
Abstract
Brillouin light scattering (BLS) spectroscopy is a non-invasive, non-contact, label-free optical technique that can provide information on the mechanical properties of a material on the submicrometre scale. Over the past decade, BLS has found increasing microscopy applications in the life sciences, driven by the observed importance of mechanical properties in biological processes, the realization of more sensitive BLS spectrometers and the extension of BLS to an imaging modality. As with other spectroscopic techniques, BLS measurements detect not only signals that are characteristic of the investigated sample, but also those of the experimental apparatus, and can be substantially affected by measurement conditions. Here we report a consensus between researchers in the field. We aim to improve the comparability of BLS studies by providing reporting recommendations for the measured parameters and detailing common artefacts. Given that most BLS studies of biological matter are still at proof-of-concept stages and use different, often self-built, spectrometers, a consensus statement is particularly timely to ensure unified advancement.
Similar content being viewed by others
Main
In the context of studying bio-relevant matter, Brillouin light scattering (BLS) spectroscopy involves measuring the frequency and lifetime of megahertz–gigahertz-frequency acoustic phonons—propagating density fluctuations—in a material. These are distinct from the higher-frequency localized optical phonons probed using, for example, Raman spectroscopy. This is made possible by the constructive and destructive interference of light inelastically scattered from propagating thermal or stimulated phonons of a given wavevector determined by the Bragg condition1. Inelastic scattering refers to light that is not of the same energy as the incident light, manifested through a detected spectrum that is different from the incident light or deviation in the arrival time of photons from the optical path length. A BLS spectrum of a homogeneous and isotropic material will consist of at least two (Stokes and anti-Stokes) peaks that reveal the amplitude, frequency and lifetime of acoustic phonons at the probed scattering wavevector(s).
Examples of the BLS spectra of distilled water, measured using different spectrometer designs, are shown in Fig. 1a–e. In each case, the spectrum of (>99% pure) cyclohexane, which has a smaller BLS frequency shift (νB) and broader linewidth (ΓB) than water, is also shown. A description of the different spectrometer designs and the detection schemes/scattering geometries can be found in Supplementary Section 1 and Supplementary Fig. 1. The frequency shift of the BLS peak in each case corresponds to the probed phonon frequency. Using the wave equation, this can be used to calculate the phonon hypersonic speed, which is related to the respective elastic modulus (Supplementary Section 2). The width of the BLS peaks is related to the imaginary (dissipative) part of the modulus, which can be expressed in terms of the longitudinal viscosity but is also dependent on extrinsic experimental conditions and commonly affected by spatial heterogeneities in the sample in a non-trivial manner.

a–e, Example BLS spectra of distilled water (black circles) and cyclohexane (red squares) measured using different spectrometer designs (normalized to unity). a, Tandem Fabry–Perot (TFP) spectrometer. b, Virtual imaged phase array (VIPA) spectrometer with electro-optic modulator reference peaks45 at 13 GHz. c, Stimulated Brillouin scattering (SBS) spectroscopy. d, Time-resolved Brillouin scattering (TRBS) microscopy. e, Impulsive SBS (I-SBS) spectroscopy. The insets in d and e show the respective measured time-resolved data. The I-SBS frequency shift depends solely on sound speed, whereas for spontaneous BLS, SBS and TRBS, the refractive index reduces the frequency difference between water and cyclohexane (see also Supplementary Section 1.2). Horizontal dashed and solid lines represent measurement and scan ranges of the respective measurements. Lines between data points are presented as a guide to the eye.
While BLS spectroscopy has been performed in research laboratories for more than half a century2, the recent surge of interest in biomedical applications can largely be attributed to its extension to an imaging modality3,4 suitable for studying biological samples4. Driven also by our growing awareness of the importance of mechanical properties for biological processes5,6, the past few decades have seen the development of a number of different approaches7,8 to obtain BLS spectra of biological matter, together with their application to diverse biological systems8,9,10. These approaches are expected to yield comparable results for the viscoelastic moduli. However, each has distinct experimental challenges, limitations and characteristic sources of uncertainty.
There are ongoing discussions on the biological relevance of BLS-derived parameters of soft tissue and cells11,12,13,14,15, but we focus here on photonics aspects necessary to extract reliable and repeatable parameters in biological matter. We also only consider Brillouin scattering at visible–near-infrared wavelengths, as X-ray16 and neutron17 Brillouin scattering are associated with distinct challenges.
Parameters that BLS measures
The two key parameters measured in BLS spectroscopy are νB and ΓB, both conventionally presented in units of megahertz or gigahertz. To obtain these parameters, one typically fits each Brillouin peak in the frequency domain with a suitable function (Supplementary Section 2.1).
Experimental data for a BLS measurement typically consist of the intensity at different frequencies spanning one or both Stokes and anti-Stokes BLS peaks, or time-series data longer than 1/ΓB with a sampling frequency >2νB. In most cases, BLS measurements do not measure the elastic scattering peak, which is suppressed, masked or otherwise avoided due to its large intensity. Below we list some physical quantities that can be extracted from the key parameters (corresponding equations are provided in the Supplementary Section 2).
-
Hypersonic acoustic speed (V) The phase velocity of the probed acoustic modes in the direction of the scattering wavevector. It is obtained from νB and, depending on the scattering geometry, the refractive index (n) of the sample in the direction of the scattering wavevector. All current implementations of BLS imaging probe the longitudinal acoustic phonon modes, but depending on the scattering geometry and detection optics, BLS may probe longitudinal, transverse or mixed transverse–longitudinal acoustic phonon modes, with the phase velocity being that of the respective modes8,18,19.
-
Longitudinal storage modulus (M′) A measure of the elastic properties subject to longitudinal boundary conditions. For isotropic materials, M′ is a combination of the real parts of the shear (G′) and bulk (K′) moduli: M′ = K′ + (4/3)G′ (Supplementary Section 2.5). In addition to all of the parameters required for calculating ν, knowledge of the mass density (ρ) is required.
-
Longitudinal loss modulus (M″) and longitudinal viscosity (ηL) Measures of the viscous (dissipative) properties subject to longitudinal boundary conditions. In addition to all parameters required for calculating M′, ΓB is required.
-
Longitudinal loss tangent (tanδ) Defined as M″/M′ = ΓB/νB, tanδ is a measure of attenuation of the longitudinal phonons that is independent of ρ and usually, to a good approximation, n.
-
Shear modulus (G) Can be obtained from the BLS frequency shift of transverse acoustic modes in a symmetry direction. Transverse acoustic phonon modes are manifested as Stokes and anti-Stokes peaks that are distinct from those of longitudinal acoustic phonons, usually with a smaller BLS frequency shift. They typically require measurements at different scattering geometries and polarizations19,20,21,22. The frequency shift of transverse acoustic phonons may be too small to measure practically in soft matter and liquids. Calculation of G requires ρ and n in the direction of the scattering wavevector. G is a complex quantity (G = G′ + G″i), with the real (G′) and imaginary (G″i) parts obtainable from the frequency shift and linewidth in an analogous manner to M′ and M″.
-
Tensile or Young’s moduli (E), K and Poisson’s ratios (σ) These can be derived from measurements of G and M and require a priori knowledge or assumptions on the symmetry of the sample19,20,21 (Supplementary Section 2). They are typically only accessible in hard matter where transverse acoustic phonon modes can be measured.
-
Landau–Placzek ratio (rLP) Defined as the ratio of the integrated Rayleigh scattering peak intensity (the component of elastic scattering from the thermal diffusivity mode23) to the integrated BLS peak intensity (defined as the area under the respective peaks)1. For homogeneous samples rLP = (cP − cV)/cV, where cP and cV are the specific heat at constant pressure and constant volume1. While rLP can provide insight into thermodynamic properties, the validity breaks down in complex materials and an accurate measure of the Rayleigh signal is challenging in the presence of other dominant elastic scattering processes, such as Tyndall scattering24. It has therefore been avoided when considering biological matter thus far.
-
Refractive index (n) Can be calculated for isotropic materials or materials with known symmetry from angle-resolved measurements or from several different scattering geometries20,25, under the assumption of a homogeneous sample, and at frequencies not in the vicinity of a structural relaxation process or phase change26.
-
Mass density (ρ) Can be calculated in stimulated BLS, with a priori knowledge of the refractive index, via the BLS gain, with which it scales linearly27.
Resolution
The spectral resolution of a BLS spectrometer may be defined (in line with guidelines of the Joint Committee for Guides in Metrology28) as the smallest change in frequency that would cause a detectable change in the corresponding measurement. Depending on the spectrometer, this can be extracted from a measurement of the width of a spectrally narrow laser line or the uncertainty of photon arrival times. While the resolution of the spectrometer is ultimately a limiting factor, spectral deconvolution29 and spectral fitting30 can be employed for highly precise measurements of BLS frequency shifts and linewidths (exceeding the accuracy with which νB can be extracted by two orders of magnitude compared with the spectral resolution of the spectrometer31). The use of the Allan variance method, introduced for IR spectroscopy, has been proposed32,33 for making unbiased comparisons of the spectral precision of different BLS spectrometer set-ups. This can be applied to BLS experiments by measuring the standard deviations of the fitting parameters over the sampling time34,35. To obtain unbiased spectrometer comparisons, one should bear in mind that spectrometers may allow optimization of this parameter by modulating (for example) the mirror spacing, acquisition/scan time, amplitude and number of acquisition points.
The spatial resolution is fundamentally limited by the effective point spread function of the microscope itself, with the exception of the axial resolution in TRBS (Supplementary Section 1.2). Given that BLS is a coherent phenomenon, the effective point spread function may be larger than that defined using incoherent probes (for example, fluorescent labels) or scattering processes. This can be understood through the dependence of the probed photon–phonon interactions on the coherence lengths of the phonons (that is, the characteristic size range over which density fluctuations are correlated)36,37. This is dependent on the sample and given, for a phonon wavelength of Λ, by ΛνB/ΓB (ref. 38). Increasing the mechanical contrast between different regions typically reduces the phonon coherence, improving the spatial resolution up to the limit of the optical resolution obtainable from incoherent scattering processes. In practice, the true spatial resolution is best determined experimentally on mock-up systems that consist of different materials with sharp interfaces and spatially scanning between them.
Reporting consensus
Below we describe parameters that we consider important to report in all BLS measurements, regardless of the spectrometer used. These are provided also in the form of a Minimal Reporting Table (Supplementary Table 1).
Spectrometer
In addition to information on the type of spectrometer, this should include its free spectral range (FSR), sampling step size (distance between voxels in raster scans and/or time increments in time-series measurements) and range (in frequency for the frequency domain or time for the time domain).
Spectral resolution
In spontaneous frequency-domain BLS measurements, this is often most easily obtained by measuring the elastic scattering peak width from a highly scattering sample (for example, a mirror flat), with the assumption that the probing laser(s) have much narrower spectral linewidths than the frequency shifts measured. In frequency-domain SBS, stimulated Rayleigh scattering can be measured as described in ref. 39, particularly the absorptive contribution to the gain spectrum, the strength of which depends on the type and purity of the sample. The different contributions to the effective spectral resolution are described in ref. 27, including the laser linewidth at a given measurement time, numerical aperture (NA), lock-in-amplifier time constant and laser frequency scanning rate. In time-domain measurements this can be estimated from the repetition rate of the pulsed laser, which acts as the frequency comb spacing in the spectral domain. The spectral resolution is generally reported in units of megahertz. Although the spectral resolution is not a direct indication of the precision with which the key parameters can be extracted if the shape of the peak is known, it is important to distinguish multiple peaks (which may occur near interfaces40) or deduce undefined/uncertain functional peak shapes (which may occur near structural transitions). In general, spectral deconvolution can be used to improve BLS frequency shift and linewidth estimates, but may increase processing time.
Spectral precision (of spectrometers)
This is distinct from the spectral resolution in that it is a measure of how precisely νB and ΓB can be determined from repeated measurements, and ultimately a measure of the stability of the microscope/spectrometer. A reasonable practical measure of this can be obtained from the spread of repeated measurements or a spatial scan of a static homogeneous sample. In certain cases, the difference in the absolute frequency shift and linewidth of the Stokes and anti-Stokes peaks may also contribute and should be considered. These parameters are generally reported as coefficients of variation on the mean (ratios of the standard deviation of the mean to the mean) and as percentages. For a heterogeneous or time-varying sample the precision can have inherent spatiotemporal variability that is not accounted for with a single spectral precision, which should be considered. The acceptable spectral precision will depend on the size of the changes in the key parameters one wishes to discern. In practice, a precision of ~0.5% for νB and 5% for ΓB often suffices to discern significant changes in the derived elastic and viscous parameters of biological samples.
Signal-to-noise and signal-to-background ratios
These are measures of how well the BLS signal compares to all other non-BLS detected signals. Various sources of background signals can exist, including residual amplified spontaneous emission (ASE), side modes of laser sources that are not perfectly clean, fluorescence/Raman signals and artefacts from elastic scattering (for example, from turbid samples like tissue or bones). In the most desirable operating conditions, all non-BLS signals would be removed and enough BLS photons would be collected to operate in the shot noise limit for the detector used. Reporting the signal-to-noise ratio (SNR) and signal-to-background ratio is therefore important to demonstrate whether a measurement is shot noise (highly preferred) or background limited. The SNR may be quantified in a manner analogous to that used in Raman spectroscopy41, as the ratio of the averaged BLS peak intensity to its standard deviation, while the signal-to-background ratio is the ratio of the averaged BLS peak intensity to the standard deviation of the noise floor in the absence of the BLS signal.
NA of excitation and detection light/scattering angle(s)
Given that νB and ΓB are dependent on the scattering angle, using high NAs (>0.4) will result in broadening of the linewidth due to the increased spread of scattering wavevectors measured42,43. At scattering angles away from backscattering, the effect of a finite NA becomes particularly pronounced and an experiment-specific trade-off between the acceptable spread in scattering wavevector, spatial resolution, laser exposure and acquisition time must be made. While conventional wisdom states that increasing the excitation and/or detection NA results in a higher lateral spatial resolution, in BLS microscopy this may not always be the case due to the finite phonon length scales38. As such, explicitly reporting both the excitation and detection NA or angles is essential for meaningful interpretation of data. In addition, for confocal implementations, reporting of the effective size of the detection pinhole (physical pinhole or fibre core diameter) in Airy units is important for understanding the confocality and probed wavevectors.
Average power/peak power at sample/pulse repetition rate
Measurements conducted at low average laser power have the advantage of reduced photodamage/phototoxicity for live cell and tissue studies. They also reduce the potential of substantial sample heating which affects the BLS spectrum. While for biological samples it is almost always desirable to use as low a laser power as possible while still maintaining an acceptable SNR, for dynamic/moving samples there may be a trade-off to capture the desired dynamics. To avoid perturbing sensitive biological processes the total laser exposure (energy deposited on sample) is often more relevant, and as such a balance between exposure time and laser power may need to be found. The acceptable power intensity is ultimately sample dependent and guidelines set out for other microscopy techniques should be followed44.
Laser wavelength(s)
Both νB and ΓB depend on the wavelength of the laser source. When selecting wavelengths, the strength of the BLS signal, the potential for photodamage and the depth within a sample one wishes to measure need to be considered. While the BLS scattering cross-section scales as λ−4 (where λ is wavelength) and the spatial resolution of a measurement increases with decreasing λ, the photodamage to biological materials is generally more substantial at shorter wavelengths.
Exposure/acquisition time
The exposure time for each spectral acquisition should be chosen to obtain a sufficient SNR. However, dynamic/moving samples may set a practical upper limit, in which case a compromise between increasing laser intensity and decreasing spectral resolution may be needed.
Sample temperature
Given the strong temperature dependence of BLS, we recommend that the sample temperature should be reported to within ±0.5 °C. This will typically cause errors in the value of νB in hydrated samples of only ~0.1% or less.
BLS spectroscopy methods
A description of the different approaches for performing BLS spectroscopy, together with measurement and reporting recommendations particular to each, are presented in Supplementary Section 1. Here we discuss aspects relevant to different spectrometer designs and the comparison of measurements on different set-ups.
Calibration spectra
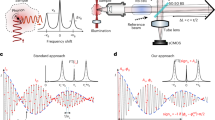
BLS spectra need accurate and robust calibration for data to be comparable across laboratories and experimental conditions, as well as over time. For instruments that need to be calibrated using a reference inelastic peak, such as VIPA architectures or single etalon systems, optimal performance is achieved by eliminating the elastic scattering and calibration is typically performed using materials with known νB to estimate the unknown dispersion parameters. The accuracy of these calibration procedures relies on the accuracy of the reference νB, which may depend on other experimental parameters such as temperature (for example, the νB of water increases by ~1% °C−1; whereas that of polystyrene decreases with increasing temperature47).
Calibration based on absolute, and highly conserved, absorption lines of atomic gas vapours such as rubidium may be employed. It has been shown that locking the laser frequency to such narrow absorption lines allows calibrated measurements that are accurate to within a few megahertz over extended periods48. Alternatively, a robust calibration strategy can involve spectrally shifting a part of the elastic scattered signal by a few gigahertz using an electro-optic modulator45,49,50 or potentially also an acousto-optic modulator. In SBS microscopy set-ups, measurements of the frequency difference between the pump and probe beams typically involve: (1) beating measurements with a fast photodetector and a frequency counter51; and (2) frequency locking one laser to a rubidium transition and measuring the wavelength of the second laser using an accurate and precise wavemeter39. One may also use a range of rubidium absorption peaks as frequency references for spectral calibration (Supplementary Section 1.2).
Fitting of BLS spectra
In cases where the presence of a mechanical relaxation process can be ruled out, the BLS peak is well represented by a damped harmonic oscillator (DHO). When spectra are measured using Fabry–Perot interferometers or monochromators, a direct DHO fit is feasible. However, for imaging spectrometers the spectral projection will be weighted by a Gaussian envelope52. This results in a slight functional correction, and fits are typically performed using Lorentzian functions. Given that νB in a DHO fit does not exactly coincide with the peak maximum, a correction (usually small) may need to be applied for broad BLS peaks when using Lorentzian fitting53.
In the ideal scenario, the detected signal is shot-noise-limited, such that the variance σ scales as \({\sigma}^{2}={N}_{\mathrm{i}}\), where Ni is the number of photons detected in a given frequency interval. Large variation in the relative photon count uncertainty across the BLS peak would require a weighted (\(\propto {\sigma }_{i}^{-2}\)) regression routine for fitting. In multi-pixel detectors there may also be additional sources of noise (due to electron amplification, for example) that increase the variance and should be considered. The fitting function may thus also include a baseline term to account for this and other physical phenomena (for example, fluorescence and Raman scattering folded into the spectral measurement region due to the finite spectrometer FSR). Rather than subtracting such contributions from the spectra, which can be statistically misleading, this contribution should explicitly be included in the fitting function.
The measured BLS spectrum is always subject to instrumental broadening and should thus ideally be deconvolved with the (spectral) instrument response function (IRF). Alternatively, a modified fitting function can be constructed by convolving the model function (DHO or Lorentzian) with the IRF. In frequency-domain spectroscopy, the IRF can be obtained by measuring the spectra of the elastically scattered probing laser through the optical set-up by replacing the sample with a mirror flat in the backscattering geometry.
Uncertainties
When reporting uncertainties for BLS measurements, following established guidelines54 is recommended. Whenever possible, repeated measurements of a sample under the same conditions should be conducted to evaluate the uncertainty caused by random error sources. This type of uncertainty (type A evaluation) is expressed as the standard deviation of the mean. For uncertainties that cannot be estimated from repeated measurements (type B evaluation), such as systematic errors, a comparison with the reported values of standard materials should be performed when possible. The combined uncertainty can then be calculated by considering the dependencies between error sources. To mitigate systematic errors, regular calibration with readily available homogeneous materials with constant νB values, and where the BLS peak is well described by a model fitting function, is recommended. To report the uncertainty of any calculated viscoelastic parameters, the uncertainties of each of the measured or assumed parameters should be evaluated separately and then combined according to the law of propagation of uncertainty.
Parameters for high-quality BLS measurements
One can distinguish between parameters that concern the probing light and the detection of the BLS signal. On the probing side, the most important parameters are: (1) the laser linewidth, which determines the upper limit of the achievable spectral resolution, and should typically be <100 MHz; (2) the long-term laser wavelength stability, which should ideally be <1 GHz of drift per hour (locking the laser wavelength to a gas absorption cell line or recalibration with control samples may be employed); and (3) the spectral noise of the laser (ASE and side modes), which can lead to considerable spectral contributions either side of the main laser line. The ASE influences the spectral noise floor from its elastic scattering by the sample or within the optical set-up. It can, owing to its spectral breadth, be challenging to filter out, potentially even requiring a combination of etalon filters. The total power outside the main laser line should typically be ~60–90 dB lower than that of the main laser line for high-quality BLS measurements.
On the detection side, it is most important to sufficiently suppress elastic scattered light. The required magnitude thereof depends on a sample’s scattering properties, although a suppression of ~60–90 dB (less in homogeneous low-scattering samples such as transparent liquids, more in highly scattering samples such as calcified bones) is desirable. At the same time, the detection of the BLS signal needs a sufficiently high SNR for high-precision fitting. If a (typical) precision of ~10 MHz is desired, a SNR of between ~30 and 50 is required, depending slightly on other spectrometer properties and acquisition parameters (spectral sampling and so on)55. Given that state-of-the-art spectrometers are often shot-noise-limited, this would correspond to a total signal that is on the order of 103–104 detected photons across the measured BLS spectrum.
While a high SNR, spectral resolution and precision will result in more reliable determination of the key parameters, how accurately they can be extracted ultimately depends on how accurately the fitting model describes the measured spectra. Finite-NA peak broadening, multiple scattering (in highly scattering samples), background noise and deconvolving with the IRF will all play a part in this.
Artefacts
Different spectrometer designs have unique sources of artefacts, which are described in the Supplementary Section 1. Below we discuss artefacts that are common to several spectrometer designs and can affect the determination of key BLS parameters.
Material (acoustic) heterogeneities
When elastic heterogeneities exist in the scattering volume, the BLS spectrum may no longer exhibit a single set of peaks (per acoustic mode). While not necessarily an artefact, it may still require explicit consideration as it can provide insight into the composition of a sample. The effect will depend on the size and the acoustic mismatch of the heterogeneities36,37,56 (Supplementary Section 2.5).
Scattering wavevector indeterminacy
Any indeterminacy in the probed scattering wavevector (q) will be reflected in both νB and ΓB and any subsequently derived parameters. Given that the probing and collection optics have a finite NA, this effect is unavoidable. In the case of low NA and in the backscattering configuration, it is greatly reduced43. However, for high-NA excitation or detection, as well as for small scattering angles57, the effect on both νB and ΓB can become substantial38,42. Given that the theoretical evaluation of the q indeterminacy may not be straightforward, it may be desirable to estimate this from measurements on materials with negligible intrinsic broadening, which can also allow us to obtain the IRF of the optical set-up.
In turbid media where there is substantial multiple scattering, the effects of q indeterminacy become more complicated, as the probed phonons are no longer defined by the external scattering geometry. Here the observed spectrum is a complex superposition of BLS spectra with different scattering geometries. Methods based on (for example) polarization gating58 may be used to separate different contributions and extract the true BLS parameters.
Sample heating effects
Temperature has a notable effect on key BLS parameters, as well as on cellular processes. In hydrated biological samples an increase in temperature will typically result in an increase in νB and decrease in ΓB. As such, it is important to consider uncertainties in the key parameters due to uncertainties in the temperature. To check whether local sample heating is non-negligible, performing equivalent measurements with different laser intensities or sample exposure times may be sufficient.
Given that some BLS approaches require intense and/or extended sample laser exposures, sample heating may become relevant. In general, this is non-trivial to calculate or measure in situ and will depend on numerous parameters. Semi-quantitative estimates can be made using the bio-heat equation59, for example.
In time-domain BLS, the temperature rise at the transducer caused by the absorption of laser pulses may also contribute to sample heating. This is most notable for large average laser powers and thermally insulating substrates. Using a sapphire substrate with 1.5 mW average laser power would result in a temperature rise of ~1 °C in the sample measurement volume60.
Laser frequency drifting or line broadening
Ideal lasers for Brillouin scattering have narrow linewidths (<10 MHz) at a single, stable frequency. Some commercial continuous-wave, single-longitudinal-mode lasers, such as those commonly used in spontaneous and stimulated frequency-domain BLS, experience frequency drifts that are predominantly caused by temperature fluctuations. Such frequency drifts can be >100 MHz for environmental temperature variations of 0.1 °C, although the amount will vary depending on the laser type and manufacturer61. To increase wavelength stability during BLS spectra acquisition, laser sources may be actively locked to a reference (for example, an Rb/I2 gas cell or a stable external cavity). Depending on the timescales associated with frequency drifts, one may perform periodic measurements of a reference material or signal with known νB and implement frequency-drift corrections during analysis45.
Elastic scattering filters
Given the tight frequency-matching requirements, stability represents the main challenge for most elastic scattering filters. The suppression of elastically scattered light can be compromised by laser frequency drifts, mechanical drifts, environmental thermal fluctuations and intrinsic ASE from the laser. As such, the use of laser lock-in or closed-loop configurations that involve a continuous readout of the elastic scattering signal are generally required for both interference- and molecular-absorption-based filters. For the latter, the multitude of absorption lines (especially in I2 absorption cells) may attenuate or distort the BLS peaks, which may need to be considered during analysis.
Spectral overlaps
BLS spectrometers operating on interferometric principles will have a finite FSR. In measuring a sample that has a νB larger than the FSR, the BLS peak(s) will appear in higher orders or overlapped orders if the spectrometer is not explicitly designed to cancel out overlapping and higher-order peaks62. For example, if a sample has νB = 45 GHz yet the spectrometer FSR is 30 GHz, the BLS anti-Stokes peak will deceptively show up at 15 GHz. This ambiguity, and the measurement of νB values larger than the FSR of a spectrometer, can be overcome by measuring the BLS peaks in higher-order ranges63. In dual-etalon TFP set-ups such spectral overlaps are eliminated by selecting different etalon FSRs64. The issue may also be mitigated in dual-VIPA set-ups by using VIPAs with different FSRs, effectively increasing the FSR of the spectrometer65. This ambiguity in νB is not an issue in stimulated and time-domain BLS.
Internal reflections in sample
For samples that are thin, highly reflective or support optical waveguiding modes, multiple reflections from interior surfaces can give rise to additional BLS peaks in non-backscattering geometries66,67. This is due to light reflected in the interior of a sample leading to two (or more) inner scattering angles. The result is the observation of two (or more) sets of BLS peaks, one associated with the experimentally chosen scattering geometry and the other corresponding to backscattering from internal reflections. The (longitudinal) modes with the largest νB will generally be those associated with backscattering, and may show angular dependence with sample rotation66. Given that transverse modes are forbidden in backscattering for high-symmetry conditions, only longitudinal modes will appear from such internal scattering and can be identified by employing crossed polarizers in the probing and detection beam paths.
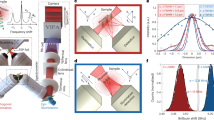
To illustrate some potential artefacts that can be found in BLS microscopy, in Fig. 2a–d we show spatial maps of the BLS frequency shift and linewidth for identical 10-μm-diameter polystyrene beads measured in four different laboratories. Experimental details can be found in the respective Minimum Reporting Tables (Supplementary Data). In Fig. 2a,b the bead has been embedded and sealed in pure (>99%) glycerol, which has a larger BLS frequency shift than polystyrene. In Fig. 2c,d the embedding media are glycerol exposed to air for ~24 h and an aqueous solution, respectively, which both have BLS frequency shifts that are less than that of polystyrene. These two scenarios (Fig. 2a,b versus Fig. 2c,d) create distinct acoustic and optical interfaces between the bead and its surroundings, which can lead to distinct imaging artefacts.

a, TRBS microscopy measurements of νB (top) and ΓB (bottom) with a 758 nm probing wavelength. b–d, Corresponding VIPA spectrometer measurements with a 660 nm probing wavelength. The polystyrene bead in a and b was embedded in glycerol (>99%), whereas the bead in c and d was measured in glycerol with a large water fraction (c) and Optiprep density gradient medium (d) respectively, which both have a lower acoustic index than the bead. Scale bars, 5 μm. Minimum Reporting Tables for measurements and representative BLS spectra can be found in Supplementary Data 1 and Supplementary Fig. 2.
The significant increase in the linewidth at the edge of the bead (Fig. 2a,b) can partly be attributed to the probing volume containing both the bead and the surrounding media (and thus contributions from phonons in both materials), effectively resulting in a broader peak. The acoustic impedance mismatch presented by the curvature of the bead surface may also result in an increased spread of scattering angles away from true backscattering. These are hard to avoid given the spherical geometry of the bead, and can also explain the intermediate values of the BLS frequency shift observed at the bead surface (Fig. 2b). Abrupt acoustic and optical interfaces generally present a challenge in BLS microscopy, which is often best addressed by fitting two Stokes and two anti-Stokes BLS peaks in their vicinity.
The region of decreased νB and increased ΓB seen at the centre of the bead (Fig. 2c,d) can be understood as a geometrical artefact of the bead acting like a lens, as would occur when the bead is immersed in a medium with a lower refractive index. As a result, measurements at this central position would be over a broader range of smaller scattering wavevectors, effectively decreasing νB and increasing ΓB, as seen in the figure. The bead represents an extreme example due to the atypically large refractive index and acoustic impedance mismatch with its surroundings (rarely encountered in soft biological matter), but serves as a useful illustration of potential geometric and material artefacts.
Comparison of absolute and relative BLS-measured values
Figure 3a,b shows the acoustic speed (V = 2πνBq−1) and kinematic longitudinal viscosity (μL = 2πΓBq−2), where q is the scattering wave vector, of distilled water obtained from measurements on different spectrometers in 15 independent laboratories around the world. Despite measuring at different wavevectors and scattering geometries, all results should yield the same values of V and μL. The values of V derived from νB are in good agreement between laboratories and spectrometer designs (typically within <0.5%). The μL values derived from ΓB, while all having similar trends with respect to temperature, show much larger variability. This is probably due to numerous compounding effects that need to be addressed on a case-by-case basis, including a lack of proper spectral deconvolution, the finite time windows used to generate spectra (for I-SBS and TRBS) and inadequate corrections for finite probing/detection NAs. For a given spectrometer design, the latter would presumably provide the largest contribution to the observed variability, as the combined linear and quadratic wavevector dependence of νB and ΓB means that even a small change in NA can result in a observable change in the measured ΓB.

a,b, V (a) and μL (b) as a function of temperature measured using different spectrometer designs in participating laboratories. The accepted values for the acoustic speed in water and values reported from acoustic spectroscopy (AS) measurements46 are also shown. Error bars show propagated uncertainties from those reported in the Minimum Reporting Tables by each laboratory. c,d, BLS-obtained μL (c) and V (d) of cyclohexane (cyc) relative to that of pure water measured using different BLS techniques after normalization to the Vienna–Hannover water standard (a set of complimentary high-resolution measurements on pure injection-grade water under controlled temperature conditions obtained from laboratories in Vienna and Hannover; Supplementary Section 2.9). The insets show absolute values for water (filled circles) and cyclohexane (open circles).
Despite the large variability in the measured μL, it is possible to obtain comparable absolute values of μL if one corrects for set-up-specific offsets by using a control sample with known values (for example). Figure 3c,d shows μL and V for >99% pure cyclohexane relative to that of injection-grade water measured with TFP, VIPA, TRBS and SBS spectrometers. In each case, a correction factor was applied (Supplementary Section 2.9) according to how the values of μL and V differed from those derived from deconvolved measurements of injection-grade water made using a TFP in Vienna and Hannover (Supplementary Table 1) and interpolated for all temperatures. The notable differences in the TRBS results may be due to the high acoustic attenuation of cyclohexane compromising the number of discernible oscillations in the time traces, and thereby the Fourier-transformed frequency spectrum.
Conclusion
BLS microscopy is an emerging imaging technique that provides a unique contrast based on the high-frequency acoustic properties of a material. These can be used to calculate the associated viscoelastic parameters and render near-optical diffraction-limited spatial maps thereof. As it is label free and minimally invasive, it offers a comparable platform to imaging modalities such as Raman microscopy. Its narrow spectral window of excitation and emission mean that BLS can also be performed simultaneously with other imaging techniques (such as Raman68 and fluorescence69), enabling multimodal imaging.
There are a number of differences between our Consensus statement and consensus statements for other spectroscopic techniques70 and techniques that explicitly probe viscoelastic properties. Most notably, many of the instruments addressed here are largely self-built, and differ both conceptually (time-domain, spontaneous, stimulated) and with respect to their unique sources of uncertainty. These complications are contrasted by there usually being only two key parameters over a narrow frequency bandwidth that are of interest. While good agreement can be obtained between laboratories and instrument designs for the parameters derived from the BLS frequency shift (such as V and M′), there is a notable variability in the reported parameters derived from the BLS linewidth (for example M″, ηL and μL), which are much more sensitive to the experimental configuration. Nevertheless, correcting using a standard sample (such as the Vienna–Hannover measurements on injection-grade water done here) can bring these values into quantitative agreement among different laboratories and spectrometer designs.
The broader implementation of BLS microscopy is, to a large extent, still limited by the lack of commercial instruments that can be integrated seamlessly in life-science research laboratories, with set-ups mostly found in laboratories that focus on optical microscopy development. However, it has the potential to fill an important gap in the toolbox of technologies that can quantitatively measure changes in the mechanical properties of biological specimens with high spatiotemporal resolution in three dimensions. BLS can also offer unique possibilities that are inaccessible to other techniques that probe mechanical properties, such as quantitatively measuring7,8 and mapping15,19,20,71 the mechanical anisotropy (different stiffness tensor components) and refractive index25, as well as combined shear and longitudinal viscoelastic parameters19,20,21,22.
In life-science and pre-clinical research, as well as ultimately in clinical applications, the value of establishing a consensus is important to compare measurements performed by different instruments and for the commercialization of spectrometers. For the latter it can serve as a blueprint for parameters that need to be documented, and a guide for designing instruments with performance parameters suited for diverse bio-applications. It may also serve as a springboard for formulating more rigorous and application-specific measurement and reporting standards, which are needed in different commercial and medical applications.
To this end, we also advocate for the establishment of a common file format to store both raw and processed BLS spectral data, together with important metadata for understanding the context of any given experiment. For this, the popular and well-accepted HDF5 file format72 could provide a versatile solution, as it allows one to store complex imagery, as well as underlying raw data, in a hierarchical and well-structured manner. A proposed specification for this file format and the code that allows one to export data to this format are available in ref. 73. We anticipate that this format will be fine-tuned in the future through input from the academic and industry communities. We expect that such a standardization in file format will facilitate collaborations and mutual comparisons of experimental results, as well as promote the development of more universal data analysis software.
While BLS is still in its infancy in regard to clinical translation, one area where it has transitioned to clinical applications is that of ophthalmology. Here it is used to identify the severity of pathologies such as keratoconus associated with spatial changes in corneal biomechanics47,74,75. As the technological sophistication of BLS continues to advance in areas such as acquisition speed76,77,78,79 and instrumentation footprint80, progress in application-specific directions, such as endoscopes81,82 and fibre-coupled probes83,84,85,86 is also being made. One can envision diverse potential clinical applications that offer improvements over current clinical mechanical sensing (such as palpation and ultrasonic elastography in terms of resolution), in vitro mechano-histopathological78,87,88,89, cellular90,91 and biofluid-based diagnostic applications84,92,93,94. For clinical translational pathways, additional sample-specific consensus points will no doubt need to be established, as has been the case for more mature technologies such as optical coherence tomography, photoacoustic imaging and Raman and infrared spectroscopy.
Applications of BLS microscopy can be expected to progress in several complementary directions: (1) BLS-measured viscoelastic parameters could provide direct insight into physical properties of fundamental biological relevance; (2) empirical or theoretically substantiated correlations with viscoelastic parameters measured at different frequencies and boundary conditions can be made; (3) BLS-measured parameters could provide a useful contrast mechanism through their empirical correlations to a biologically relevant process/state (for example, a specific pathology). Here they might themselves be interpreted as presenting a complex chemo-physical property, related to mechanical properties insofar as the measured hypersonic acoustic quantities are concerned. Regardless of the field’s trajectory, it is currently in a serendipitous position, with the technology being ahead of our ability to appreciate the full biological importance of the measured parameters. To this end, establishing consensus early on is particularly important to ensure meaningful and consistent progress on all fronts.
Data availability
All data are contained in the Consensus Statement, Supplementary Information or Supplementary Data 1. A template for the proposed Minimum Reporting Table and the Vienna–Hannover pure water values used for instrument comparisons are included in Supplementary Table 1 and are available via Figshare at https://doi.org/10.6084/m9.figshare.27794910 (ref. 95) and https://doi.org/10.6084/m9.figshare.27794913 (ref. 96), respectively. Completed Minimum Reporting Tables for data included in the main text are provided in Supplementary Data 1 and are available via Figshare at https://doi.org/10.6084/m9.figshare.27794916 (ref. 97).
Code availability
Any specialized codes used for calculating and fitting BLS spectra for the different techniques described is detailed in publications on the respective techniques.
References
Berne, B. J. & Pecora, R. Dynamic Light Scattering: With Applications to Chemistry, Biology, and Physics (Dover, 2000).
Dil, J. G. Brillouin scattering in condensed matter. Rep. Prog. Phys. 45, 285–334 (1982).
Koski, K. J. & Yarger, J. L. Brillouin imaging. Appl. Phys. Lett. 87, 061903 (2005).
Scarcelli, G. & Yun, S. H. Confocal Brillouin microscopy for three-dimensional mechanical imaging. Nat. Photon. 2, 39–43 (2007).
Massey, A. et al. Mechanical properties of human tumour tissues and their implications for cancer development. Nat. Rev. Phys. 6, 269–282 (2024).
Tran, R. et al. Biomechanics of haemostasis and thrombosis in health and disease: from the macro- to molecular scale. J. Cell. Mol. Med. 17, 579–596 (2013).
Antonacci, G. et al. Recent progress and current opinions in Brillouin microscopy for life science applications. Biophys. Rev. https://doi.org/10.1007/s12551-020-00701-9 (2020).
Palombo, F. & Fioretto, D. Brillouin light scattering: applications in biomedical sciences. Chem. Rev. 119, 7833–7847 (2019).
Elsayad, K., Palombo, F., Dehoux, T. & Fioretto, D. Brillouin light scattering microspectroscopy for biomedical research and applications: introduction to feature issue. Biomed. Opt. Express 10, 2670–2673 (2019).
Kabakova, I. et al. Brillouin microscopy. Nat. Rev. Methods Primers 4, 8 (2024).
Wu, P. J. et al. Water content, not stiffness, dominates Brillouin spectroscopy measurements in hydrated materials. Nat. Methods 15, 561–562 (2018).
Scarcelli, G. & Yun, S. H. Reply to ‘Water content, not stiffness, dominates Brillouin spectroscopy measurements in hydrated materials. Nat. Methods 15, 562–563 (2018).
Gutmann, M. et al. Beyond comparison: Brillouin microscopy and AFM-based indentation reveal divergent insights into the mechanical profile of the murine retina. J. Phys. Photon 6, 035020 (2024).
Yan, G., Monnier, S., Mouelhi, M. & Dehoux, T. Probing molecular crowding in compressed tissues with Brillouin light scattering. Proc. Natl Acad. Sci. USA 119, e2113614119 (2022).
Keshmiri, H. et al. Brillouin light scattering anisotropy microscopy for imaging the viscoelastic anisotropy in living cells. Nat. Photon. 18, 276–285 (2024).
Eisenberger, P., Alexandropoulos, N. G. & Platzman, P. M. X-ray Brillouin scattering. Phys. Rev. Lett. 28, 1519–1522 (1972).
Verkerk, P. Neutron brillouin scattering. Neutron News 1, 21 (1990).
Bottani, C. E. & Fioretto, D. in Advances in Physics: X Vol. 3, 607–633 (Taylor and Francis Ltd, 2018).
Koski, K. J., Akhenblit, P., McKiernan, K. & Yarger, J. L. Non-invasive determination of the complete elastic moduli of spider silks. Nat. Mater. 12, 262–267 (2013).
Palombo, F. et al. Biomechanics of fibrous proteins of the extracellular matrix studied by Brillouin scattering. J. R. Soc. Interface 11, 20140739 (2014).
Czibula, C. et al. The elastic stiffness tensor of cellulosic viscose fibers measured with Brillouin spectroscopy. J. Phys. Photon. 6, 035012 (2024).
Kim, M. et al. Shear Brillouin light scattering microscope. Opt. Express 24, 319–328 (2016).
Boon, J. P. & Yip, S. Molecular Hydrodynamics (Dover, 1991).
Figgins, R. Inelastic light scattering in liquids: Brillouin scattering. Contemp. Phys. 12, 283–297 (1971).
Fiore, A., Bevilacqua, C. & Scarcelli, G. Direct three-dimensional measurement of refractive index via dual photon-phonon scattering. Phys. Rev. Lett. 122, 103901 (2019).
Pochylski, M. Structural relaxation in the wave-vector dependence of the longitudinal rigidity modulus. Biomed. Opt. Express 10, 1957–1964 (2019).
Remer, I., Shaashoua, R., Shemesh, N., Ben-Zvi, A. & Bilenca, A. High-sensitivity and high-specificity biomechanical imaging by stimulated Brillouin scattering microscopy. Nat. Methods 17, 913–916 (2020).
JCGM Publications: Guides in Metrology JCGM https://www.bipm.org/en/committees/jc/jcgm/publications
Vanderwal, J., Mudare, S. M. & Walton, D. Deconvolution of Brillouin spectra. Opt. Commun. 37, 33–36 (1981).
Huang, J. et al. Processing method of spectral measurement using F-P etalon and ICCD. Opt. Express 20, 18568–18578 (2012).
Meng, Z. & Yakovlev, V. V. Precise determination of Brillouin scattering spectrum using a virtually imaged phase array (VIPA) spectrometer and charge-coupled device (CCD) camera. Appl. Spectrosc. 70, 1356–1363 (2016).
Allan, D. W. Statistics of atomic frequency standards. Proc. IEEE 54, 221–230 (1966).
Adib, G. A., Sabry, Y. M. & Khalil, D. Allan variance characterization of compact Fourier transform infrared spectrometers. Appl. Spectrosc. 77, 734–743 (2023).
Coker, Z. et al. Assessing performance of modern Brillouin spectrometers. Opt. Express 26, 2400–2409 (2018).
Ballmann, C. W., Meng, Z. & Yakovlev, V. V. Nonlinear Brillouin spectroscopy: what makes it a better tool for biological viscoelastic measurements. Biomed. Opt. Express 10, 1750–1759 (2019).
Mattarelli, M., Vassalli, M. & Caponi, S. Relevant length scales in Brillouin imaging of biomaterials: the interplay between phonons propagation and light focalization. ACS Photon. 7, 2319–2328 (2020).
Passeri, A. A. et al. Size and environment: the effect of phonon localization on micro-Brillouin imaging. Biomater. Adv. 147, 213341 (2023).
Caponi, S., Fioretto, D. & Mattarelli, M. On the actual spatial resolution of Brillouin imaging. Opt. Lett. 45, 1063–1066 (2020).
Ballmann, C. W. et al. Stimulated Brillouin scattering microscopic imaging. Sci. Rep. 5, 18139 (2015).
Bevilacqua, C., Sanchez-Iranzo, H., Richter, D., Diz-Munoz, A. & Prevedel, R. Imaging mechanical properties of sub-micron ECM in live zebrafish using Brillouin microscopy. Biomed. Opt. Express 10, 1420–1431 (2019).
McCreery, R. L. Raman Spectroscopy for Chemical Analysis (Wiley, 2005).
Mattana, S. et al. Non-contact mechanical and chemical analysis of single living cells by microspectroscopic techniques. Light Sci. Appl. 7, 17139–17139 (2018).
Antonacci, G., Foreman, M. R., Paterson, C. & Török, P. Spectral broadening in Brillouin imaging. Appl. Phys. Lett. 103, 221105 (2013).
Wäldchen, S., Lehmann, J., Klein, T., van de Linde, S. & Sauer, M. Light-induced cell damage in live-cell super-resolution microscopy. Sci. Rep. 5, 15348 (2015).
Testi, C. et al. Electro-optic modulator source as sample-free calibrator and frequency stabilizer for Brillouin microscopy. Preprint at https://arxiv.org/abs/2412.20516 (2024).
Holmes, M. J., Parker, N. G. & Povey, M. J. W. Temperature dependence of bulk viscosity in water using acoustic spectroscopy. J. Phys. Conf. Ser. 269, 012011 (2011).
Shao, P. et al. Spatially-resolved Brillouin spectroscopy reveals biomechanical abnormalities in mild to advanced keratoconus in vivo. Sci. Rep. 9, 7467 (2019).
Zhang, H., Asroui, L., Randleman, J. B. & Scarcelli, G. Motion-tracking Brillouin microscopy for in-vivo corneal biomechanics mapping. Biomed. Opt. Express 13, 6196–6210 (2022).
Sussner, H. & Vacher, R. High-precision measurements of Brillouin scattering frequencies. Appl. Opt. 18, 3815–3818 (1979).
Caponi, S. et al. Electro-optic modulator for high resolution Brillouin scattering measurements. Rev. Sci. Instrum. 72, 198–200 (2001).
Remer, I., Cohen, L. & Bilenca, A. High-speed continuous-wave stimulated Brillouin scattering spectrometer for material analysis. J. Vis. Exp. https://doi.org/10.3791/55527 (2017).
Shijun, X., Weiner, A. M. & Lin, C. A dispersion law for virtually imaged phased-array spectral dispersers based on paraxial wave theory. IEEE J. Quantum Electron. 40, 420–426 (2004).
Cardinali, M.A., Caponi, S., Mattarelli, M., Loré, D. & Fioretto, D. Brillouin micro spectroscopy and morpho mechanics of a hybrid lens. In SIF Congress 2023 330 (Società Italiana di Fisica, 2023).
Evaluation of Measurement Data—Guide to the Expression of Uncertainty in Measurement (Joint Committee for Guides in Metrology, 2008); https://www.bipm.org/documents/20126/2071204/JCGM_100_2008_E.pdf/cb0ef43f-baa5-11cf-3f85-4dcd86f77bd6
Török, P. & Foreman, M. R. Precision and informational limits in inelastic optical spectroscopy. Sci. Rep. 9, 6140 (2019).
Cardinali, M. A., Caponi, S., Mattarelli, M. & Fioretto, D. Brillouin scattering from biomedical samples: the challenge of heterogeneity. J. Phys. Photon. 6, 035009 (2024).
Battistoni, A., Bencivenga, F., Fioretto, D. & Masciovecchio, C. Practical way to avoid spurious geometrical contributions in Brillouin light scattering experiments at variable scattering angles. Opt. Lett. 39, 5858–5861 (2014).
Mattarelli, M., Capponi, G., Passeri, A. A., Fioretto, D. & Caponi, S. Disentanglement of multiple scattering contribution in Brillouin microscopy. ACS Photon. 9, 2087–2091 (2022).
Hristov, J. Bio-heat models revisited: concepts, derivations, nondimensalization and fractionalization approaches. Front. Phys. https://doi.org/10.3389/fphy.2019.00189 (2019).
Pérez-Cota, F. et al. Picosecond ultrasonics for elasticity-based imaging and characterization of biological cells. J. Appl. Phys. https://doi.org/10.1063/5.0023744 (2020).
Antonacci, G. et al. Birefringence-induced phase delay enables Brillouin mechanical imaging in turbid media. Nat. Commun. 15, 5202 (2024).
Vaughan, M. The Fabry-Perot Interferometer: History, Theory, Practice and Applications 1st edn (Routledge, 1989).
Soltwisch, M., Sukmanowski, J. & Quitmann, D. Brillouin scattering on noncrystalline ZnCl2. J. Chem. Phys. 86, 3207–3215 (1987).
Sandercock, J. R. Brillouin scattering study of SbSI using a double-passed, stabilised scanning interferometer. Opt. Commun. 2, 73–76 (1970).
Berghaus, K., Zhang, J., Yun, S. H. & Scarcelli, G. High-finesse sub-GHz-resolution spectrometer employing VIPA etalons of different dispersion. Opt. Lett. 40, 4436–4439 (2015).
Takagi, Y. & Gammon, R. W. Brillouin scattering in thin samples: observation of backscattering components by 90° scattering. J. Appl. Phys. 61, 2030–2034 (1987).
Zhang, Y., Reed, B. W., Chung, F. R. & Koski, K. J. Mesoscale elastic properties of marine sponge spicules. J. Struct. Biol. 193, 67–74 (2016).
Scarponi, F. et al. High-performance versatile setup for simultaneous Brillouin-Raman microspectroscopy. Phys. Rev. 7, 031015 (2017).
Elsayad, K. et al. Mapping the subcellular mechanical properties of live cells in tissues with fluorescence emission-Brillouin imaging. Sci. Signal. 9, rs5 (2016).
Fales, A. M., Ilev, I. K. & Pfefer, T. J. Evaluation of standardized performance test methods for biomedical Raman spectroscopy. J. Biomed. Opt. 27, 074705 (2021).
Webb, J. N., Zhang, H., Sinha Roy, A., Randleman, J. B. & Scarcelli, G. Detecting mechanical anisotropy of the cornea using Brillouin microscopy. Transl. Vis. Sci. Technol. 9, 26 (2020).
HDF5 (HDF Group, 2024); https://www.hdfgroup.org/solutions/hdf5
HDF5 and Brillouin Scattering Code Repositories (BioBrillouin, 2024); https://www.biobrillouin.org/hdf5
Yun, S. H. & Chernyak, D. Brillouin microscopy: assessing ocular tissue biomechanics. Curr. Opin. Ophthalmol. 29, 299–305 (2018).
Randleman, J. B. et al. Subclinical keratoconus detection and characterization using motion-tracking Brillouin microscopy. Ophthalmology 131, 310–321 (2024).
Bevilacqua, C. et al. High-resolution line-scan Brillouin microscopy for live imaging of mechanical properties during embryo development. Nat. Methods 20, 755–760 (2023).
Zhang, J., Nikolic, M., Tanner, K. & Scarcelli, G. Rapid biomechanical imaging at low irradiation level via dual line-scanning Brillouin microscopy. Nat. Methods 20, 677–681 (2023).
Shaashoua, R. et al. Brillouin gain microscopy. Nat. Photon. 18, 836–841 (2024).
Krug, B., Koukourakis, N. & Czarske, J. W. Impulsive stimulated Brillouin microscopy for non-contact, fast mechanical investigations of hydrogels. Opt. Express 27, 26910–26923 (2019).
Fiore, A. & Scarcelli, G. Single etalon design for two-stage cross-axis VIPA spectroscopy. Biomed. Opt. Express 10, 1475–1481 (2019).
Kabakova, I. V., Xiang, Y., Paterson, C. & Török, P. Fiber-integrated Brillouin microspectroscopy: towards Brillouin endoscopy. J. Innov. Opt. Health Sci. 10, 1742002 (2017).
Xiang, Y. et al. Background-free fibre optic Brillouin probe for remote mapping of micromechanics. Biomed. Opt. Express 11, 6687 (2020).
Pruidze, P., Chayleva, E., Weninger, W. J. & Elsayad, K. Brillouin scattering spectroscopy for studying human anatomy: towards in situ mechanical characterization of soft tissue. J. Eur. Opt. Soc. Rapid Publ. 19, 31 (2023).
Illibauer, J. et al. Diagnostic potential of blood plasma longitudinal viscosity measured using Brillouin light scattering. Proc. Natl Acad. Sci. USA 121, e2323016121 (2024).
La Cavera, S. et al. Label-free Brillouin endo-microscopy for the quantitative 3D imaging of sub-micrometre biology. Commun. Biol. 7, 451 (2024).
La Cavera, S., Pérez-Cota, F., Smith, R. J. & Clark, M. Phonon imaging in 3D with a fibre probe. Light Sci. Appl. 10, 91 (2021).
Ryu, S., Martino, N., Kwok, S. J. J., Bernstein, L. & Yun, S.-H. Label-free histological imaging of tissues using Brillouin light scattering contrast. Biomed. Opt. Express 12, 1437–1448 (2021).
Martinez-Vidal, L. et al. Progressive alteration of murine bladder elasticity in actinic cystitis detected by Brillouin microscopy. Sci. Rep. 14, 484 (2024).
Mattana, S., Caponi, S., Tamagnini, F., Fioretto, D. & Palombo, F. Viscoelasticity of amyloid plaques in transgenic mouse brain studied by Brillouin microspectroscopy and correlative Raman analysis. J. Innov. Opt. Health Sci. https://doi.org/10.1142/S1793545817420019 (2017).
Fasciani, A. et al. MLL4-associated condensates counterbalance polycomb-mediated nuclear mechanical stress in Kabuki syndrome. Nat. Genet. 52, 1397–1411 (2020).
Cikes, D. et al. PCYT2-regulated lipid biosynthesis is critical to muscle health and ageing. Nat. Metab. 5, 495–515 (2023).
Steelman, Z., Meng, Z., Traverso, A. J. & Yakovlev, V. V. Brillouin spectroscopy as a new method of screening for increased CSF total protein during bacterial meningitis. J. Biophoton. 8, 408–414 (2015).
Adichtchev, S. V. et al. Brillouin spectroscopy of biorelevant fluids in relation to viscosity and solute concentration. Phys. Rev. E 99, 062410 (2019).
Windberger, U., Sparer, A. & Elsayad, K. The role of plasma in the yield stress of blood. Clin. Hemorheol. Microcirc. https://doi.org/10.3233/ch-231701 (2023).
Elsayad, K. Proposed Minimum Reporting Table for Brillouin light scattering microscopy. Figshare https://doi.org/10.6084/m9.figshare.27794910 (2024).
Elsayad, K. Vienna-Hannover pure water BLS values. Figshare https://doi.org/10.6084/m9.figshare.27794913 (2024).
Elsayad, K. Filled in Minimum Reporting Tables for Brillouin light scattering microscopy measurements on different instruments. Figshare https://doi.org/10.6084/m9.figshare.27794916 (2024).
Acknowledgements
P.B. and K.E. acknowledge funding from the Austrian Science Fund, FWF (grant no. P34783) and the Medical University of Vienna. C.B., J.L. and R.P. acknowledge funding from the EMBL, an ERC Consolidator Grant (grant no. 864027, Brillouin4Life) and the German Center for Lung Research (DZL). S.C., D.F. and M.Mattarelli acknowledge funding from the European Union NextGenerationEU (grant no. ECS00000041, VITALITY, CUP J97G22000170005 and CUP B43C22000470005; Prin PNRR grant P2022RH4HH, COMBINE-CUP B53D2302864 0001 and PRIN 2022 CODIR, CUP J53D23001370006). J.C., N.K. and D.K. acknowledge funding from the DFG (grant nos. Cz55/44 and CZ 55/40-2). T.H. acknowledges funding from ERC SYNERGY grant HYDROSENSING (ERC10118769) and NFR grant WALLINTEGRITY (NFR315325). D.H. and T.L. acknowledge funding from the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) under Germany’s Excellence Strategy within the Cluster of Excellence PhoenixD (EXC 2122, Project ID 390833453). H.J.-R. acknowledges funding from the Canada Research Chairs programme (grant no. CRC-2021-00456). I.K. acknowledges funding from the ARC Centre of Excellence in Quantum Technology (grant no. CE230100021) and ARC Centre of Excellence in Optical Microcombs for Breakthrough Science (grant no. CE230100006). K.K. acknowledges funding from the National Science Foundation (grant no. DMR-2202472). S.L. acknowledges funding from the Royal Academy of Engineering Research Fellowships scheme (grant no. RF-2324-23-223) and Nottingham Research Fellowship scheme. J.M. acknowledges funding from an EIC-2022-PathfinderOpen grant (VIRUSong, grant no. 101099058). M.Monegan acknowledges funding from Research Ireland (grant no. 13/RC/2073_P2) and Research Ireland Frontiers for the Future Project (grant no. 22/FFP-P/11394). D.R.O. acknowledges funding from the National Institutes of Health (grant no. EY022359). F.P.C acknowledges funding from a Royal Academy of Engineering Research Fellowship (RF\_201718\_17144), the Engineering and Physical Sciences Research Council (EP/W031876/1, EP/Y002857/1), and Cancer Research UK (DRCMDP-Jun24/100011). E.P. and C.T. acknowledge funding from an EIC-2022-PathfinderOpen grant (ivBM-4PAP, grant no. 101098989). G.R. and C.T. acknowledge funding from an ERC-2019-Synergy Grant (ASTRA, grant no. 855923). G.S. acknowledges funding from the National Science Foundation (grant no. DBI-1942003) and the National Institutes of Health (grant nos. R21CA258008, R01EY028666, R01EY030063, R01EY032537 and R21EY035483). P.T. acknowledges funding from the Nanyang Technological University, the Ministry of Education (MOE) and the National Research Foundation (NRF). V.Y. acknowledges funding from The Air Force Office of Scientific Research 1189 (grant nos. FA9550-20-1-0366, FA9550-20-1-0367 and FA9550-23-1-0599), the National Institutes of Health (grant nos. R01GM127696, R01GM152633, R21GM142107 and 1R21CA269099) and support from NASA, BARDA, the NIH and USFDA under contract/agreement no. 80ARC023CA002. S.H.-Y. acknowledges funding from the National Institutes of Health (grant nos. R01EY033356 and R01EY034857). J.Z. acknowledges funding from the National Science Foundation (grant no. CBET-2339278) and National Institutes of Health (grant nos. K25HD097288 and R21HD112663). F.P. acknowledges funding from the UK Engineering and Physical Sciences Research Council (grant nos. EP/M028739/1 and NS/A000063/1) and Cancer Research UK (grant no. NS/A000063/1). A.B. acknowledges support from the Israel Science Foundation (grant no. 2576/21). We acknowledge funding from the European Cooperation in Science and Technology (COST) Association (COST Action BioBrillouin grant number CA16124 and COST Innovators Grant BLiSS-BMD grant no. IG16124).
Author information
Authors and Affiliations
Contributions
C.B., Y.A., G.A., J.A., S.C., S. C.-L., Y.F., D.K., S.L., T.L., J.L., H.M., F. P.-C., E.P., F.S., C.T. and L.V. performed experiments. P.B., C.B., G.A. S.C. S.C.-L., J.C., T.D., D.F., T.H., D.H., I.K., K.K., N.K., D.K., S.L., T.L., J.M., M.Mattarelli, D.R.O., F.P-C., E.P., R.P., G.R., G.S., F.S., C.T., V.Y., J.Z. F.P., A.B. and K.E wrote sections of the article. All authors provided suggestions and critical feedback. P.B. and K.E. compiled these into the current form of the article.
Corresponding authors
Ethics declarations
Competing interests
F.S. and J.S. are employed by Tablestable Ltd, who manufacture BLS spectrometers. E.P. is employed by CREST Optics S.p.A., who manufacture optical microscopy solutions. S.C.-L. was employed by LightMachinery Inc, who manufacture Brillouin spectrometers, while engaged in this research project. T.J. is affiliated with CellSense Technologies GmbH, who manufacture BLS microscopes. G.A. owns shares in Specto Srl, who manufacture BLS spectrometers. G.S. has intellectual property related to Brillouin technology (Massachusetts General Hospital and University of Maryland), owns shares in Intelon Optics and is a consultant for Intelon Optics and CellSense technologies. R.P. is an advisor to CellSense, which commercializes Brillouin microscopes. K.E. serves as a scientific advisor for Specto Srl.
Peer review
Peer review information
Nature Photonics thanks Benjamin Eggleton, Kirill Larin and Fan Yang for their contribution to the peer review of this work.
Supplementary information
Supplementary Information
Supplementary Text and Figs. 1–5.
Supplementary Data 1
Minimum Reporting Tables for BLS measurements.
Supplementary Table 1
Minimum Reporting Table template and Vienna–Hannover pure water values.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Bouvet, P., Bevilacqua, C., Ambekar, Y. et al. Consensus statement on Brillouin light scattering microscopy of biological materials. Nat. Photon. 19, 681–691 (2025). https://doi.org/10.1038/s41566-025-01681-6
Received:
Accepted:
Published:
Version of record:
Issue date:
DOI: https://doi.org/10.1038/s41566-025-01681-6
This article is cited by
-
Stabilized real-time Brillouin microscopy reveals fractal organization of protein condensates in living cells
Nature Communications (2026)
-
Cellular mechanical properties in response to environmental viscosity imaged by Brillouin Microscopy
Communications Biology (2025)
