, Georgia (JGI project Gs0128817). HeimdallR3 was first identified in the metatranscriptomic fragment Ga0182069_1327250, and the complete open reading frame (ORF) could be recovered by recruiting raw metagenomic and metatranscriptomic reads from read runs included in the project with bowtie2 (v.2.3.5.1)34 using Ga0182069_1327250 as seed and assembling the recruited reads with spades v.3.15.5. The resulting contig was further extended in both directions by iterative read recruitment with bowtie2. The highest coverage for the final scaffold was obtained from the metagenomic run SRR7152995 and metatranscriptomic run SRR6980959. ACB-G35 (GenBank accession number MBA4694301.1) from MAG MCMED-G35 (Archaea, ‘Ca. Thermoplasmatota’, ‘Ca. Poseidoniia’, ‘Ca. Poseidoniales’, ‘Ca. Poseidoniaceae’, species MGIIa-K1 sp003602415)35 was selected as a representative of the Archaea clade B family.
‘Ca. K. pelagium’ pan-genome
The pan-genome of ‘Ca. K. pelagium’ was obtained from the MAGs GCA_002728275.1, GCA_008081315.1, GCA_010028315.1, GCA_011525205.1, GCA_018623815.1, GCA_036161275.1, GCA_938030875.1, OceanDNA-a2, OceanDNA-a3, OceanDNA-a4, OceanDNA-a5, OceanDNA-a6, OceanDNA-a7 and TARA_SAMEA2620081_METAG_EEAABNBB with SuperPang (v.1.3.0)36 using a relaxed identity threshold of 80% and a k-mer size of 101. A total of 15 pan-genomic contigs with an overall length of 1,524,797 bp were found to contain regions identified as core and were used to represent the ‘Ca. K. pelagium’ pan-genome. Gene prediction and annotation were performed using the NCBI Prokaryotic Genome Annotation Pipeline (v.2024-04-27.build7426)37.
Genome phylogeny
Genomes chosen for phylogenetic reconstruction of the ‘Ca. Kariarchaeaceae’ were obtained from GenBank, OceanDNA38 and Ocean Microbiomics Database39 on the basis of the taxonomic assignment in GTDB (r.220)40 and source databases. To exclude contaminating scaffolds in the ‘Ca. K. pelagium’ assemblies, scaffolds contributing to the pan-genomic contigs with core regions (see above) were selected. Genes were predicted with Prodigal (v.2.6.3)36 on the basis of the filtered assemblies (for ‘Ca. K. pelagium’) or entire assemblies (other members of the family). The predicted amino-acid sequences were used to reconstruct concatenation phylogeny with PhyloPhlAn (v.3.02)41 using the ‘phylophlan’ marker database, USEARCH (v.11.0.667)42 for marker identification, MAFFT (v.7.475)43 for alignment, TrimAl (v.1.4.1)44 for alignment trimming and RAxML (v.8.2.12)45 with 1,000 rapid bootstrap iterations for phylogenetic reconstruction. Only markers appearing in at least 60% of the genomes were included in the phylogenetic reconstruction. RAxML was run with automatic model selection: the best-fitting model with CAT approximation selected by the Bayesian information criterion (BIC) was VT (with empirical frequencies). The scaffold containing HeimdallR3 was placed on the resulting tree using the following strategy: homologues of two genes located on the scaffold were found in at least four other ‘Ca. Kariarchaeaceae’ genomes using BLASTp from NCBI BLAST+ (v.2.15.0)46 (E-value threshold of 1 × 10−15): gene G1 of unknown function and G2 coding for a DnaJ-like protein, and were recruited for phylogenetic placement. MAFFT alignments of the protein sequences of G1 and G2 were added to the concatenated alignment of the marker genes, and the resulting extended alignment was used to evaluate the species tree with RAxML to obtain model parameters for pplacer and update the branch lengths. The model used for the evaluation was obtained with ProtTest3 (v.3.4.2)47 among models supported by pplacer (LG and WAG): the best-fitting model according to BIC was WAG with Γ-distributed rates and empirical frequencies. The extended alignment and the updated species tree were used to place the HeimdallR3 scaffold using pplacer (v.1.1.alpha)48. The species tree shown in the main text corresponds to the RAxML tree evaluated using the extended alignment. The tree was midpoint-rooted but the position of the root was found to be in agreement with the phylogenetic reconstruction in GTDB.
Rhodopsin phylogeny
The branching pattern of the early-diverging families of the proteorhodopsin–xanthorhodopsin superclade, including HeimdallR1, receives weak support values in phylogenetic reconstructions and varies between studies. Therefore, to visualize phylogenetic relationships among the rhodopsins, we used a phylogenetic network. Rhodopsin sequences representative of the different families in the proteorhodopsin–xanthorhodopsin superclade were aligned with MAFFT (--auto) and trimmed with TrimAl (-gt 0.9). NeighborNet network was obtained with SplitsTree (v.4.17.0)49 on the basis of uncorrected p distances. Presence and type of fenestration were checked by folding the proteins with AlphaFold 3 (ref. 50). A distance-based tree of the HeimdallR family was obtained by collecting HeimdallR protein sequences at least 210 residues in length, clustering them at 100% identity level with CD-HIT (v.4.8.1)51, aligning with MAFFT (automatic mode) and reconstructing a neighbour-joining phylogeny in MEGA (v.10.2.5)52 under the Dayhoff model with gamma-distributed substitution rates (shape parameter of 1.0) with 100 bootstrap replicates.
Global distribution of HeimdallRs
Ocean Microbial Reference Catalog v.2 (OM-RGC v.2)21 and proteins assigned to pfam01036 family in aquatic metagenome and metatranscriptome assemblies in JGI Integrated Microbial Genomes and Microbiomes (IMG/M)53 were used to extract quantitative data on the distribution of HeimdallRs and other marine archaeal proton pump clades. For the IMG/M data, only samples from marine and estuarine environments no deeper than 200 m were considered. Sequences belonging to marine archaeal proton pumps were identified by searching the databases with BLASTp using representative sequences from the proteorhodopsin–xanthorhodopsin superclade with the following empirical thresholds: an E-value threshold of 1 × 10−10, minimum identity of 50% and a bitscore threshold of 90. Proteins with best matches to one of the three families were assigned to taxa with mmseqs2 (v.14.7e284)54 using a pre-built database of taxonomically classified rhodopsin sequences extracted from GTDB representative genome assemblies. Sequences classified to Archaea were retained. The accuracy of this rhodopsin clade classification strategy was verified by BLAST searches, alignment and phylogenetic analysis. Length-adjusted abundances for the genes were taken directly from the gene atlas in the case of OM-RGC v.2 and approximated by dividing the scaffold read depths by scaffold lengths in the case of IMG/M. Relative abundances of HeimdallRs were obtained by dividing their abundance by the total abundance of the three archaeal proton pump clades. Relative abundances with respect to all regular microbial rhodopsins were obtained by dividing HeimdallR abundances by the total abundance of rhodopsin genes identified in the same samples by searching protein sequences against the Pfam profile PF01036.24 with hmmsearch from HMMER (v.3.4)55.
Analysis of the global distribution of ‘Ca. K. pelagium’ and variation across the genome
The contigs that constituted the core pan-genome of ‘Ca. K. pelagium’ (see above) were used as a reference to recruit contigs from marine metagenomic and metatranscriptomic datasets in Logan (v.1.1)56 with minimap2 (v.2.28)57 using default settings. The mappings were filtered to retain only hits with <20% sequence divergence based on minimap2’s ‘de’ tag. The fraction of the pan-genome covered by the mapped contigs was used as a measure of ‘Ca. K. pelagium’ abundance at each location. For the analysis of genome-wide variation, metagenomic contigs were treated as a single pool of mapped sequences and depth of coverage calculation and variant calling were performed with DepthOfCoverage and Mutect2 from gatk4 (v.4.6.1.0)58, respectively. The variants were subsequently filtered with FilterMutectCalls, and single-nucleotide polymorphisms as well as short multinucleotide variations (up to nine nucleotides) were used to obtain numbers of variable positions per nucleotide calculated for sufficiently long ORFs (at least 300 bp) and for sliding windows of 200 bp.
Carotenoids
Lutein (PHR1699), β-carotene (PHR1239) and fucoxanthin (F6932) were purchased from Sigma-Aldrich. Diatoxanthin was obtained from Phaeodactylum tricornutum strain CCAP 1055/5 (Culture Collection of Algae and Protozoa – Scottish Association for Marine Science, Scotland, UK). The extract from dried biomass of P. tricornutum was obtained with 80% ethanol. For the separation of diatoxanthin from the extract of P. tricornutum biomass, a high-performance countercurrent chromatography (HPCCC) system was used in a two-step procedure. The mobile phase consisted of the lower phase of a two-phase solvent system (n-heptane, ethyl acetate, ethanol and water in a ratio of 5:4:5:3, v/v/v/v), while the stationary phase was the upper phase. In the first HPCCC separation step, an amount of 120 mg of the algal extract was processed with the HPCCC using the mobile phase at a flow rate of 3 ml min−1, resulting in 0.25 mg of the fraction containing diatoxanthin. The same separation process was repeated 10 times, and the collected target fractions were pooled, finally yielding 2.5 mg of the diatoxanthin fraction after removal of the solvent by rotary evaporation under reduced pressure at 28 °C. To increase the purity of the obtained fraction (2.5 mg), a second HPCCC separation step was performed at a mobile phase flow rate of 1.5 ml min−1, yielding 1.5 mg of the diatoxanthin fraction. The diatoxanthin fraction obtained by the two-step HPCCC was finally purified by gel permeation chromatography using Sephadex LH-20 gel and a mobile phase of 100% methanol. The collected diatoxanthin fraction was evaporated using a rotary evaporator under reduced pressure at 28 °C and yielded 0.57 mg of the compound.
Mediterranean Sea chromophore extract
Water (450 l) was sampled on 25 January 2023 at 8:00 in the Mediterranean Sea near Michmoret harbour (32° 32.410144’ N, 34° 34.846542’ E). The water sample was then filtered on a 0.22 µm Durapore PVDF membrane filter (Milliporem, GVWP14250) after pre-filtration through a mesh net. The sample-containing membranes were then freeze dried using a lyophiliser (Coolsafa 110-4, ScanVac) for ~48 h. Chromophore extraction was done directly on the dried membranes with hexane59. Briefly, dried samples were resuspended in 10 ml acetone by applying extensive pipetting and vortexing. Hexane and 10% NaCl were added to the mixture in a 2:2:1 ratio (acetone:hexane:10% NaCl). The mixture was vortexed and then centrifuged at 3,000 g at 4 °C for 3 min. The hexane (top) layer was then transferred to a separate Falcon tube and the process was repeated until the hexane phase became colourless. Combined hexane fractions were then dried using N2 gas and either reconstituted in 1 ml absolute ethanol or lyophilised for further characterization.
N2-dried lyophilised extracts (10 mg) were resuspended in 1 ml of methanol, agitated for 2 min in 2‐ml screw‐top polystyrene tubes with 0.5 g of 0.5-mm glass beads under N2 in a Genie disruptor and incubated overnight at −20 °C. The chromatographic analysis of the pigments in the extracts was performed in a Merck Hitachi HPLC equipped with a diode array detector according to the method described in ref. 60. The column used was an RP-18, the flow rate was 1 ml min−1, and 100 µl of the sample was injected. The mobile phases used were: solvent A (ethyl acetate 100%) and solvent B (acetonitrile:H2O, 9:1 v/v). The gradient applied was: 0–16 min 0–60% A; 16–30 min 60% A; and 30–35 min 100% A. Standards were supplied by MERCK-SIGMA or DHI.
Expression of HeimdallR family representatives
The genes of HeimdallR2 and HeimdallR3 were first optimized using JCat61 for expression in E. coli and cloned into pET21a (+) vector with a C-terminal six-His tag using NdeI and XhoI restriction sites. The point mutation in HeimdallR1 G141F, prepared to block the fenestration in HeimdallR1, was obtained using the NEB Q5 site-directed protocol (https://nebasechanger.neb.com/) with primers 5′-TGTGGTTTGGtttACCCTGAGCGGC-3′ and 5′-CGCATGCCGTCAACG-3′.
E. coli C43(DE3) cells harbouring the pET21a (+) HeimdallR1/HeimdallR2/HeimdallR3/HeimdallR1-G141F cloned plasmid were grown overnight at 37 °C in LB medium supplemented with ampicillin (50 μg ml−1). The next day, the overnight culture was inoculated at a 1:20 dilution into M9 medium containing 50 μg ml−1 ampicillin. This was grown at 220 r.p.m. and 37 °C until an optical density at 600 nm (OD600) of ~0.6. The expression of the protein was then induced with 0.25 mM isopropyl β-d-thiogalactopyranoside (IPTG) in the presence of 10 μM all-trans retinal (Toronto Research Chemicals) at 37 °C for 4 h.
Expression of ACB-G35 rhodopsin
The gene for ACB-G35 rhodopsin was first optimized for the E. coli expression system and cloned into pET21a (+) vector with a C-terminal six-His tag, using NdeI and XhoI restriction sites.
E. coli C43 cells harbouring the pET21a (+) ACB-G35 rhodopsin cloned plasmid were grown at 37 °C in LB medium supplemented with ampicillin (50 μg ml−1) overnight. The next day, the overnight culture was inoculated at a 1:100 dilution in LB medium containing 50 μg ml−1 ampicillin. This was grown at 220 r.p.m. and 37 °C until an OD600 of ~0.6. The expression protein was induced by 1 mM IPTG in the presence of 10 μM all-trans retinal (Toronto Research Chemicals) at 37 °C for 4 h.
Rhodopsin purification
The rhodopsin-expression E. coli cultures were centrifuged at 5,000 × g for 15 min at 4 °C and kept at −80 °C overnight. The pellet was thawed on ice and resuspended in a buffer containing 50 mM Tris-HCl pH 8.0, 5 mM MgCl2 and 0.1 mM PMSF (P7626, Sigma-Aldrich). The sample was disrupted by using a microfluidizer for 10 passes at 60 psi. Then, the sample was centrifuged at 5,000 × g for 15 min at 4 °C to pellet undisrupted cells or large cell debris. Membranes were collected by centrifuging the sample at 37,000 × g for 1.5 h at 4 °C, and resuspended in a buffer containing 50 mM MES-NaOH pH 6.0, 300 mM NaCl, 5 mM imidazole, 5 mM MgCl2, 10% glycerol and 2% n-dodecyl-β-d-maltopyranoside (DDM) final concentration. The sample was incubated overnight at 4 °C with gentle rotation, and a second centrifugation at 37,000 × g for 1.5 h at 4 °C was performed. The supernatant was then incubated with Ni-Beads (31103, Cube Biotech) for 1 h. Beads were washed on a gravity column using a buffer containing 50 mM MES-NaOH pH 6.0, 300 mM NaCl, 10% glycerol, 0.05% DDM and 5 mM imidazole. Protein was eluted from the column using a buffer containing 50 mM MES-NaOH pH 6.0, 300 mM NaCl, 10% glycerol, 0.05% DDM and 250 mM imidazole. Eluted protein was washed with storage buffer (50 mM MES-NaOH pH 6.0, 300 mM NaCl, 10% glycerol and 0.05% DDM) using Amicon 3-kDa cut-off centrifugal filters (UFC800324, Millipore). The protein was then flash frozen in liquid N2 and stored at −80 °C.
Binding of the Mediterranean Sea chromophore extract to rhodopsin proteins
Purified rhodopsin protein (1 mg) was mixed with the Mediterranean Sea chromophore extract at a ratio of at least 1:3 OD (rhodopsin:carotenoid mixture) and incubated overnight with gentle rotation at 4 °C. Ethanol, the solvent used for carotenoid resuspension, was kept below 1% throughout the incubation step to ensure minimal denaturation of the rhodopsin protein. Then, 0.5 ml of Ni-Beads (31103, Cube Biotech) was added to the mixture and incubated with gentle rotation for 1 h at 4 °C, and loaded onto a gravity column where the bound protein was washed with 20 ml (equivalent of 40 column volume) of storage buffer (50 mM MES-NaOH pH 6.0, 300 mM NaCl, 10% glycerol and 0.05% DDM) at room temperature. The rhodopsin protein was then eluted using 3 ml of buffer containing storage buffer and 300 mM imidazole, followed by an additional step of buffer exchange using 18 ml of storage buffer.
Binding of commercial xanthophyll to rhodopsin proteins
The purified rhodopsin was mixed with carotenoids fresh stock dissolved in dimethylsulfoxide (DMSO) and incubated overnight with gentle rotation at 4 °C. The molar quantities of each component were estimated on the basis of their respective absorption spectra, utilizing molar extinction coefficients of ε = 45,000–50,000 M−1 cm−1 at the absorption peak of rhodopsin and ε = 145,100 M−1 cm−1 at 445 nm for carotenoids. The volume of DMSO was adjusted to ensure it did not exceed 5% of the solution volume of rhodopsin. Subsequently, ~0.5 ml of Ni-Beads (31103, Cube Biotech) was added to the mixture for 1 h at 4 °C. The rhodopsins were then washed extensively using a storage buffer and eluted using the same buffers used for the initial purification.
Absorption spectroscopic measurements
Absorption spectral measurements of HeimdallR1, HeimdallR2, HeimdallR3, ACB-G35 and Kin4B8-xanthorhodopsin with and without carotenoids were taken with a Shimadzu (UV-1800) spectrophotometer. The absorption spectral measurements of HeimdallR1 and ACB-G35 with carotenoid extract were taken with a BioTek Synergy MX plate reader.
Fluorescence spectroscopic measurements
Fluorescence emission and excitation spectral measurements were taken on a Jobin Yvon-Spex Fluorolog-3 spectrofluorometer. The spectrofluorometer is equipped with 450 W Xe-lamp as the light source, double-grating monochromator in the excitation, single-grating in emission positions, and a photomultiplier tube detector (R928P). All measurements were done at pH 5.5 and at a slit width of 10 nm in the excitation and emission wavelengths. Emission was monitored at 720 nm and excitation was varied between 400 nm and 650 nm. The absorbance of all samples was kept at OD < 0.5. Fluorescence excitation spectral profile was corrected by the absorbance spectra profile for each nm due to the inner filter effect: \({F}_{{\rm{ideal}}}={F}_{{\rm{flu}}}{10}^{({A}_{{\rm{ex}}}+{A}_{{\rm{em}}})/2}\), where \({F}_{{\rm{ideal}}}\) is the ideal fluorescence intensity after we consider the inner filter effect, \({F}_{{\rm{flu}}}\) is the fluorescence intensity measured, \({A}_{{\rm{ex}}}\) is the absorbance at the \({F}_{{\rm{flu}}}\) excitation point and \({A}_{{\rm{em}}}\) is the absorbance at the emission point (always at 720 nm in our case)62.
Protein expression and purification of HeimdallR1 for photocycle measurements
E. coli C43(DE3) cells harbouring the HeimdallR1-cloned plasmid were grown at 37 °C in LB medium supplemented with ampicillin (50 μg ml−1) overnight. The next day, the overnight culture was inoculated in M9 medium containing 50 μg ml−1 ampicillin. This was grown at 200 r.p.m. and 37 °C (until OD600 of ∼0.6). Expression of the C-terminal 6× His-tagged protein was induced by 0.1 mM IPTG in the presence of 10 μM all-trans retinal (Toronto Research Chemicals) at 37 °C for 4 h. The collected cells were sonicated (Ultrasonic Homogenizer VP-300N, TAITEC) for disruption in a buffer with 50 mM Tris-HCl pH 8.0 and 5 mM MgCl2. The membrane fraction was collected by ultracentrifugation (CP80NX, Eppendorf Himac Technologies) at 142,000 × g for 1 h. The proteins were solubilized in a buffer containing 50 mM MES-NaOH pH 6.5, 300 mM NaCl, 5 mM imidazole, 5 mM MgCl2 and 3% DDM (ULTROL Grade; Calbiochem, Sigma-Aldrich). Solubilized proteins were separated from the insoluble fractions by ultracentrifugation at 142,000 × g for 1 h. Proteins were purified using a Co-NTA affinity column (HiTrap TALON crude, Cytiva). The resin was washed with a buffer containing 50 mM MES-NaOH pH 6.5, 300 mM NaCl, 50 mM imidazole, 5 mM MgCl2 and 0.1% DDM. Proteins were eluted in a buffer containing 50 mM Tris-HCl pH 7.0, 300 mM NaCl, 300 mM imidazole, 5 mM MgCl2 and 0.1% DDM. Eluted protein was immediately concentrated using a 50 ml centrifugal ultrafiltration filter with a molecular weight cut-off of 30 kDa (Amicon Ultra-4, Millipore) and the buffer exchanged with another buffer containing 50 mM HEPES-NaOH pH 7.0, 150 mM NaCl, 10% glycerol and 0.1% DDM.
Purification of carotenoid-binding HeimdallR1
Carotenoid-binding HeimdallR1 was obtained by mixing purified HeimdallR1 with carotenoids (lutein or fucoxanthin). First, carotenoids were dissolved in DMSO at twice the molar amount of HeimdallR1 to be bound. The molar amounts of each were estimated by measuring the absorption spectrum (Extended Data Fig. 6a) using a molar extinction coefficient of ε = 45,000 M−1 cm−1 at the absorption maximum of rhodopsin and ε = 145,100 M−1 cm−1 at 445 nm for carotenoids. The volume of DMSO was adjusted so that it did not exceed 2% of the rhodopsin solution volume. After preparing the carotenoid solution as described above, it was added to HeimdallR1 and left on ice and in the dark for 2 h, gently mixing the solution every 15 min. To remove the excess of carotenoids not bound to HeimdallR1, it was purified again using the same method as the purification of the original HeimdallR1 protein. The purified protein was immediately concentrated using a 50 ml centrifugal ultrafiltration filter with a molecular weight cut-off of 30 kDa (Amicon Ultra-4, Millipore) and the buffer exchanged with another buffer containing 20 mM HEPES-NaCl pH 7.0, 100 mM NaCl and 0.05% DDM.
Laser-flash photolysis
For laser-flash photolysis spectroscopy, HeimdallR1 with/without lutein or fucoxanthin was solubilized in 20 mM HEPES-NaCl pH 7.0, 100 mM NaCl and 0.05% DDM. The OD of the rhodopsin was adjusted to ~0.4–0.5 (protein concentration of ~0.2–0.25 mg ml−1) for HeimdallR1 and HeimdallR1 with fucoxanthin and ~0.2 (protein concentration of ~0.1 mg ml−1) for HeimdallR1 with lutein at the λamax. The laser-flash photolysis measurement was conducted as previously described16,63. Nanosecond pulses from an optical parametric oscillator (λexc = 550 (without lutein), 535 (with lutein) and 540 nm (with fucoxanthin), 4.5 mJ pulse−1 cm−2, 1.1 Hz (basiScan, Spectra-Physics)) pumped by the third harmonics of Nd-YAG laser (λ = 355 nm, INDI40, Spectra-Physics) were used for the excitation of HeimdallR1 with or without lutein and with or without fucoxanthin. The transient absorption spectra were obtained by monitoring the intensity change of white light from a Xe-arc lamp (L9289-01, Hamamatsu Photonics) passed through the sample with an ICCD linear array detector (C8808-01, Hamamatsu Photonics). To increase the signal-to-noise ratio, 40–60 spectra were averaged, and singular-value decomposition analysis was applied. To measure the time evolution of transient absorption change at specific wavelengths, the output of a Xe-arc lamp (L9289-01, Hamamatsu Photonics) was monochromated by monochromators (S-10, Soma Optics), and the change in intensity after the photoexcitation was monitored with a photomultiplier tube (R10699, Hamamatsu Photonics). To increase signal-to-noise ratio, 200–400 signals were averaged. The signals were globally fitted using a multi-exponential function to determine the lifetimes and absorption spectra of each photointermediate.
To measure the fold change in transient absorption change between with and without xanthophylls, nanosecond pulses from an optical parametric oscillator (basiScan, Spectra-Physics) pumped by the third harmonics of Nd-YAG laser (λ = 355 nm, INDI40, Spectra-Physics) were used for the excitation of HeimdallR1 at different wavelengths (λexc = 425, 450, 465, 480, 545 and 590 nm for lutein, and λexc = 440, 465, 580, 490, 530, 540, 550 and 600 nm for fucoxanthin). The pulse energy was adjusted to 0.55 mJ cm−2 and 0.56 mJ cm−2 for lutein and fucoxanthin, respectively, to keep the linearity between the number of the absorbed photons and the transient absorption change. Quantum yield (QY) based on fold change in transient absorption change between with and without xanthophylls was calculated using the following equations:
For lutein:
For fucoxanthin:
HPLC analysis of retinal isomers
Retinal configuration was analysed by HPLC using purified HeimdallR1 or HeimdallR1 with lutein or fucoxanthin in a buffer containing 20 mM HEPES-NaCl pH 7.0, 100 mM NaCl, 0.05% DDM. Before the measurements, the OD of the rhodopsin was adjusted to ~0.2 (protein concentration of ~0.1 mg ml−1), and the proteins were stored at 4 °C overnight in the dark for dark-adapted (DA in Fig. 4) samples. The HPLC system was equipped with a silica column particle size of 3 μm, 150 × 6.0 mm (Pack SIL, YMC), pump (PU-4580, JASCO) and UV–Vis detector (UV-4570, JASCO). The solvent was composed of 15% (v/v) ethyl acetate and 0.15% (v/v) ethanol in hexane and with a flow rate of 1.0 ml min−1. To denature the protein, 280 μl of 90% methanol solution was added to the 75 μl sample. Retinal oxime formed by the hydrolysis reaction with 25 μl of 2 M hydroxylamine solution was extracted with 800 μl of hexane, and 200 μl of the solution was injected into the HPLC system. For measurements under light illumination (Light in Fig. 4), the sample solutions were illuminated at λ = 550 ± 10 nm for HeimdallR1 and HeimdallR1 with fucoxanthin and at λ = 540 ± 10 nm for HeimdallR1 with lutein (Bandpass, AGC Techno Glass) for 1 min, followed by denaturation and hydrolysis of the retinal chromophore under illumination. For measurements of light-adapted (LA in Fig. 4) samples, the sample solution was illuminated at λ = 550 ± 10 nm for HeimdallR1 and HeimdallR1 with fucoxanthin and at λ = 540 ± 10 nm for HeimdallR1 with lutein for 1 min, and after waiting for 1 min, denaturation and hydrolysis reactions of the retinal chromophore were conducted. The molar compositions of the retinal isomers were calculated from the areas of the corresponding peaks in the HPLC patterns. The molar composition of the retinal isomers in the sample was determined with the molar extinction coefficients at 360 nm for each isomer (all-trans-15-syn: 54,900 M−1 cm−1; all-trans-15-anti: 51,600 M−1 cm−1; 13-cis-15-syn, 49,000 M−1 cm−1; 13-cis-15-anti: 52,100 M−1 cm−1; 11-cis-15-syn: 35,000 M−1 cm−1; 11-cis-15-anti: 29,600 M−1 cm−1). Three independent measurements were performed to estimate experimental error.
pH titration
To investigate the pH dependence of the absorption spectra of HeimdallR1 with/without lutein and with/without fucoxanthin, the OD of the rhodopsin was adjusted to ~0.5 (protein concentration of ~0.25 mg ml−1) and solubilized in a 6-mix buffer (trisodium citrate, MES, HEPES, MOPS, CHES, CAPS (10 mM each, pH 7.0), 100 mM NaCl and 0.05% DDM). The pH was adjusted to the desired value by the addition of small aliquots of 1–5 N HCl and NaOH. Absorption spectra were recorded using a UV–Vis spectrometer (V-750, JASCO). The measurements were performed at every 0.3–0.6 pH value.
Low-temperature UV–Vis and FTIR spectroscopic analysis
Samples for low-temperature UV–Vis and FTIR spectroscopy were prepared as described for photocycle measurements with small modifications. The proteins were solubilized using 1% DDM (Anatrace). Then, the samples, HeimadallR1 with lutein, with fucoxanthin, or without xanthophylls, were reconstituted into POPE:POPG membrane as described previously7.
Low-temperature UV–Vis spectroscopy and FTIR spectroscopy were performed as described previously7. Briefly, lipid-reconstituted HemidallR1 was placed onto a BaF2 window making dry films. Hydrated films with 1 µl H2O or D2O were fixed to a cryostat (Optistat, Oxford Instruments) attached to UV–Vis (V-750, JASCO) and FTIR (Cary670, Agilent) spectrometers. K intermediates of HemidallR1 were generated by irradiating 530 nm light for 30 s using an interference filter (KL53, Toshiba) at 77 K, which was reverted to the original state by >590 nm for 30 s with a cut-off filter (R-61 cut-off filter, Toshiba). Light-activated difference spectra were obtained by subtracting spectra before light irradiation from spectra after light irradiation at 77 K. Averages of 100 experiments were conducted for the spectra of HeimadallR1 with lutein, with fucoxanthin, or without xanthophylls.
Proton-pumping measurements
The protocol was modified from ref. 64. In brief, 200 ml of rhodopsin-expressing E. coli cells was centrifuged at 3,600 g for 10 min and resuspended into 20 ml of 30 mM Tris-HCl pH 8.0 and 20% sucrose. Lysozyme (200 µg) (L6876, Sigma-Aldrich) was added to the cell suspension and gently rotated for 1 h at room temperature. The resulting spheroplasts were centrifuged at 3,600 g for 15 min at room temperature and the pellet was resuspended with 6 ml of unbuffered solution (10 mM NaCl, 10 mM MgSO4·7H2O and 100 μM CaCl2), added with 100 µM lutein, diatoxanthin or fucoxanthin to ~2 ml (a third of the volume), and incubated overnight at 4 °C with gentle rotation. Spheroplasts were then washed three times with unbuffered solution (10 mM NaCl, 10 mM MgSO4·7H2O and 100 μM CaCl2).
Samples were kept in the dark until the pH stabilized, and illuminated using a Max-303 compact xenon lamp (Asahi Spectra). The light intensity at the sample location was 860 µmol m−2 s−1 for the different carotenoids experiments (Figs. 5a), and 2,000, 860, 590 and 515 µmol m−2 s−1 for the different light intensity experiments (Fig. 5b). The light intensity measurements were performed using a LI-COR Biosciences LI-250A light metre. pH was monitored using a LAQUA F-72G pH/ION metre (HORIBA) equipped with a 9618S-10D pH microelectrode. To explore the effect of xanthophyll antennas on proton pump activity, pH measurements were converted to proton concentration. An initial analysis of the data determined that proton concentrations were changing in a near-linear manner in the range between 40 and 70 s after illumination onset, hence the rate of proton pumping was approximated by the slope of the regression line for Δ[H+] over time in this range: \(a+b\times t\) (where a and b are intercept and slope, respectively, and t is time). The experiment was conducted in a pairwise manner: a single preparation of spheroplasts derived from a single E. coli colony (all prepared from the same transformation stock expressing the rhodopsin gene) was used to obtain proton-pumping rates for the protein with (br+a) and without the carotenoid antenna (br). In total, at least four independent spheroplast preparations were utilized for each protein assay with and without a carotenoid antenna. Spheroplast samples exhibiting low rhodopsin expression where proton-pumping activity was near or below the detection threshold of the pH metre used were excluded from the analysis. A total of nine samples were excluded due to insufficient proton-pumping activity. Fold change was calculated as the ratio between the two rates: br+a/br. The resulting log2-fold changes (LFC) were used to analyse the influence of the presence/absence of the fenestration, light intensity and type of antenna using linear models in R (v.4.4.1)65. Since spheroplasts were prepared in batches, batch was incorporated as a random effect and mixed models were fit with lme4 (v.1.1-31)66. The design formula used for the light intensity experiment (with lutein as the antenna) was as follows: \({\rm{LFC}} \sim {\rm{intensity}}* {\rm{fenestration}}+(1{|\rm{batch}})\). For the experiment with varying antenna types (at 860 µmol m−2 s−1), the formula was \({\rm{LFC}} \sim {\rm{antenna}}\; {\rm{type}}* {\rm{fenestration}}+(1{|\rm{batch}})\). Type-II analysis of deviance tables was conducted with the Anova function in the car package (v.3.1-1)67. Distribution of the residuals was checked visually using Q–Q plots and while they were normally distributed in the light-intensity analysis, outliers in the antenna-type analysis were found to cause moderate deviation from the expected distribution of the residuals. Removal of these outliers improved the fit but did not lead to qualitative changes in the results of the analysis.
Protein expression and purification for structural analysis
pET21a-HeimdallR1 was transfected in E. coli C41 (Rosetta). The transformant was grown in LB supplemented with 50 µg ml−1 ampicillin at 220 r.p.m. at 37 °C. When the OD600 reached 0.6, expression was induced using 1 mM IPTG. The induced culture was grown at 120 r.p.m. for 4 h at 37 °C in the presence of 10 µM all-trans retinal. The collected cells were disrupted by sonication in buffer containing 20 mM Tris-HCl pH 8.0, 200 mM NaCl and 10% glycerol. The crude membrane fraction was collected by ultracentrifugation at 180,000 × g for 1 h. The membrane fraction was solubilized in buffer containing 20 mM Tris-HCl pH 8.0, 200 mM NaCl, 1.5% DDM, 10% glycerol and 10 mM imidazole for 1 h at 4 °C. The supernatant was separated from the insoluble material by ultracentrifugation at 140,000 g for 30 min and incubated with Ni-NTA resin (Qiagen) for 30 min. The resin was washed with 10 column volumes of wash buffer containing 20 mM Tris-HCl pH 8.0, 500 mM NaCl, 0.03% DDM, 10% glycerol and 20 mM imidazole. The resin was incubated overnight (more than 12 h) with 2 column volumes of wash buffer containing 150 µM fucoxanthin. Then, the resin was washed with 5 column volumes of wash buffer. The protein was eluted in buffer containing 20 mM Tris-HCl pH 8.0, 500 mM NaCl, 0.03% DDM, 10% glycerol and 300 mM imidazole. The eluate was dialysed against buffer containing 20 mM Tris-HCl pH 8.0, 150 mM NaCl and 0.03% DDM. The protein bound to fucoxanthin was concentrated to 50 mg ml−1 using a centrifugal filter device (Amicon 10-kDa MW cut-off) and frozen until crystallization.
X-ray crystallographic analysis of HeimdallR1
Since cryo-electron microscopy did not provide structural data, X-ray crystallographic analysis was performed. The protein bound to fucoxanthin was reconstituted into monoolein at a weight ratio of 1:1.5 (protein to lipid). The protein-laden mesophase was dispensed into 96-well glass plates in 30-nl drops and overlaid with 800 nl precipitant solution using a Gryphon robot (ARI) as previously described68. Crystals of HeimdallR1 were grown at 20 °C in precipitant conditions containing 30% PEG300, 100 mM sodium acetate pH 3.9 and 100 mM sodium nitrate. The crystals were collected directly from the lipid cubic phase using micromeshes (MiTeGen) and frozen in liquid nitrogen without adding any extra cryoprotectant.
X-ray diffraction data were collected at the SPring-8 beamline BL32XU with an EIGER X 9M detector (Dectris) using a wavelength of 1.0 Å. Small-wedge (10° per crystal) datasets were collected using a 15 × 10 μm2 beam with the ZOO system69, an automatic data-collection system developed at SPring-8. The collected images were processed using KAMO70 with XDS71, and 132 datasets were indexed with consistent unit cell parameters. After correlation coefficient-based clustering using normalized structure factors followed by merging using XSCALE72 with outlier rejections implemented in KAMO, 115 datasets were selected for the downstream analyses, because they gave the highest inner-shell and outer-shell half-dataset correlation coefficients (CC1/2). The HeimdallR1 structure was determined by molecular replacement with PHASER71 using the model obtained with AlphaFold2 (ref. 73). Subsequently, the model was rebuilt and refined using Coot74 and phenix.refine75. The final model of HeimdallR1 contained all the residues (1–246), retinal, 3 monoolein molecules, 2 nitrate ions and 121 water molecules. The fucoxanthin molecule was not modelled due to the absence of the corresponding density.
The structure of ACB-G35 was predicted with AlphaFold 3 (ref. 50) after removing the signal peptide. Retinal was used as a covalent modification of the lysine in TM7.
QM/MM simulations of HeimdallR1 with lutein and fucoxanthin
The structures of the carotenoids were docked by visual inspection of HeimdallR1 and by comparison to zeaxanthin-bound Kin4B8 (PDB: 8I2Z)7. The structures were further optimized using the hybrid QM/MM method27. Retinal linked to K231 was considered in the QM region along with lutein or fucoxanthin. A region of 5 Å around the retinal protonated Schiff base and the carotenoid was considered in the active region during optimization. The QM/MM boundary was placed between Cδ and Cε of the lysine sidechain. The QM part was described using Grimme’s GFN2-xTB method76, whereas the remaining part of the protein within the active region was treated using the AMBER ff14SB force field77 (MM part). The water molecules were described using the TIP3P model78. To model non-bonded interactions, the force switching scheme was implemented on the coulomb interaction with a cut-off of 12 Å79. Optimization was performed using the limited-memory Broyden–Fletcher–Goldfarb–Shanno (L-BFGS) method using the ORCA 5.0 programme80. Excitation energies and natural transition orbitals were computed at the RI-ADC(2)81 level in combination with the cc-pVDZ82 basis set using the Turbomole programme package83.
Statistics and reproducibility
No sample-size calculations were performed. After initial optimization trials, the biophysical experiments (flash photolysis, pH titration, FTIR) were generally performed once given the high reproducibility of such experiments and their mutual support. Absorption spectra for HeimdallR1 with different carotenoids were obtained several times in three different laboratories under different conditions. The HPLC experiment was performed on three technical replicates. Spectral measurements of HeimdallR1 with carotenoids were performed in three different laboratories in different settings, and no indication of qualitative differences in the behaviour of the protein were observed. For the proton-pumping activity experiments, the spheroplast preparations were done in batches, hence batch was included as a random effect in the mixed models; data exclusion criteria are outlined in the corresponding section in Methods. The investigators were not blinded to allocation during experiments and outcome assessment.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
All data are available in the main text or the Supplementary Information. Results of the bioinformatic analyses are deposited in figshare at https://doi.org/10.6084/m9.figshare.26906593 (ref. 84). Atomic coordinates of the crystal structure of HeimdallR1 have been deposited in the Protein Data Bank under 9JTQ. Sequences of the codon-optimized constructs used for expression of HeimdallR1, HeimdallR2, HeimdallR3 and ACB-G35 have been deposited in GenBank under accession numbers PV059850, PV059851, PV059852 and PV059853, respectively. Source data are provided with this paper.
Code availability
The code used for the bioinformatic analyses is available from Figshare at https://doi.org/10.6084/m9.figshare.26906593 (ref. 84).
References
Rozenberg, A., Inoue, K., Kandori, H. & Béjà, O. Microbial rhodopsins: the last two decades. Annu. Rev. Microbiol. 75, 427–447 (2021).
Finkel, O. M., Béjà, O. & Belkin, S. Global abundance of microbial rhodopsins. ISME J. 7, 448–451 (2013).
DeLong, E. F. & Béjà, O. The light-driven proton pump proteorhodopsin enhances bacterial survival during tough times. PLoS Biol. 8, e1000359 (2010).
Munson-McGee, J. H. et al. Decoupling of respiration rates and abundance in marine prokaryoplankton. Nature 612, 764–770 (2022).
Balashov, S. P. et al. Xanthorhodopsin: a proton pump with a light-harvesting carotenoid antenna. Science 309, 2061–2064 (2005).
Imasheva, E. S., Balashov, S. P., Choi, A. R., Jung, K.-H. & Lanyi, J. K. Reconstitution of Gloeobacter violaceus rhodopsin with a light-harvesting carotenoid antenna. Biochemistry 48, 10948–10955 (2009).
Chazan, A. et al. Phototrophy by antenna-containing rhodopsin pumps in aquatic environments. Nature 615, 535–540 (2023).
Luecke, H. et al. Crystallographic structure of xanthorhodopsin, the light-driven proton pump with a dual chromophore. Proc. Natl Acad. Sci. USA 105, 16561–16565 (2008).
Kopejtka, K. et al. A bacterium from a mountain lake harvests light using both proton-pumping xanthorhodopsins and bacteriochlorophyll-based photosystems. Proc. Natl Acad. Sci. USA 119, e2211018119 (2022).
Rinke, C. et al. A phylogenomic and ecological analysis of the globally abundant Marine Group II archaea (Ca. Poseidoniales ord. nov.). ISME J. 13, 663–675 (2019).
Frigaard, N.-U., Martinez, A., Mincer, T. J. & DeLong, E. F. Proteorhodopsin lateral gene transfer between marine planktonic Bacteria and Archaea. Nature 439, 847–850 (2006).
Iverson, V. et al. Untangling genomes from metagenomes: revealing an uncultured class of marine Euryarchaeota. Science 335, 587–590 (2012).
Pinhassi, J., DeLong, E. F., Béjà, O., González, J. M. & Pedrós-Alió, C. Marine bacterial and archaeal ion-pumping rhodopsins: genetic diversity, physiology, and ecology. Microbiol. Mol. Biol. Rev. 80, 929–954 (2016).
Tully, B. J. Metabolic diversity within the globally abundant Marine Group II Euryarchaea offers insight into ecological patterns. Nat. Commun. 10, 271 (2019).
Bulzu, P.-A. et al. Casting light on Asgardarchaeota metabolism in a sunlit microoxic niche. Nat. Microbiol. 4, 1129–1137 (2019).
Inoue, K. et al. Schizorhodopsins: a family of rhodopsins from Asgard archaea that function as light-driven inward H+ pumps. Sci. Adv. 6, eaaz2441 (2020).
Eme, L. et al. Inference and reconstruction of the heimdallarchaeial ancestry of eukaryotes. Nature 618, 992–999 (2023).
Vosseberg, J. et al. The emerging view on the origin and early evolution of eukaryotic cells. Nature 633, 295–305 (2024).
Liu, Y. et al. Expanded diversity of Asgard archaea and their relationships with eukaryotes. Nature 593, 553–557 (2021).
Varghese, N. J. et al. Microbial species delineation using whole genome sequences. Nucleic Acids Res. 43, 6761–6771 (2015).
Vernette, C. et al. The Ocean Gene Atlas v2.0: online exploration of the biogeography and phylogeny of plankton genes. Nucleic Acids Res. 50, W516–W526 (2022).
Ernst, O. P. et al. Microbial and animal rhodopsins: structures, functions, and molecular mechanisms. Chem. Rev. 114, 126–163 (2014).
Kirk, J. T. O. in Primary Productivity and Biogeochemical Cycles in the Sea (eds Falkowski, P. G. et al.) 9–29 (Springer, 1992).
Hirschi, S., Kalbermatter, D., Ucurum, Z., Lemmin, T. & Fotiadis, D. Cryo-EM structure and dynamics of the green-light absorbing proteorhodopsin. Nat. Commun. 12, 4107 (2021).
Gushchin, I. et al. Structural insights into the proton pumping by unusual proteorhodopsin from nonmarine bacteria. Proc. Natl Acad. Sci. USA 110, 12631–12636 (2013).
Balashov, S. P. et al. Breaking the carboxyl rule: lysine 96 facilitates reprotonation of the Schiff base in the photocycle of a retinal protein from Exiguobacterium sibiricum. J. Biol. Chem. 288, 21254–21265 (2013).
Field, M. J., Bash, P. A. & Karplus, M. A combined quantum mechanical and molecular mechanical potential for molecular dynamics simulations. J. Comput. Chem. 11, 700–733 (1990).
Béjà, O., Spudich, E. N., Spudich, J. L., Leclerc, M. & DeLong, E. F. Proteorhodopsin phototrophy in the ocean. Nature 411, 786–789 (2001).
Man, D. et al. Diversification and spectral tuning in marine proteorhodopsins. EMBO J. 22, 1725–1731 (2003).
Gómez-Consarnau, L. et al. Light stimulates growth of proteorhodopsin-containing marine Flavobacteria. Nature 445, 210–213 (2007).
Liu, R. et al. Metagenomic insights into Heimdallarchaeia clades from the deep-sea cold seep and hydrothermal vent. Environ. Microbiome 19, 43 (2024).
Tully, B. J., Graham, E. D. & Heidelberg, J. F. The reconstruction of 2,631 draft metagenome-assembled genomes from the global oceans. Sci. Data 5, 170203 (2018).
Rodriguez-R, L. M., Tsementzi, D., Luo, C. & Konstantinidis, K. T. Iterative subtractive binning of freshwater chronoseries metagenomes identifies over 400 novel species and their ecologic preferences. Environ. Microbiol. 22, 3394–3412 (2020).
Langmead, B. & Salzberg, S. L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359 (2012).
Haro-Moreno, J. M. et al. Dysbiosis in marine aquaculture revealed through microbiome analysis: reverse ecology for environmental sustainability. FEMS Microbiol. Ecol. 96, fiaa218 (2020).
Puente-Sánchez, F., Hoetzinger, M., Buck, M. & Bertilsson, S. Exploring environmental intra-species diversity through non-redundant pangenome assemblies. Mol. Ecol. Resour. 23, 1724–1736 (2023).
Tatusova, T. et al. NCBI prokaryotic genome annotation pipeline. Nucleic Acids Res. 44, 6614–6624 (2016).
Nishimura, Y. & Yoshizawa, S. The OceanDNA MAG catalog contains over 50,000 prokaryotic genomes originated from various marine environments. Sci. Data 9, 305 (2022).
Paoli, L. et al. Biosynthetic potential of the global ocean microbiome. Nature 607, 111–118 (2022).
Parks, D. H. et al. A complete domain-to-species taxonomy for Bacteria and Archaea. Nat. Biotechnol. 38, 1079–1086 (2020).
Asnicar, F. et al. Precise phylogenetic analysis of microbial isolates and genomes from metagenomes using PhyloPhlAn 3.0. Nat. Commun. 11, 2500 (2020).
Edgar, R. C. Search and clustering orders of magnitude faster than BLAST. Bioinformatics 26, 2460–2461 (2010).
Katoh, K. & Standley, D. M. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol. 30, 772–780 (2013).
Capella-Gutiérrez, S., Silla-Martínez, J. M. & Gabaldón, T. trimAl: a tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics 25, 1972–1973 (2009).
Stamatakis, A. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30, 1312–1313 (2014).
Camacho, C. et al. BLAST+: architecture and applications. BMC Bioinformatics 10, 421 (2009).
Darriba, D., Taboada, G. L., Doallo, R. & Posada, D. ProtTest 3: fast selection of best-fit models of protein evolution. Bioinformatics 27, 1164–1165 (2011).
Matsen, F. A., Kodner, R. B. & Armbrust, E. V. pplacer: linear time maximum-likelihood and Bayesian phylogenetic placement of sequences onto a fixed reference tree. BMC Bioinformatics 11, 538 (2010).
Huson, D. H. & Bryant, D. Application of phylogenetic networks in evolutionary studies. Mol. Biol. Evol. 23, 254–267 (2006).
Abramson, J. et al. Accurate structure prediction of biomolecular interactions with AlphaFold 3. Nature 630, 493–500 (2024).
Fu, L., Niu, B., Zhu, Z., Wu, S. & Li, W. CD-HIT: accelerated for clustering the next-generation sequencing data. Bioinformatics 28, 3150–3152 (2012).
Tamura, K., Stecher, G. & Kumar, S. MEGA11: Molecular Evolutionary Genetics Analysis version 11. Mol. Biol. Evol. 38, 3022–3027 (2021).
Chen, I.-M. A. et al. IMG/M v.5.0: an integrated data management and comparative analysis system for microbial genomes and microbiomes. Nucleic Acids Res. 47, D666–D677 (2019).
Steinegger, M. & Söding, J. MMseqs2 enables sensitive protein sequence searching for the analysis of massive data sets. Nat. Biotechnol. 35, 1026–1028 (2017).
Eddy, S. R. A new generation of homology search tools based on probabilistic inference. Genome Inform. 23, 205–211 (2009).
Chikhi, R., Raffestin, B., Korobeynikov, A., Edgar, R. & Babaian, A. Logan: planetary-scale genome assembly surveys life’s diversity. Preprint at bioRxiv https://doi.org/10.1101/2024.07.30.605881 (2024).
Li, H. Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics 34, 3094–3100 (2018).
McKenna, A. et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 20, 1297–1303 (2010).
Ahmed, F. et al. Profiling of carotenoids and antioxidant capacity of microalgae from subtropical coastal and brackish waters. Food Chem. 165, 300–306 (2014).
Young, A. J., Orset, S. & Tsavalos, A. J. in Handbook of Photosynthesis (ed. Pessarakli, M.) 597–622 (Marcel Dekker, 1997).
Grote, A. et al. JCat: a novel tool to adapt codon usage of a target gene to its potential expression host. Nucleic Acids Res. 33, W526–W531 (2005).
Lakowicz, J. R. Principles of Fluorescence Spectroscopy (Springer, 2006).
Inoue, K. et al. A light-driven sodium ion pump in marine bacteria. Nat. Commun. 4, 1678 (2013).
Wang, W.-W., Sineshchekov, O. A., Spudich, E. N. & Spudich, J. L. Spectroscopic and photochemical characterization of a deep ocean proteorhodopsin. J. Biol. Chem. 278, 33985–33991 (2003).
Ihaka, R. & Gentleman, R. R: a language for data analysis and graphics. J. Comput. Graph. Stat. 5, 299–314 (1996).
Bates, D., Mächler, M., Bolker, B. & Walker, S. Fitting linear mixed-effects models using lme4. J. Stat. Softw. 67, 1–48 (2015).
Fox, J. & Weisberg, S. An R Companion to Applied Regression (Sage, 2019).
Shihoya, W. et al. Crystal structure of heliorhodopsin. Nature 574, 132–136 (2019).
Hirata, K. et al. ZOO: an automatic data-collection system for high-throughput structure analysis in protein microcrystallography. Acta Crystallogr. D Struct. Biol. 75, 138–150 (2019).
Yamashita, K., Hirata, K. & Yamamoto, M. KAMO: towards automated data processing for microcrystals. Acta Crystallogr. D Struct. Biol. 74, 441–449 (2018).
Kabsch, W. XDS. Acta Crystallogr. D Biol. Crystallogr. 66, 125–132 (2010).
McCoy, A. J. et al. Phaser crystallographic software. J. Appl. Crystallogr. 40, 658–674 (2007).
Jumper, J. et al. Highly accurate protein structure prediction with AlphaFold. Nature 596, 583–589 (2021).
Emsley, P., Lohkamp, B., Scott, W. G. & Cowtan, K. Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 66, 486–501 (2010).
Afonine, P. V. et al. Towards automated crystallographic structure refinement with phenix.refine. Acta Crystallogr. D Biol. Crystallogr. 68, 352–367 (2012).
Bannwarth, C., Ehlert, S. & Grimme, S. GFN2-xTB—an accurate and broadly parametrized self-consistent tight-binding quantum chemical method with multipole electrostatics and density-dependent dispersion contributions. J. Chem. Theory Comput. 15, 1652–1671 (2019).
Maier, J. A. et al. ff14SB: improving the accuracy of protein side chain and backbone parameters from ff99SB. J. Chem. Theory Comput. 11, 3696–3713 (2015).
Jorgensen, W. L., Chandrasekhar, J., Madura, J. D., Impey, R. W. & Klein, M. L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 79, 926–935 (1983).
Steinbach, P. J. & Brooks, B. R. New spherical-cutoff methods for long-range forces in macromolecular simulation. J. Comput. Chem. 15, 667–683 (1994).
Neese, F. Software update: the ORCA program system—version 5.0. WIREs Comput. Mol. Sci. 12, e1606 (2022).
Hättig, C. in Advances in Quantum Chemistry Vol. 50 (ed. Jensen, H. J. Å.) 37–60 (Academic Press, 2005).
Dunning, T. Gaussian basis sets for use in correlated molecular calculations. I. The atoms boron through neon and hydrogen. J. Chem. Phys. 90, 1007–1023 (1989).
Furche, F. et al. Turbomole. WIREs Comput. Mol. Sci. 4, 91–100 (2014).
Rozenberg, A. Structural insights into light harvesting by antenna-containing rhodopsins in marine Asgard archaea. Bioinformatic Data. figshare https://doi.org/10.6084/m9.figshare.26906593 (2025).
Matsui, Y. et al. Specific damage induced by X-ray radiation and structural changes in the primary photoreaction of bacteriorhodopsin. J. Mol. Biol. 324, 469–481 (2002).
Po, H. N. & Senozan, N. M. The Henderson–Hasselbalch equation: its history and limitations. J. Chem. Educ. 78, 1499 (2001).
Larkin, M. A. et al. Clustal W and Clustal X version 2.0. Bioinformatics 23, 2947–2948 (2007).
Robert, X. & Gouet, P. Deciphering key features in protein structures with the new ENDscript server. Nucleic Acids Res. 42, W320–W324 (2014).
Acknowledgements
We thank S. Balashov, M. Sheves and G. Yahel for discussing rhodopsins and oceanography with us, and R. Edrei for help with spectroscopic measurements. O.B. is grateful to the Atmosphere and Ocean Research Institute (Univ. Tokyo, Japan) for the stay during the work on the paper. This work was supported by the European Commission, under Horizon Europe’s research and innovation programme (Bluetools project, Grant Agreement No. 101081957 to O.B.), the Nancy and Stephen Grand Technion Energy Program (GTEP, to O.B.), the Israel Science Foundation (Research Center grant 3131/20 to I.S. and O.B., and grant 1207/24 to O.B.), the German-Israeli Foundation for Scientific Research and Development (GIF NEXUS grant I-1560-207.9/2023 to I.S. and O.B.), Deutsche Forschungsgemeinschaft (DFG SFB 1078 to I.S.), Swiss National Science Foundation (SNF Sinergia grant 213507 to I.S.), the Agencia Estatal de Investigación (grant PID2022-140995OB-C21 by MICIU/AEI/10.13039/501100011033 and ERDF/EU to R.L.), the Czech Ministry of Education (OP JAK project Photomachines reg. no. CZ.02.01.01/00/22_008/0004624 to M. Koblížek), the Institute for Fermentation Osaka (W.S.), Research Foundation for Opto-Science and Technology (W.S.), JSPS KAKENHI Grants-in-Aid (grants JP21H04969 to H.K., JP23H04404 and 24H02268 to K.I., JP19H05777 to W.S., JP23KJ0721 to Y. Matsuzaki, JP24K23232 to T.T., and JP24KJ0909 to S.M.), JST CREST (grants JPMJCR1753 to H.K., JPMJCR22N2 to K.I., and JPMJCR20E2 to O.N.), the MEXT Promotion of Development of a Joint Usage/Research System Project: Coalition of Universities for Research Excellence Program (CURE) (grant JPMXP1323015482 to H.K. and K.I.), and the Platform Project for Supporting Drug Discovery and Life Science Research (Basis for Supporting Innovative Drug Discovery and Life Science Research (BINDS)) from AMED, grant numbers JP24ama121001 (support number S01859001, W.S.), JP24ama121002 (support number 3272, O.N.) and JP24ama121012 (support number 6234, K.I.). M.d.C.M. thanks the Azrieli Foundation for the award of the Azrieli International Postdoctoral Fellowship (cohort 2022-2023). O.B. holds the Louis and Lyra Richmond Chair in Life Sciences.
Author information
Authors and Affiliations
Contributions
G.T. and A.C. conceived the project. G.T. performed environmental carotenoid extractions and binding to rhodopsins, and measured light-dependent proton-pumping activity. M. Konno, R.A.-Y. and M.d.C.M. performed expression and purification of HeimdallR1 and carotenoid-binding HeimdallR1. M.d.C.M. performed HPLC analysis of retinal isomers, pH titration and experimental data analysis. M.d.C.M. and K.I. performed laser-flash photolysis. A.R. performed bioinformatics. S.L. and M. Konno performed molecular biology. A.M.-M. and R.L. performed carotenoid characterization from environmental samples and rhodopsin-bound carotenoids. M. Koblížek, D.B.-P. and J.C. purified diatoxanthin from P. tricornutum. S.I., Y. Mizuno, K.K. and H.K. performed low-temperature UV–Vis and FTIR spectroscopy. Y. Matsuzaki, T.T., S.M., W.S. and O.N. performed structural analyses. P.N. and I.S. performed MD simulations, hybrid QM/MM simulations and interpreted the results of the simulations. O.B. coordinated the project and wrote the paper with input from all authors.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Microbiology thanks Takefumi Morizumi and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Peer reviewer reports are available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data
Extended Data Fig. 1 Environmental marine xanthophylls bind to HeimdallR1.
HPLC profile of Mediterranean Sea chromophore extract (blue) and HeimdallR1 bound chromophores (red). Main peaks correspond to fucoxanthin (2), diatoxanthin (3), lutein (5), β-carotene (8), and uncharacterized (1,4,6,7,9). Chromophores were registered at 450 nm.
Extended Data Fig. 2 Spectroscopic characterization of Kin4B8-xanthorhodopsin and HeimdallR1-G141F with hydroxylated carotenoids.
a, Absorbance change (up) and fluorescence excitation spectra (bottom) of Kin4B8-xanthorhodopsin upon incubation with (orange) or without (purple) diatoxanthin. b, Absorbance change (up) and fluorescence excitation spectra (bottom) of Kin4B8-xanthorhodopsin upon incubation with (orange) or without (purple) fucoxanthin. c, Absorbance change (up) and fluorescence excitation spectra (bottom) of HeimdallR1-G141F upon incubation with (orange) or without (purple) lutein; emissions were recorded at 720 nm.
Extended Data Fig. 3 Rhodopsin genes and genes involved in biosynthesis of carotenoids in “Ca. Kariarchaeum”.
a, Neighbour-joining tree of (near-)complete HeimdallR amino-acid sequences. The three clades are highlighted with color and proteins used for expression are indicated with asterisks. For proteins coming from MAGs, the corresponding scaffold names are provided. Numbers next to edges refer to bootstrap support values > 50. b, Rhodopsins and enzymes with putative roles in carotenoid biosynthesis in the “Ca. K. pelagium” core pan-genome. Top: genomic locations of the rhodopsin genes, heimdallarchaeial rhodopsin (heimdallR) and heliorhodopsin (heR1-3) genes. Middle: genomic locations of genes involved in carotenoid biosynthesis – phytoene synthase (crtB), phytoene desaturase (crtI) and two additional genes of unknown function located in the same putative operon; a putative lycopene elongase (lyeJ, prenyltransferase cl00337), carotenoid desaturase (crtD, phytoene dehydrogenase-related protein COG1233), and a putative carotenoid glycosyltransferase (crtQ?). Coordinates are provided for the corresponding genomic regions with respect to the pan-genome scaffolds. Bottom: reactions that might be catalyzed by the corresponding enzymes.
Extended Data Fig. 4 Global distribution of “Ca. Kariarchaeum pelagium” and patterns of variation across its genome.
a, Distribution of “Ca. K. pelagium” based on Logan contigs from marine metagenomes and metatranscriptomes recruited to the “Ca. K. pelagium” core pan-genome. Abundances are expressed as fractions of the pan-genome covered by the mapped contigs with a minimum coverage of 0.5%. Locations of “Ca. K. pelagium” MAGs are indicated with labels coloured according to the heimdallR allele type. Locations of the other “Ca. Kariarchaeum” are indicated for reference: MAG FT_008 (sp016839545) and scaffold Kari_Gs0128817 (sp. nov.). b, Per-nucleotide depth of coverage of Logan metagenomic contigs along the core pan-genome of “Ca. K. pelagium” (upper) and number of variable positions per nucleotide for genes across the pan-genome. Only genes with ORF longer than 300 bp were analyzed. Asterisks indicate outlier genes with values exceeding upper quartile + 1.5×interquartile range. c, Number of variable positions per nucleotide for sliding windows of 200 bp across the pan-genomic scaffold containing the heimdallR gene (scaffold 11). In panels b and c, upper and lower quartiles (dashed lines) and median (solid lines) are indicated.
Extended Data Fig. 5 Biophysical characterization of diverse marine HeimdallRs with lutein, diatoxanthin or fucoxanthin.
Absorbance change (up) and fluorescence excitation spectra (bottom) of HeimdallR1, 2 and 3 upon incubation with (orange) or without (purple) lutein (a), diatoxanthin (b), and fucoxanthin (c); emissions were recorded at 720 nm. The results for HeimdallR1 (also shown in Figs. 1 and 3) are shown for reference.
Extended Data Fig. 6 Absorption spectra of purified HeimdallR1 with and without lutein or fucoxanthin and pH dependence of the absorption.
a, UV–vis absorption spectra: HeimdallR1 alone (left, purple), HeimdallR1 complexed with lutein (middle, yellow), and HeimdallR1 complexed with fucoxanthin (right, red). To estimate the λamax of the retinal in HeimdallR1, the absorption spectra of pure xanthophylls (blue) were subtracted from those of HeimdallR1 complexed with each xanthophyll (green). Since the peak positions of pure xanthophylls slightly differ from those complexed with HeimdallR1, the spectra of pure xanthophylls were manually shifted so that the peak positions coincides between the spectra of pure xanthophylls and HeimdalR1-xanthophyll complexes. Pictures of purified proteins are shown next to the corresponding results. b, Absorption spectra (top) and the different absorption spectra (bottom), and c, pH titration curves for the calculation of pKa were measured depending on acidic (left) and alkaline (right) pH changes. The red-shifted (red arrow) and blue-shifted (blue arrow) absorption spectra at each pH are indicated by arrows. The pH titration curves were analyzed using the Henderson–Hasselbalch equation86. d, Color protein changes upon acid-basic titration.
Extended Data Fig. 7 Influence of lutein or fucoxanthin on the retinal photoisomerization in HeimdallR1 at 77 K.
UV-visible (a, b) and FTIR (c-h) spectra obtained for lipid-reconstituted HeimdallR1 with (red; H(F)) or without (black; H(-)) fucoxanthin (top) and HeimdallR1 with (red; H(L)) or without (black; H(-)) lutein (middle) are compared to those for lipid-reconstituted Kin4B8-xanthorhodopsin with (green; K(L)) or without (black; K(-)) lutein (bottom) previously published7. a, UV-visible absorption spectra of H(F) (red) or H(-) (black) (top), H(L) (red) or H(-) (middle), and K(L) (green) or K(-) (black) (bottom) at 77 K. One division of the y-axis corresponds to 1.0 absorbance units. b, Difference UV-visible spectra upon illumination of H(F) (red) or H(-) (black) (top), H(L) (red) or H(-) (black) (middle), and K(L) (green) or K(-) (black) (bottom) at 77 K. Hydrated films of lipid-reconstituted protein were illuminated at 540 nm light, which forms the red-shifted K intermediate. One division of the y-axis corresponds to 0.08 absorbance units. c, Light-minus-dark difference FTIR spectra upon illumination of H(F) (red) or H(-) (black) (top), H(L) (red) or H(-) (black) (middle), and K(L) (green) or K(-) (black) (bottom) at 77 K. Hydrated films of lipid-reconstituted protein with H2O were first illuminated at 540 nm light (solid lines), which forms the K intermediate, and the K intermediate was then reverted by illumination at >590 nm light (dotted lines). Spectral acquisition was repeated to improve signal-to-noise ratio. Positive and negative signals originate from the K intermediate and unphotolyzed state, respectively. One division of the y-axis corresponds to 0.0032 absorbance units. d, Enlarged spectra of the C = C stretching frequency region of the retinal chromophore (1600-1450 cm−1) from (c). Negative peaks are different between H(F) (1534 cm−1) and H(-) (1532 cm−1) (top) or between H(L) (1534 cm−1) and H(-) (1532 cm−1) (middle), but not for K(L) and K(-). One division of the y-axis corresponds to 0.003 absorbance units. e, Enlarged spectra of the C-C stretching frequency region of the retinal chromophore (1250-1150 cm−1) from (c). Spectra are identical with and without xanthophylls. One division of the y-axis corresponds to 0.002 absorbance units. f, Enlarged spectra of the hydrogen out-of-plane (HOOP) vibrational region of the retinal chromophore (1020-945 cm−1) from (c). Positive peaks are different between H(F) (986 cm−1) and H(-) (985 cm−1) (top) or between H(L) (986 cm−1) and H(-) (985 cm−1) (middle), but not for the 960-cm−1 band (bottom). One division of the y-axis corresponds to 0.0015 absorbance units. g, Spectral comparison of the HOOP bands between H2O (red or green) and D2O (blue) hydrations. Down-shifts of the positive peaks at 986 cm−1 (top, middle) and 960 cm−1 (bottom) show that these HOOP bands originate from the Schiff base region. One division of the y-axis corresponds to 0.0015 absorbance units. h, Enlarged spectra of the amide-I region of the peptide backbone (1680-1575 cm−1) from (c). Spectra are identical with and without xanthophylls. One division of the y-axis corresponds to 0.001 absorbance units.
Extended Data Fig. 8 Structural features of HeimdallR1.
a, Size-exclusion chromatography profile of the HeimdallR1 bound to fucoxanthin. The relative absorbance at 280 nm, 460 nm and 550 nm is shown in the chromatogram (left panel). The peak fraction (indicated by a black bar) was analyzed by SDS-PAGE (right panel). b, HeimdallR1 crystals in lipidic mesophase. c, Crystal packing of HeimdallR1. d, Key rhodopsin proton pump motifs in HeimdallR1. Black dashed lines indicate hydrogen-bonding interactions. Red spheres indicate water molecules. e–g, Structural comparison of HeimdallR1 with Kin4B8-xanthorhodopsin (PDB ID: 8I2Z) and green proteorhodopsin (GPR; PDB ID: 7B03), from the membrane plane (e), the extracellular side (f) and the intracellular side (g). Notably, ICL3 of HeimdallR1 forms a membrane-extending α-helix. h, Interaction between ICL3 and TM6 in HeimdallR1. Black dashed lines indicate hydrogen-bonding interactions. Red spheres indicate water molecules. i, Sequence alignment of HeimdallRs around ICL3. The sequence alignment was created using ClustalW87 and ESPript 3.088 servers. Boxes indicate α-helices.
Extended Data Fig. 9 Simulation features of HeimdallR1 bound to lutein and fucoxanthin.
a, HeimdallR1. b, HeimdallR1 with lutein. c, HeimdallR1 with fucoxanthin. d, Kin4B8-xanthorhodopsin with zeaxanthin. ATR — all-trans-retinal. Black dashed lines indicate hydrogen bonds.
Extended Data Fig. 10 Electronic transitions in HeimdallR1/xanthophyll complexes.
Energy level diagram along with the nature of the excited states (LE: local excitation; CT: charge transfer), and natural transition orbitals (isovalue = 0.01 a.u.) displaying the dominant orbitals involved in the electronic transitions of the first three excited states in HeimdallR1/xanthophyll with lutein (a) and fucoxanthin (b). The subscript denotes excitation in retinal (R), lutein (L), fucoxanthin (F) or transition from retinal to the respective xanthophyll. The transition orbitals are shown only for those with contribution greater than 5% towards the excited state (transition contribution between each pair of orbitals is marked in the figure).
Supplementary information
Supplementary Information
Supplementary Figs. 1–3 and Tables 1–3.
Source Data Supplementary Fig. 1
Source data Supplementary Fig. 1.
Source Data Supplementary Fig. 2
Source data Supplementary Fig. 2.
Source Data Supplementary Fig. 3
Source data Supplementary Fig. 3.pdb.
Source data
Source Data Fig. 1
Source data Fig. 1.
Source Data Fig. 2
Source data Fig. 2.
Source Data Fig. 3
Source data Fig. 3.
Source Data Fig. 4
Source data Fig. 4.
Source Data Fig. 5
Source data Fig. 5.
Source Data Fig. 6
Source data Fig. 6.pdb.
Source Data Supplementary Fig. 4
Source data Supplementary Fig. 4. Lutein energies.
Source Data Extended Data Fig. 1
Source data ED Fig. 1.
Source Data Extended Data Fig. 2
Source data ED Fig. 2.
Source Data Extended Data Fig. 3a
Source data ED Fig. 3a.nex.
Source Data Extended Data Fig. 4
Source data ED Fig. 4.
Source Data Extended Data Fig. 5
Source data ED Fig. 5.
Source Data Extended Data Fig. 6
Source data ED Fig. 6.
Source Data Extended Data Fig. 7
Source data ED Fig. 7.
Source Data Extended Data Fig. 8
Source data ED Fig. 8.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Tzlil, G., Marín, M.d.C., Matsuzaki, Y. et al. Structural insights into light harvesting by antenna-containing rhodopsins in marine Asgard archaea. Nat Microbiol 10, 1484–1500 (2025). https://doi.org/10.1038/s41564-025-02016-5
Received:
Accepted:
Published:
Issue date:
DOI: https://doi.org/10.1038/s41564-025-02016-5
This article is cited by
-
Carotenoids bind rhodopsins and act as photocycle-accelerating pigments in marine Bacteroidota
Nature Microbiology (2025)
