
Abstract
Current approaches used to track stem cell clones through differentiation require genetic engineering1,2 or rely on sparse somatic DNA variants3,4, which limits their wide application. Here we discover that DNA methylation of a subset of CpG sites reflects cellular differentiation, whereas another subset undergoes stochastic epimutations and can serve as digital barcodes of clonal identity. We demonstrate that targeted single-cell profiling of DNA methylation5 at single-CpG resolution can accurately extract both layers of information. To that end, we develop EPI-Clone, a method for transgene-free lineage tracing at scale. Applied to mouse and human haematopoiesis, we capture hundreds of clonal differentiation trajectories across tens of individuals and 230,358 single cells. In mouse ageing, we demonstrate that myeloid bias and low output of old haematopoietic stem cells6 are restricted to a small number of expanded clones, whereas many functionally young-like clones persist in old age. In human ageing, clones with and without known driver mutations of clonal haematopoieis7 are part of a spectrum of age-related clonal expansions that display similar lineage biases. EPI-Clone enables accurate and transgene-free single-cell lineage tracing on hematopoietic cell state landscapes at scale.
Similar content being viewed by others

Main
Lineage tracing using genetic or physical labels has been an important tool in developmental and stem cell biology for more than a century1,2. More recently, genetic barcoding compatible with single-cell RNA sequencing (scRNA-seq) has provided information on the cellular output of hundreds of stem cell clones together with cell-state information on the stem cell itself8,9,10,11,12. Such methods require complex genetic engineering and therefore have limited applications, for example, in humans or during native ageing. Thus, methods are needed that rely on endogenous clonal markers (for example, somatic mutations) and allow tracing of various stem cell clones in parallel. Whole-genome sequencing can reconstruct cellular phylogenies3 but has limited throughput. It also lacks information about cell states, which precludes clonal tracking across cellular differentiation landscapes. Conversely, spontaneous mitochondrial DNA (mtDNA) mutations can be captured together with cell-state information by scRNA-seq or ATAC–seq4,13,14. Although mtDNA variants can be clonally informative, it is unclear whether mtDNA variants can reconstruct cellular phylogenies15,16.
Clonal signals have been identified in bulk DNA methylation data obtained from cancer and healthy tissues17,18. Somatic epimutations, defined as spontaneous but heritable losses and gains of DNA methylation, have been explored as a potential clonal label in cancer19,20. However, differentiation-associated changes in DNA methylation may mask clone-associated differences21,22. Furthermore, current single-cell DNA methylation methods23,24 suffer from data sparsity, which makes it challenging to exploit the stochasticity of epimutations at individual CpGs.
A compelling case for the use of lineage tracing is haematopoiesis, whereby, in humans, 50,000–200,000 stem cell clones generate blood throughout life3. Ageing induces clonal expansion with substantial loss of clonal diversity. In mice, much of our understanding of clonal behaviour in ageing either comes from transplantation experiments25 or mathematical modelling26, which may not recapitulate steady-state haematopoiesis or lacks the resolution of single-cell lineage analysis. In humans, literature focuses on the role of driver mutations in clonal haematopoiesis (CH), but clonal expansions without (known) drivers are common with age and are associated with an increased all-mortality risk27. So far, the lineage output of clones with or without (known) CH driver mutations have not been compared because of a lack of suitable methods.
Here we develop EPI-Clone, a method that exploits the targeted single-cell readout of DNA methylation at single-CpG resolution to track clones while providing detailed cell-state information. EPI-Clone builds on single-cell targeted analysis of the methylome (scTAM-seq), which is implemented on the Mission Bio Tapestri platform to read out methylation states of several hundred CpGs in thousands of single cells at a time, with a dropout rate of around 7%5. scTAM-seq uses a methylation-sensitive restriction enzyme to selectively digest unmethylated CpGs and thus generates sequencing reads only from methylated CpGs. We applied EPI-Clone to lineage-barcoded cells and in native human and mouse haematopoiesis to characterize the decline in clonal complexity and the functional properties of age-expanded clones in mouse and human ageing.
A DNA methylation map of haematopoiesis
We performed a series of experiments, which, for clarity, are defined as follows: scTAM-seq applied to eight different settings in mice (experiments M.1–M.8; Extended Data Fig. 1a); scTAM-seq applied to two human cohorts (A.1–A.7 and B.1–B.5); and experiments demonstrating the combination of scTAM-seq with RNA-seq and mitochondrial lineage tracing from the same cell (X.1 and X.2). An overview of all data is provided in Supplementary Table 1.
To create a ground-truth dataset of clonal identity and DNA methylation, we labelled mouse haematopoietic stem cells (HSCs) with lentiviral barcodes using the LARRY system8. Labelled HSCs were transplanted into lethally irradiated recipient mice and the mice were profiled 5 months later, a time point at which all blood populations should be reconstituted. Sorted haematopoietic stem and progenitor cells (HSPCs) from bone marrow (sorted as LIN−KIT+ (LK) cells with additional enrichment of LIN−SCA1+KIT+ (LSK) cells) were profiled by scTAM-seq (experiment M.1, the main LARRY experiment; Fig. 1a, Extended Data Fig. 1a, Supplementary Table 1 and Supplementary Fig. 1). The experiment was repeated (experiment M.2, replicate LARRY experiment) and we profiled LK and LSK bone marrow from untreated mice (experiment M.3, native haematopoiesis). Specifically, we analysed methylation of 453 CpGs that were selected as differentially or variably methylated from bulk HSPC DNA methylation data22 (Fig. 1b, Methods and Extended Data Fig. 1b,c). The LARRY barcode was read out directly from the DNA by including a LARRY-specific amplicon in our targeting panel for scTAM-seq. Finally, the expression of 20 surface proteins (Supplementary Table 2) was simultaneously profiled using oligonucleotide-tagged antibodies to obtain independent information on cellular differentiation. In summary, for experiments M.1–M.3, we profiled DNA methylation at 453 CpGs and the expression of 20 surface proteins across HSPCs. In experiments M.1 and M.2, we also profiled LARRY barcodes from the same cells.

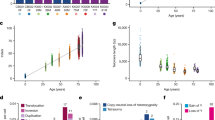
a, Schematic of experiments M.1 (LARRY main experiment) and M.2 (replicate LARRY experiment). b, Overview of the 453 CpGs covered by our scTAM-seq panel in mice. Variably methylated CpGs were selected from bulk whole-genome bisulfite sequencing data22. DMC, differentially methylated CpG; IMC, intermediately methylated CpG; WSH, within-sample heterogeneity (see Extended Data Fig. 1c for definition). c, UMAP of DNA methylation data for HSPCs from experiments M.1–M.3. Batch correction was applied before UMAP. Colours highlight groups identified from unsupervised clustering. Annotations are based on d–f. d, DNA methylation UMAP as in c, highlighting the average, relative methylation state of cells across all CpGs that are methylated in HSCs or MPP3/MPP4 cells in bulk-sequencing data22. e, Enrichment analysis of TFBSs near CpGs specifically unmethylated in a cell-type cluster. See the section ‘Data integration and annotation of cell states’ in the Methods. f, Normalized surface-protein expression of SCA1, KIT, CD135, CD201, CD48 and CD150. The CD135–CD201 and CD48–CD150 plots only show LSK cells. Colour indicates cell states per c. g, UMAP of DNA methylation data from HSPCs from experiment M.1. Colour indicates cell states per c. h, Same UMAP as in g, highlighting clones as defined from LARRY barcodes. LARRY barcodes were read out from DNA as part of scTAM-seq. i, Scatter plot depicting, for n = 453 CpGs, the average methylation rate, the statistical association with surface-protein expression and the statistical association with the LARRY clonal barcode (P value from a two-sided chi-squared test). The CpGs in the upper and lower central rectangle were defined as static or dynamic CpGs, respectively. j, Bar chart depicting the percentage of static and dynamic CpGs annotated as enhancer or heterochromatin. DMC, differentially methylated cytosine; IMC, intermediately methylated cytosine; MEP, megakaryocyte–erythroid progenitor cells; WSH, within-sample heterogeneity. The scTAM-seq schematic in a was adapted from ref. 5 under a Creative Commons licence CC BY 4.0.
We applied Seurat’s default batch-correction method to integrate methylation data from 28,782 cells across the three experiments. We thereby obtained a low-dimensional embedding in which most variation was driven by differentiation along four trajectories (Fig. 1c). To annotate cell states from the DNA methylation data, we used three layers of information: (1) bulk methylation profiles (Fig. 1d and Supplementary Fig. 2a); (2) the methylation states of important lineage-specific transcription-factor-binding sites (TFBSs; Fig. 1e and Supplementary Fig. 2b); and (3) the expression of surface proteins (Fig. 1f and Supplementary Fig. 2c,d). We identified cell-state-specific demethylation of CpGs that neighboured crucial TFBSs, including GATA2 (an erythroid factor), EBF1 (a lymphoid factor) and SPI1 (a myeloid factor) (Fig. 1e and Supplementary Fig. 2b). scTAM-seq data revealed a cluster of HSCs and early multipotent progenitors (MPP1, also called short-term or active HSCs), several additional MPP subsets (MPP2, MPP3 and MPP4), myeloid, erythroid and B cell progenitors, as well as two subsets of megakaryocyte progenitors (MKPs). As we also performed scRNA-seq on different cells obtained from the same samples, we could compare low-dimensional uniform manifold approximation and projection (UMAP) generated by DNA methylation with a UMAP generated from transcriptomic data (Extended Data Fig. 2a). We observed an overall similar topology (Extended Data Fig. 2b) with the four main differentiation trajectories. Overall, through data integration of several experiments, we obtained a DNA-methylation-based map of mouse HSC differentiation at single-CpG resolution. This map contains two orders of magnitude more cells than two previous, single-cell bisulfite sequencing datasets of the haematopoietic system28,29.
DNA methylation encodes clones and cell states
Computational batch-correction methods, by definition, remove most individual-specific signals (Extended Data Fig. 2c). As clonal information is individual-specific, we computed a UMAP display of the data from experiment M.1 only. This analysis revealed that DNA methylation jointly captures two layers of information: differentiation state and clonal identity. Specifically, although cells clustered according to differentiation states (Fig. 1g), they also clustered by their clonal identity as defined through LARRY barcodes (Fig. 1h). To use this clone-specific signal for lineage tracing, we sought to determine whether clonal identity and differentiation affect different subsets of CpGs. We tested for the association of every CpG with the expression of any surface protein and thereby identified differentiation-associated, dynamic CpGs. Performing dimensionality reduction using only these dynamic CpGs (Extended Data Fig. 2d) or only the expression of surface proteins (Extended Data Fig. 2e,f) resulted in a similar landscape to that obtained by batch correction. This finding indicates that dynamic CpGs and surface antigens independently capture differentiation state well. The remaining, static CpGs were frequently associated with clonal identity, as defined through LARRY barcodes (Fig. 1i). Dynamic CpGs were enriched in enhancer elements, whereas the static CpGs were preferentially located in heterochromatic regions (Fisher test P = 2.2 × 10−5; Fig. 1j). Moreover, static CpGs were enriched in late-replicating domains (Fisher test P = 0.001; Extended Data Fig. 2g). In summary, clonal identity and differentiation state affect the methylation of different sets of CpGs in haematopoietic cells, which creates a valuable tool to read out both processes simultaneously at the single-cell level.
The EPI-Clone algorithm
We focused on exploiting static CpGs to analyse clonal identity. To this end, we developed EPI-Clone, which is divided into three steps: (1) identification of static CpGs, as described above; (2) identification of cells from expanded clones by using cell density in the DNA methylation space defined by the static CpGs; and (3) clustering of cells from the expanded clones (Fig. 2a and Methods).

a, Schematic overview of EPI-Clone. See the main text for details. Exp, expression. b, UMAP of DNA methylation computed on static CpGs only for experiment M.1, which highlights clonal identity as defined by LARRY barcodes. Only cells carrying a LARRY barcode are shown and cells with a relative clone size (rel. size; defined using LARRY) less than 0.25% are shown in grey. c, Same UMAP as in b, but highlighting the cell states as defined in Fig. 1c. d, UMAP highlighting cells that were selected as part of expanded clones based on local density in PCA space. e, Receiver-operating characteristics curve visualizing the performance of classifying cells into expanded and non-expanded clones based on local density in PCA space spanned by the static CpGs. LARRY clone size was used as the ground truth, whereby clone sizes larger than 0.25% were considered expanded. TPR, true positive rate; FPR, false positive rate. f, Heatmap depicting the overlap between LARRY barcode and methylation-based clonal clusters identified by EPI-Clone. The row labelled with an asterisk contains all LARRY clones with a clone size less than 0.25%. g, Schematic of experiment M.5: LARRY mature immune cell experiment. h, UMAP of DNA methylation for cells from expanded clones in experiment M.5. Cells are coloured by LARRY barcode. The static CpGs identified from experiment M.1 were used. i, Same UMAP representation as in h, but highlighting the cell-state annotation as defined in Supplementary Fig. 4. Of note, most of the clones identified using EPI-Clone were specific for T cells, B cells or myeloid cells, in line with the result from LARRY (Supplementary Fig. 4d). j, ARI values between the ground-truth clonal label (LARRY) and the clones identified by EPI-Clone stratified by cell type.
Using this algorithm, expanded LARRY clones with relative clone sizes larger than 0.25% clustered separately, with no influence of cell state (Fig. 2b,c and Supplementary Fig. 3). By contrast, cells from small LARRY clones with relative sizes less than 0.25% were interspersed between clusters (Fig. 2d). EPI-Clone identified cells that belong to expanded clones on the basis of the high local density in principal component analysis (PCA) space spanned by the static CpGs (Fig. 2b,d). EPI-Clone correctly identified cells from expanded clones with an area under the receiver operating characteristic curve (AUC) of 0.79 when using the LARRY clone sizes as ground truth (Fig. 2e). Subsequently, EPI-Clone clustered cells from expanded clones by clonal identity, achieving an adjusted rand index (ARI) of 0.88 relative to ground-truth LARRY barcodes (Fig. 2f). Quantitatively and qualitatively similar results were obtained from a biological replicate that used the same parameters and cut-off values in the EPI-Clone analysis (Extended Data Fig. 3; AUC = 0.68, ARI = 0.82). These results demonstrate that epimutational clonal signals are stably maintained in blood stem and progenitor cells over long periods of time (5 months from transplant to analysis).
We next asked whether EPI-Clone can determine clonal identity in mature immune cells. To that end, we collected mature immune (lymphoid and myeloid) cells from bone marrow and spleen (experiment M.5; Fig. 2g, Supplementary Table 1 and Supplementary Fig. 4) and profiled surface-antigen expression and DNA methylation at the same CpGs as in experiments M.1–M.3. Using the static CpGs defined from experiment M.1, EPI-Clone again produced clonal clustering that recapitulated ground-truth clonal labels (Fig. 2h). We separately computed ARI values between EPI-Clone results and LARRY barcodes. ARI values were higher than 0.7 for monocytes, neutrophils, other myeloid cells, CD8+ T cells and one B cell subset, higher than 0.4 for CD4+ T cells and low for macrophages and a second B cell subset (Fig. 2i,j). Most T cells and B cells belonged to lymphoid-dominated (LARRY and EPI-Clone) clones (Fig. 2i and Supplementary Fig. 4d), which implicated an origin in lymphoid-biased or restricted progenitors30. In a separate experiment (M.4), we profiled mature myeloid cells from lung, bone marrow and peripheral blood, and found that myeloid cell types, except macrophages, retained this clonal mark also outside of the bone marrow (Extended Data Fig. 4). These results show that clonal information encoded in the DNA methylation state is maintained in most lineages until terminal differentiation and 10 months after the lentiviral labelling event (Discussion).
Finally, we asked whether EPI-Clone can be applied to tissues other than blood. We used the same CpG panel to sorted endothelial cells (ECs) from lung of an aged mouse. ECs share a common developmental origin with blood (experiment M.6; Extended Data Fig. 5a). Using the dynamic CpGs defined in haematopoiesis and CD31, SCA1 and podoplanin protein-expression information, we identified two previously described types of capillaries and lymphatic ECs31 (Extended Data Fig. 5b–f). Using the same set of static CpGs as in haematopoiesis, EPI-Clone revealed cell-state-independent, yet statistically supported, clusters containing all three cell types (Extended Data Fig. 5g,h). We conclude that a similar set of static and dynamic CpGs defines clones and differentiation states, respectively, in endothelia and haematopoiesis (Extended Data Fig. 5i).
In summary, DNA methylation patterns at static CpGs constitute a broadly applicable clonal barcode.
HSC-expanded clones in mouse ageing
EPI-Clone can provide joint information on the cell state of progenitors, clonal identity and clonally derived progeny. Therefore, it is an ideal method to characterize the clonal dynamics of native (unperturbed) haematopoiesis. In contrast to the transplantation setting, native haematopoiesis has been described as polyclonal32,33, whereby several thousand clones contribute to blood formation. To investigate whether EPI-Clone also identifies clones in native haematopoiesis, we applied it to bone marrow samples from two untreated, young mice (experiment M.7, 12 weeks old; Supplementary Fig. 5a). Approximately 50% of cells were part of large clones (defined as a relative size larger than 1%) that individually made up 1–4% of total HSPCs (Fig. 3a,c). These clone sizes are in line with a study that genetically barcoded adult haematopoietic clones in situ33 (Fig. 3c). The remaining cells were classified as belonging to small and non-expanded clones. A limitation of EPI-Clone is that only cells belonging to expanded clones can be assigned to their clone of origin. Cells belonging to very small clones (<0.25% of cells after transplant and <1% in native haematopoiesis) could be identified as not belonging to expanded clones, but their clonal identity could not be inferred with the cell numbers used here.

a, DNA methylation UMAP based on the static CpGs for a native, young (12 weeks old) mouse from experiment M.7. b, DNA methylation UMAP based on the static CpGs for an old mouse (100 weeks old). In a and b, three outlier clusters with size <1% were removed to improve visualization. c, Comparison of clone sizes for old and young mice (two biological replicates), and a young mouse from a previous study33. Clones with a relative size less than 1% are shown in grey. d, Comparison of HSC/MPP1 output and myeloid output for the 20 clones with the highest HSC/myeloid output between young and old mice (2 replicates). e, Bubble plot visualizing the frequency of HSC/MPP1 cells per clone for old and young mice. f, Differentiation UMAP defined on the basis of dynamic CpGs, highlighting example clones with different behaviour for old and young mice. For a, b, e and f, data from replicate 1 is shown, see Supplementary Fig. 7 for replicate 2. g, Comparison of the ratio between lymphoid and myeloid output per clone identified using EPI-Clone. P values calculated using two-sided Wilcoxon tests. h, Experimental design for the transplantation experiment (M.8). i,j, Boxplots of post-transplant clone sizes, comparing clones with different pre-transplant differentiation bias calculated as the ratio of mature versus immature cells per clone (i) and different pre-transplant immature clone sizes (j). Tertile T1 has the lowest mature output (i) and smallest clone size (j). k, Boxplot showing the distribution of pairwise cosine observed (Obs.) versus expected (Exp.) distances (before and after transplant) computed using the cell-type distribution of each clone. Observed data are compared with a null model created by randomly shuffling the clonal identities of post-transplant clones (1,000 times). P values of i–k are from two-sided Wilcoxon tests. For d,e,g and i–k, see the section ‘Data visualization’ in the Methods for a definition of boxplot elements and further detail. The scTAM-seq schematic in h was adapted from ref. 5 under a Creative Commons licence CC BY 4.0.
We next applied EPI-Clone to study ageing by comparing the data from young mice (12 weeks old) to 100-week-old mice in two biological replicates (experiment M.7; Fig. 3b and Supplementary Fig. 5a). We observed weak shifts in cell-type proportions between the young and the old mice, a result that confirmed previous observations34 (Supplementary Fig. 5b–e). When comparing the EPI-Clone result, we observed more expanded clones in the old mice than in the young mice (Fig. 3c and Supplementary Figs. 6 and 7). Expanded clones in the old mice were individually also larger than in the young mice (Fig. 3c; two-sided Wilcoxon test P = 0.012). This gradual loss of clonality with age resembles certain properties of human HSC ageing (see below).
Next, we measured the distribution of cell types for each clone across the various stem and progenitor clusters. In the old mice, we observed several expanded clones that contained mostly HSCs across both of our replicates (Fig. 3d–f and Supplementary Fig. 7d,e; Kolmogorov–Smirnov test P < 0.05), which were not present in the young mice. These HSC-expanded clones contained large numbers of stem cells apparently incapable of proceeding with differentiation and contained little progeny. Old mice showed a moderate increase in the number of myeloid-biased clones, which was in contrast to results from classical transplantation experiments35,36,37,38 (Fig. 3d and Supplementary Figs. 6 and 7). However, the rare HSC-expanded clones were mostly myeloid-biased (Fig. 3g; Wilcoxon test P = 0.01 (replicate 1) and P = 0.076 (replicate 2)).
To determine the long-term stability of the HSC-expanded clonal behaviour, we performed a transplantation assay using an aged donor mouse. We used EPI-Clone to compare the clonal composition of the haematopoietic system in the native state (before transplant) and after transplant, and used LARRY barcoding as an additional control during transplantation (experiment M.8; Fig. 3h and Extended Data Fig. 6a). Clonal identities defined using EPI-Clone remained stable during transplantation (Extended Data Fig. 6b–e). HSCs with abundant progeny before transplant showed poor engraftment, a result in line with serial transplantation studies using lentiviral barcoding8,33 (Fig. 3i and Extended Data Fig. 6f). Notably, HSC-expanded clones also engrafted poorly, and we identified non-expanded HSCs as the major drivers of transplantation haematopoiesis (Fig. 3j). Clones with quantifiable output before and after transplant showed a stable lineage bias that was inherited after transplantation (Fig. 3k and Extended Data Fig. 6g).
In summary, our data demonstrate age-related loss of clonal complexity in mouse ageing that is accompanied by an emergence of HSC-expanded clones with low engraftment capacity. We propose that these rare but expanded clones drive the increase in stem cell number and decrease in output that had typically been associated with aged haematopoiesis in transplantation studies39,40,41 and in Cre-lox-based native lineage-tracing studies42. Our transplant data support the idea that HSCs that do not expand with age persist and drive regeneration.
EPI-Clone in human bone marrow
To relate these results to human ageing, we next adapted EPI-Clone for use on human samples. We designed a panel that targeted 448 CpGs with variable methylation between or within blood progenitor populations (Methods and Extended Data Fig. 7a,b). We also included 147 genomic regions commonly mutated in CH and 20 regions that targeted chromosome Y to serve as a partial ground truth for clones identified by EPI-Clone.
We collected CD34+-enriched total bone marrow (TBM) samples from seven donors of different ages (donors A.1–A.7). We also assembled a dataset of CD34+ cells from bone marrow from nine donors (donors B.1–B.5 and X.1, and donors A.1, A.3 and A.4, for whom >1,000 CD34+ cells had been captured from TBM) (Fig. 4a and Supplementary Table 1). Three of the TBM donors had previously been characterized for CH mutations43, and we de novo identified CH mutations or loss of the Y chromosome (LoY) for four additional donors from scTAM-seq data (Methods). In total, we identified ten CH mutations and one LoY event in our cohort. Samples were stained with an antibody panel targeting 45 surface proteins to provide phenotypic characterization. Across all donors, we profiled 135,432 single cells using scTAM-seq.

a, Summary of donor characteristics (Supplementary Table 1). Dots connected by dashed lines denote samples that were analysed as part of the TBM and the CD34+ dataset. b, Integrated UMAP of dynamic CpG and surface-protein data for all donors from the TBM and CD34+ datasets. Cell states were annotated based on the expression of surface proteins (Extended Data Fig. 7c–e). c, UMAPs computed per donor on a consensus set of static CpGs, highlighting cells containing the specified CH mutations. See Extended Data Fig. 7f–h and Methods for how consensus static CpGs were identified. The donors are sorted by increasing age. d, UMAPs as in c, highlighting clones identified using EPI-Clone. e, Scatter plot displaying the percentage of cells from each identified clone displaying CH mutations. The identified clones (x axis) are sorted by size. Dots in colours correspond to the clones dominated by a CH mutation, see c for colour scheme. f,g, Scatter plot relating donor age (f) and the presence of GMPs (g) to the number of clones identified by EPI-Clone in the TBM cohort and CD34+ cohort, respectively. P value calculated with a two-sided t-test computed from a generalized linear model of the Poisson family, using the number of cells observed as a weight. Dot size denotes the number of cells analysed (see b for a scale). h, Boxplot depicting clone sizes stratified into clones carrying CH mutations and clones for which no CH mutation was identified. See the section ‘Data visualization’ in the Methods for a definition of boxplot elements.
We followed the same analytical strategy as for the mouse experiments, but with minor adaptations (Methods). Specifically, we detected expanded clones using a statistical criterion (CHOIR44; Extended Data Fig. 3g), and we identified cell types and differentiation states using a combination of both dynamic CpGs and surface proteins (Fig. 4b and Extended Data Fig. 7c–e). We then used all myeloid cells to identify a consensus set of static CpGs across individuals (Extended Data Fig. 7f–h). To assess the fidelity of static CpGs to identify clones, we exploited the CH mutations and LoY events as a clonal ground-truth. CH clones clustered together in static CpG UMAPs in all cases (Fig. 4c and Extended Data Fig. 8a). EPI-Clone recapitulated the CH clones in all donors except A.5, which was covered with substantially fewer cells than the rest of the TBM cohort (Fig. 4d,e and Extended Data Fig. 8b). Quantitatively, the epimutational clones dominated by CH mutant cells were on average 78.8% mutant and those dominated by wild-type cells were on average 95.4% wild-type (Fig. 4e). These numbers probably underestimate the true overlap between the identified clones and CH clones owing to allelic dropout of CH mutations. We observed a stronger separation of clones identified using our algorithm and better overlap with CH mutations in older donors than in young donors. This result suggests that EPI-Clone most accurately identifies clones in haematopoietic systems of reduced clonal complexity. Besides the CH clones, EPI-Clone identified a total of 67 other clonal expansions in the seven TBM donors, a result that highlights the capacity of this algorithm to recapitulate clonal expansions driven by known and unknown drivers.
We included natural killer (NK) cells and immature B cells in our analysis and used CH mutations to validate that these cells also clustered by clone (Extended Data Fig. 8c,d). When T cells and mature B cells were included, they associated with lymphoid-dominant clusters, a finding in line with the results from mice (Fig. 2i and Extended Data Fig. 8e) and indicating their distinct clonal origins compared with the other cells. In donor A.4, in whom a large CH clone contributed to T cells, mutant T cells clustered with the remaining CH-derived cells (Extended Data Fig. 8e). Together with the results from the mouse LARRY experiment, this finding constitutes evidence that the identified clones remain stable from HSCs to myeloid, T cells, NK cells and immature B cells.
To establish a conservative estimate for a minimum clone size of EPI-Clone in humans, we determined the smallest CH clone identified using this method. The clone DNMT3A(C666Y) in donor A.4 had 145 cells or a relative size of 1% in the myeloid compartment. Furthermore, we observed that several large CH clones (for example, DNMT3A(R659H) in donor A.4; Fig. 4d and Supplementary Fig. 8) had diversified into two clones with a similar but distinguishable static CpG profile. This result suggests that over decades, epimutations can continue to accrue phylogenetic information. In conclusion, these analyses demonstrate the ability of EPI-Clone to identify expanded haematopoietic clones of a wide range of sizes in human bone marrow and blood.
Clonal expansions in human ageing
We leveraged the ability of EPI-Clone to trace both CH clones, which are well characterized in humans28,43, and clones without known driver mutations (non-CH clones) to functionally compare these two types of clonal expansions in our TBM and CD34+ cohorts. Owing to their putatively distinct clonal origins, we excluded T cells and mature B cells from this analysis. As expected3, in the TBM cohort, we observed an age-dependent accumulation of expanded CH and non-CH clones (Fig. 4f). Notably, in the CD34+ cohort, which was mostly sampled from individuals aged 50–60 years, we identified a correlation between the fraction of granulocyte–macrophage progenitors (GMPs) in the sample and the accumulation of expanded clones (Fig. 4g), which suggested that cues that enhance myelopoiesis also lead to more clonal expansions.
CH clones tended to be more expanded than non-CH clones, but were not always among the largest ones (Fig. 4h). Expanded clones were significantly depleted (compared with cells from non-expanded clones) from the B cell and erythroid lineages (Fig. 5a,b and Extended Data Fig. 8f), which implicated a link between myelopoiesis and expansion even for non-CH clones. Compared with non-CH clones, CH clones were significantly enriched in HSCs and MPPs but depleted from the B cell and erythroid lineages (Fig. 5b and Extended Data Fig. 8f,g). These results highlight a stem-cell bias in age-expanded clones that is conserved across mice and humans, and they support a model whereby CH clones are part of a spectrum of such age-expanded clones.

a, Scatter plot depicting the fraction of immature B cells per clone relative to the fraction of immature B cells in non-expanded clones from the same patient. Grey dots are clones with no known driver mutation, dots in colour are clones with a CH mutation (see Fig. 4c for the colour scheme). b, Dot plot depicting P values for enrichments and depletion of cell types in expanded versus non-expanded and CH versus non-CH clones. For this analysis, cell-type composition of clones (for example, the percentage of clone CD34+) were transformed using a logit transform and P values were computed using a mixed-effect model, using donor as a random effect and clone type (expanded or non-expanded or CH or non-CH) as a fixed effect (Extended Data Fig. 8f,g). c, Schematic of the scTAMARA-seq protocol (for the X.1 experiment; Extended Data Fig. 9). d, Clones discovered using EPI-Clone were identified on CD34+ cells from donor X.1 using DNA methylation data. Subsequently, genes with differential expression between clones and correlation with the percentage of HSC/MPPs in the clone were identified. Adjusted P values were calculated using two-sided tests for Pearson correlation, adjusted for multiple testing. e, Schematic of experiment X.2 (scTAMito-seq; Extended Data Fig. 10). f, Scatter plot depicting the presence of six mitochondrial variants in the different clones identified using EPI-Clone from X.2. Cells were scored as positive for the variant if at least 5% of reads supported the variant. The enrichment of variants in the identified clones was determined by a two-sided binomial test. The identified clones were classified as B cell, T cell or NK cell clones if at least 80% of cells were from a single lineage or as multilineage clones otherwise. g, Like f, but for the mt:7076A>G variant.
To resolve transcriptional differences between clones in the HSC and MPP (HSC/MPP) compartment, we added targeted RNA-seq to the scTAM-seq protocol (single-cell targeted analysis of the methylome and RNA (scTAMARA-seq); Fig. 5c and Extended Data Fig. 9a). To that end, we combined SDR-seq45, a recently described targeted RNA-seq protocol for Mission Bio Tapestri, with scTAM-seq. We profiled one of the CD34+ bone marrow samples (X.1) and obtained high-quality DNA methylation and RNA-seq data from 2,745 cells (Extended Data Fig. 9b–e). scRNA-seq data confirmed the accuracy of DNA-methylation-based cell-state annotation and showed an increased resolution of transcriptomic data at the level of erythromyeloid progenitors (Extended Data Fig. 9f,g). We then investigated the gene-expression pattern of distinct clones. HSC/MPP-biased clones expressed low levels of TAL1, SLC40A1 and CDC45 at the HSC/MPP level and high levels of CEBPA, which suggested that clonal fate biases are correlated with gene-expression changes at early stem and progenitor states (Fig. 5d). These results further demonstrate the compatibility of EPI-Clone with targeted RNA-seq from the same cell.
EPI-Clone and mitochondrial variants
In the field, there is controversy regarding the potential of other somatic events, in particular low-heteroplasmy mtDNA variants, for lineage tracing14,15,16. To perform a direct experimental comparison, we used EPI-Clone to analyse peripheral blood from a 38-year-old healthy donor (X.2) that had previously been characterized by a state-of-the-art single-cell mitochondrial lineage tracing method, mt-scATAC-seq13,46. We identified 44 clones from this sample, which displayed prominent clonal expansions of NK cells and T cells (Extended Data Fig. 10a). By including a mitochondrial targeting panel into scTAM-seq, we achieved a median coverage of 176 reads per cell on the mitochondrial genome (Fig. 5e and Extended Data Fig. 10b,c). Of the 23 mtDNA variants previously identified46 (Supplementary Table 3) in this donor and covered in scTAM-seq, 5 had clear phylogenetic relationships with the clones identified using EPI-Clone. That is, they were either subclones of single clones or were parental to several of the identified clones (Fig. 5f), and one variant was observed in two clones. A highly abundant variant (mt:7076A>G) was strongly enriched or depleted in 17 T cell or NK cell clones identified using EPI-Clone, but was observed in approximately 50% of cells of the remaining, mostly multilineage or B cell, clones identified (Fig. 5g). This variant was probably present before epimutational patterns were established and repeatedly underwent selection throughout development and adulthood. Therefore, T cell clones with a recent history of expansion may or may not carry the variant, whereas multilineage clones that expanded before selection of the variant contain a mix of mutant and wild-type cells. Finally, the remaining 16 low-heteroplasmy mitochondrial variants did not segregate with clones identified using EPI-Clone (Extended Data Fig. 10d,e). These findings are in line with a recent report15 observing that only some observed mitochondrial variants carry phylogenetic information, and illustrate the complexity of mitochondrial genetics, for which selection of variants can happen repeatedly during differentiation46. These results also provide additional orthogonal validation of the value of EPI-Clone outside the setting of CH.
Discussion
In summary, DNA methylation at a few hundred CpGs is sufficient to simultaneously identify clones and cell states of haematopoietic cells and ECs, whereas individual CpGs are either informative of cell states or clones. Somatic epimutations seem to be a stable, long-term lineage tracer. Indeed, 5–10 months had elapsed between introduction of the ground-truth clonal label and collection of cells after transplantation. In humans, previous studies have indicated that decades pass between the initial acquisition of CH or LoY and the observation of expanded clones in age3.
This result raises the question of where and how clonal epimutations arise. We found that they randomly occur but remain stable over many cell divisions. Moreover, their numbers do not increase during differentiation (Supplementary Fig. 9a) and they are enriched for heterochromatic and late-replicating domains. We propose that some developmental events that are characterized by rapid cellular proliferation and/or a remodelling of the DNA methylome, such as the specification of HSCs47, essentially randomize the methylation state of CpGs in heterochromatic and late-replicating regions. A potential explanation of this effect is that in rapidly dividing cells, DNMT1 may not act sufficiently to copy the DNA methylation state to the nascent DNA strand (Supplementary Fig. 9b). Consistent with this idea, a recent study of bulk methylome profiles from blood cells in monozygotic twins suggested that clone-associated variation of the methylome may be established during embryonic development48. In the case of some large CH clones, we observed additional diversification of epimutational patterns.
We therefore propose that variably methylated CpGs in non-regulatory genomic regions can act as a digital barcode of clonal origin. The digital and stochastic nature of epimutations makes single-cell methods that are capable of mapping the methylation state of single CpGs at high confidence, such as scTAM-seq, a powerful tool for lineage tracing. While this article was under review, a method termed MethylTree49 demonstrated identification of clonal identity from sparse whole-genome, single-cell DNA methylation data. Compared with MethylTree, our approach is more scalable, less expensive and less computationally intense. Conversely, scTAM-seq requires the design of a species-specific targeting panel.
The robustness of EPI-Clone is best evidenced by its capacity to identify high-resolution clonal patterns in native haematopoiesis. We demonstrated that both native human and mouse haematopoiesis shifts from highly polyclonal to oligoclonal blood production, and we investigated clone function in these two species using a coherent, unified method. Expanded clones in mice tended to be more numerous, but individually smaller, and poorly contribute to haematopoiesis in transplants. This observation seems to be in line with the larger and more polyclonal stem cell compartment in humans, but a much longer period of clonal selection and drift. In our human data, oligoclonal blood production became detectable at an age of around 50 years and manifested itself as an inevitable and potentially clock-like process after the age of 60 years.
Our data further put CH mutations into a perspective with clonal expansions without known drivers. That is, CH clones are more strongly biased towards the myeloid lineage and towards an expansion of stem cells, but together with non-CH clones form part of a spectrum of age-related clonal expansions that display similar functional properties. In aged mice, we similarly detected large HSC-expanded clones that had reduced regenerative capacity. Together with recent transplantation studies of human HSCs50, this result suggests that there is conservation of the processes that drive haematopoietic ageing and decline in clonal complexity, and it highlights that CH mutations might not be the main driver of this process. Epidemiological studies have demonstrated an increased mortality risk in carriers of driver-free expanded clones27. These results call for a broader investigation of age-related decline in clonality instead of a strict focus on CH.
Methods
Methods summary
An overview of all experiments performed for this study is included in Extended Data Fig. 1a and Supplementary Table 1. For the mouse experiments with an available ground truth from the LARRY lentiviral barcoding system (experiments M.1, M.2, M.4, M.5), LARRY barcoding vectors were constructed and lentiviruses were produced (see the section ‘Lentiviral barcoding using the LARRY system’). Stem cells were then collected from mice, transduced with the LARRY lentiviruses and transplanted, and different cellular compartments were collected 5–10 months later for profiling by scTAM-seq (see the section ‘Experimental procedures (mouse study)’). Additional experiments were performed on biological material from non-treated mice of different ages (experiments M.3 and M.6–M.8; see the section ‘Experimental procedures (mouse study)’). For the human study, primary bone marrow samples were analysed (see the section ‘Experimental procedures (human study)’).
All biological material was analysed by scTAM-seq5 (see the section ‘Single-cell DNA methylation profiling with scTAM-seq’). scTAM-seq is a targeted method for DNA methylation analysis based on the Mission Bio Tapestri platform. Specifically, up to 1,000 amplicons 200–400 base pairs in length are amplified from the genomes of single cells. Before this amplification step, scTAM-seq includes a digestion step with a methylation-sensitive restriction enzyme, HhaI. Therefore, CpG dinucleotides in HhaI sites are only effectively amplified if methylated. The selection of the target amplicons comprising individual CpGs is a crucial step in this protocol (see the sections ‘Mouse panel design for scTAM-seq’ and ‘Human panel design for scTAM-seq’). Relevant genetic information (LARRY barcodes or CH mutations) can be read out from gDNA by scTAM-seq in the same cells, specifically by covering these regions with amplicons not containing HhaI cut sites. Surface-antigen expression was read out through the inclusion of oligonucleotide-barcoded antibodies in the protocol. We also included dedicated experiments demonstrating the combination of scTAM-seq with RNA-seq from the same single cell (experiment X.1, see the section ‘Combined profiling of DNA methylation and RNA in the same cell’) or mitochondrial genome sequencing (experiment X.2, see the section ‘Combined profiling of DNA methylation and mitochondrial variants’).
Key steps in the data analyses (see the section ‘Bioinformatic analysis (mouse)’) were to define cell states through data integration and subsequently to identify clones using the EPI-Clone algorithm. This algorithm first identifies CpGs with no surface-antigen association as potentially clone-associated or ‘static’ CpGs, and subsequently performs clustering and dimensionality reduction exclusively on these CpGs (see the section ‘EPI-Clone’). For the analyses of the human data, the same overall strategy was used. Additional steps and adjustments included mutation calling and definition of a consensus set of static CpGs across donors (see the section ‘Bioinformatic analysis (human)’).
A detailed protocol for performing scTAM-seq for clonal tracing with EPI-Clone is available from protocols.io51.
Lentiviral barcoding using the LARRY system
Construction of lentiviral pLARRY vectors
Barcode libraries were constructed according to a previously established protocol (https://www.protocols.io/view/barcode-plasmid-library-cloning-4hggt3w). First, the T-Sapphire or eGFP coding sequences and the EF1a promoter sequence were PCR-amplified from pEB1-T-Sapphire and pLARRY-eGFP with primers homologous to the vector insertion site in a custom lentiviral plasmid backbone (Vectorbuilder) using Gibson assembly (Gibson assembly master mix, NEB, E2611L). After magnetic bead purification, ligated vectors were transformed into NEB10-beta electroporation ultracompetent Escherichia coli cells (NEB 10-beta electrocompetent E. coli, NEB, C3020K) and grown overnight on LB plates supplemented with 50 μg ml–1 carbenicillin (carbenicillin disodium salt, Thermo Scientific Chemicals, 11568616). Colonies were scraped using LB medium and pelleted by centrifugation. Plasmid maxipreps were performed using an Endotoxin-Free Plasmid Maxi kit (Macheray Nagel), following the manufacturer’s protocol. pEB1-T-Sapphire was a gift from P. Cluzel (Addgene plasmid 103977). pLARRY-eGFP was a gift from F. Camargo (Addgene plasmid 140025).
Barcode lentivirus library generation and diversity estimation
To barcode pLARRY plasmids and generate a library, a spacer sequence flanked by EcoRV restriction sites was cloned into the plasmid after the WPRE element of the vector. Custom PAGE-purified single-strand oligonucleotides with a pattern of 20 random-bases (GTTCCANNNNTGNNNNCANNNNGTNNNNAGNNNN) and surrounded by 25 nucleotides homologous to the vector insertion site were synthesized by IDT DNA Technologies. The assembly of these components and subsequent purification steps were carried out through Gibson assembly (Gibson assembly master mix, NEB, E2611L). Six electroporations of the bead-purified ligations were performed into NEB10-beta E. coli cells (NEB 10-beta electrocompetent E. coli, New England Biolabs, C3020K) using a Gene Pulser electroporator (Bio-Rad). After transformation, the cells were incubated at 37 °C for 1 h at 220 r.p.m. After incubation, the transformed cells were plated in six large LB–ampicillin agar plates overnight at 30 °C. Colonies from all six plates were collected by scraping with LB–ampicillin and then grown for an additional 2 h at 225 r.p.m. and 30 °C. Cultures were pelleted by centrifugation, and plasmids were isolated using an Endotoxin-Free Plasmid Maxi kit (Macheray-Nagel), following the manufacturer’s protocol.
For estimating diversity, LARRY barcode amplicon libraries were prepared by PCR amplification of the lentiviral library maxiprep using flanking oligonucleotides carrying TruSeq read1 and read2 adaptors using 10 ng of the library (Supplementary Table 4). We used the minimal number of cycles that we could detect by quantitative PCR to avoid PCR amplification bias (10–12 cycles). After bead purification, 10 ng of the first PCR product was used as a template for a second PCR to add Illumina P5 and P7 adaptors and indexes (Supplementary Table 4). Two independent PCRs were sequenced on an Illumina NovaSeq 6000 S4 platform (Novogene) to confirm diversity after correction of errors through collapsing with a Hamming distance of 4. After collapsing, libraries were confirmed to contain at least 50 million different barcodes, with enough diversity for uniquely labelling up to 100,000 HSCs with a minimal false-positive rate.
Lentivirus production and barcode labelling
Lentivirus production and HSPC transduction were performed as previously described8.
Experimental procedures (mouse study)
Mice and animal guidelines
All procedures involving animals adhered to the pertinent regulations and guidelines. Approval and oversight for all protocols and strains of mice were granted by the Institutional Review Board and the Institutional Animal Care and Use Committee at Parque Científico de Barcelona under protocols CEEA-PCB-22-001-ARF-P1 and CEEA-PCB-22-002-ARF-P2. The study followed all relevant ethical regulations. CD45.1 (CD45.1, B6.SJL-Ptprca Pep3b/BoyJ, 002014, The Jackson Laboratory) mice were used as transplantation recipients for CD45.2 (BL6/J) donor cells. Mice were kept under specific-pathogen-free conditions for all experiments. We used 12–100-week-old male and female mice for our experiments. Neither randomization nor blinding was used. Experiments were performed with one or two biological replicates of mice, and no statistical methods were used for sample size choice. To minimize distress, euthanasia was performed by administering isoflurane inhalation, followed by cervical dislocation to ensure the animals were fully deceased.
LARRY lentiviral barcoding and transplantation
Following euthanasia, bone marrow was collected from the femur, tibia, pelvis and sternum through mechanical crushing, ensuring the retrieval of most of the cells. The collected bone marrow cells were then sieved through a 40 μm strainer and cleansed with a cold ‘Easy Sep’ buffer containing PBS, 2% FBS, 1 mM EDTA and penicillin–streptomycin followed by lysis of red blood cells using RBC lysis buffer (BioLegend, 420302). At first, mature lineage cells were selectively depleted using a Lineage Cell Depletion kit, mouse (Miltenyi Biotec, 130-110-470), and the resulting LIN– (lineage-negative) fraction was then enriched for KIT expression using CD117 MicroBeads (Miltenyi Biotec, 130-091-224). These KIT-enriched cells were washed, blocked with FcX and stained with the following fluorescently labelled antibodies: APC anti-mouse CD117, clone ACK2 (BioLegend, 105812); PE/Cy7 anti-mouse Ly6a (SCA1) (BioLegend, 108114); Pacific Blue anti-mouse Lineage cocktail (BioLegend, 133310); PE anti-mouse CD201 (EPCR) (BioLegend, 141504); PE/Cy5 anti-mouse CD150 (SLAM) (BioLegend, 115912); and APC/Cy7 anti-mouse CD48 (BioLegend, 103432). For transplants, EPCR+LIN–SCA1+KIT+CD48–CD150+ HSCs were sorted by FACS with a BD FACSAria Fusion with a 70 µm nozzle.
In vitro cultures of HSCs were done under self-renewing F12-PVA-based conditions as previously described52. To culture HSCs, 96-well flat-bottom plates from Thermo Scientific were coated with a layer of 100 ng ml–1 fibronectin (bovine fibronectin protein, 1030-FN) for 30 min at room temperature. After the sorting process, HSCs were transferred into 200 µl complete HSC medium supplemented with 100 ng ml–1 recombinant mouse TPO (PeproTech Recombinant Murine TPO, 315-14) and 10 ng ml–1 recombinant mouse SCF (PeproTech Recombinant Murine SCF, 250-03) and grown at 37 °C with 5% CO2. During lentiviral library transduction, the first medium change took place 24 h after transduction. Three days after labelling, the cultured HSCs were collected and subsequently transplanted into conditioned CD45.1 mice. The CD45.1 recipient mouse was preconditioned with a lethal X-ray radiation dose, administered as two separate sessions amounting to 5 Gy each, with a 4-h interval between them. To assess the engraftment of donor cells, the percentage of CD45.2+ peripheral blood leukocytes (and the percentage of fluorescent-protein-labelled cells) was determined. All mice demonstrated stable long-term engraftment until the experimental end point. Engraftment analysis, along with the measurement of labelling frequency, was carried out using BD FACS Fusion.
Collection of cells for single-cell characterization
In all single-cell experiments, unless described otherwise in the subsequent sections, transplanted or untreated mice were euthanized at specified ages and time points after transplant, and a KIT-enriched cell fraction was isolated from the femur, tibia, pelvis and sternum, per the protocol described above. This KIT-enriched cell population was stained with FcX block to prevent nonspecific binding and subsequently stained again with the following panel of fluorescently labelled antibodies: APC anti-mouse CD117 (clone ACK2, BioLegend, 105812); PE/Cy7 anti-mouse Ly6a (SCA1) (BioLegend, 108114); and Pacific Blue anti-mouse Lineage cocktail (BioLegend, 133310). In all mouse experiments, cells were also labelled with a custom TotalSeq-B antibody cocktail (Supplementary Table 2). After staining, distinct cellular compartments were sorted as illustrated in Supplementary Fig. 1 and profiled by scTAM-seq (see below).
LARRY experiments
For validating EPI-Clone using a ground-truth genetic lineage-tracing experiment, we performed two experiments: the main LARRY experiment (M.1) and the LARRY replicate experiment (M.2) (Figs. 1 and 2 and Extended Data Figs. 2 and 3). For M.1, two donor mice were killed, and HSCs were labelled with LARRY constructs containing a GFP label in one case and LARRY constructs containing a Sapphire label in the other case. Subsequently, labelled cells from each donor were transplanted into two recipient mice each. Accordingly, the dataset contains cells from four mice that contain two sets of clones, labelled with GFP and Sapphire, respectively. GFP and Sapphire clones did not mix on EPI-Clone UMAPs (Extended Data Fig. 3f), which further demonstrates that clones identified using EPI-Clone are individual-specific. We profiled all four recipient mice after allowing full blood reconstitution over 5 months. We also repeated this experiment again for validating the computational method (experiment M.2) using only one donor mouse. For both experiment M.1 and experiment M.2, we collected LSK and LK cells from the bone marrow and mixed them at 60,000 (LK) plus 50,000 (LSK) before analysing the cells using the Tapestri platform (Supplementary Table 1).
Native haematopoiesis
In this experiment (M.3; Fig. 1), we killed a 12-week-old wild-type BL6/J (CD45.2) mouse, extracted 120,000 LK cells and subjected them to scTAM-seq (Supplementary Table 1 and Supplementary Fig. 1).
Mature myeloid cell experiment
For profiling tissue-resident myeloid cells (experiment M.4; Extended Data Fig. 4), a single LARRY-transplanted mouse was anaesthetized 10 months after transplantation and perfused. Subsequently, lungs were extracted from the chest cavity, and a single-cell suspension was prepared using a protease and DNAse solution from a Lung Dissociation kit (Miltenyi Biotech, 130–095-927) followed by mechanical dissociation using gentleMACS ‘C’ columns (Miltenyi Biotech, 130–093-237) according to the manufacturer’s instructions. The dissociated cells were filtered using a 70 μm strainer and centrifuged at 400g for 5 min at room temperature. The supernatant was removed by aspiration and red blood cell lysis was performed using RBC lysis buffer (BioLegend, 420302). Cells were then washed with FACS buffer and pelleted at 400g for 5 min at 4 °C. The supernatant was removed, and the pellet was resuspended in FACS buffer before being passed through a 40 μm strainer and stained for the mature myeloid cell marker. Cells were stained with the following fluorescently labelled antibodies: PerCP/Cyanine5.5 anti-mouse/human CD11b (BioLegend, 101227; clone M1/70) and PE/Cyanine7 anti-mouse CD45.2 (BioLegend, 109829; clone 104). Cells were also labelled with TotalSeq-B antibody cocktail. We then sorted CD45.2+CD11b+LARRY(GFP)+ immune cells from lung. In parallel, we also sorted and stained LSK and LK cells and mature CD11b+ populations from both bone marrow and peripheral blood, followed by single-cell profiling.
Mature immune cell experiment
For this experiment (M.5; Fig. 2 and Supplementary Fig. 4), a single LARRY-transplanted mouse was euthanized 5 months after transplantation, and cells from the spleen and bone marrow were collected as described above. After red blood cell lysis, equal amounts of cells from both organs were pooled, washed and blocked with FcX. The cells were then stained with the following fluorescently labelled antibodies: Pacific Blue anti-mouse FcεRIα (BioLegend, 134313; clone MAR-1); PE/Cyanine5 anti-mouse CD19 (BioLegend, 115509; clone 6D5); Brilliant Violet 605 anti-mouse CD11c (BioLegend, 117333; clone N418); PerCP/Cyanine5.5 anti-mouse/human CD11b (BioLegend, 101227; clone M1/70); APC/Cyanine7 anti-mouse Ly-6G (BioLegend, 127623; clone 1A8); APC anti-mouse CD3 (BioLegend, 100235; clone 17A2); and PE/Cyanine7 anti-mouse CD115 (CSF-1R) (BioLegend, 135523; clone AFS98). We then sorted the following populations from LARRY+ live cells based on their surface markers: T cells (CD3+CD19–), B cells (CD3–CD19+), neutrophils (CD11b+CD3–CD19–Ly6G+), monocytes (CD11b+CD3–CD19–Ly6G–CD115+) and eosinophils and basophils (CD11b+CD3–CD19–FcεR1a+).
Lung EC experiment
For profiling ECs from 100-week-old mice (experiment M.6; Extended Data Fig. 5), dissociated lung cells were collected as described above. The resultant cell population was then enriched for CD31 expression using CD31 MicroBeads (mouse, 130-097-418, Miltenyi Biotec) per the manufacturer’s guidelines. These CD31-enriched cells were then washed, blocked with FcX and stained with the following fluorescently labelled antibodies: PE anti-mouse CD31 (BioLegend, 102507; clone MEC13.3) and PE/Cyanine7 anti-mouse CD45.2 (BioLegend, 109829; clone 104). Following staining, CD31+ and CD45.2– cells were sorted as illustrated in Extended Data Fig. 5a and Supplementary Table 1.
Native haematopoiesis experiments in old and young mice
For this experiment (M.7; Fig. 3 and Supplementary Figs. 5–7), the KIT-enriched cell fraction was stained and subsequently sorted to collect LSK and LK populations as described above. Samples were collected from two young (12-week-old) BL6/J (CD45.2) mice and two aged (100-week-old) BL6/J (CD45.2) mice.
LARRY transplant experiments in old mice
For this experiment (M.8; Fig. 3 and Extended Data Fig. 6), half of the HSC population from a 100-week-old mouse that was profiled as part of experiment M.7 were labelled with LARRY lentivirus and transplanted into lethally irradiated mice. Six months after transplant, mice were euthanized, and a KIT-enriched cell fraction was isolated from the femur, tibia, pelvis and sternum, following the protocol outlined above. This KIT-enriched cell population was stained with FcX block to prevent nonspecific binding and subsequently stained again with the following panel of fluorescently labelled antibodies: APC anti-mouse CD117 (clone ACK2, BioLegend, 105812); PE/Cy7 anti-mouse Ly6a (SCA1) (BioLegend, 108114); and Pacific Blue anti-mouse Lineage cocktail (BioLegend, 133310). After staining, LK and LSK cells were sorted as described above.
Experimental procedures (human study)
Human samples and their previous characterization by genomic assays
Bone marrow samples were obtained from different sources. Samples A.1, A.6 and A.7 were bone marrow aspirates from healthy volunteers collected at the Heidelberg University Hospital after informed written consent. This study was approved by the Ethics Committee of the Medical Faculty of Heidelberg University (S-480/2011). Sample A.4 was a TBM sample obtained through the Banc de Sang i Teixits (Barcelona, Spain) and approved by the Ethics Committee of the Hospital Clinic de Barcelona (HCB/2023/0367). Samples B.1–B.5 and X.1 were commercially available samples of purified CD34+ cells from organ donors (Ossium Health). No genomic characterization was performed on these samples before this study. Samples A.2, A.3 and A.5 were collected after informed written consent from individuals undergoing elective total hip replacement surgery at the Nuffield Orthopaedic Centre under the ‘Mechanisms of Age-Related Clonal Haematopoiesis’ (MARCH) study. This study was approved by the Yorkshire and The Humber–Bradford Leeds Research Ethics Committee (NHS REC ref: 17/YH/0382). These samples were screened for somatic mutations with a variant allele frequency of ≥0.01 by targeted DNA sequencing of a panel covering 97 genes (347 kb) recurrently mutated in myeloid malignancies and CH, as previously described43. Samples with somatic mutations in DNMT3A and PPM1D were selected for analyses. Finally, sample X.2. was a peripheral blood sample collected and characterized by mt-scATAC–seq as previously described46. Informed consent was given and approved for genomics profiling by the Stanford Institutional Review Board (number 14734).
All experiments involving human samples were approved by the corresponding ethics committees and were in accordance with the Declaration of Helsinki.
Bone marrow samples were thawed and stained using CD34 and CD3 sorting antibodies (BioLegend, 343517) and a pool of oligonucleotide-conjugated antibodies from the TotalSeq-D Heme Oncology Cocktail from BioLegend (MB53-0053) as well as additional TotalSeq-D antibodies from BioLegend (Supplementary Table 6). Samples were then sorted for CD34+ and CD34– populations and subjected to scTAM-seq (see below). For details on sorting, see Supplementary Table 1.
Multiplexing
Samples B.1 and B.5, B.2 and B.4, and A.2 and A.5 were in pairs, multiplexed into single Tapestri lanes. Demultiplexing was performed on the basis of germline single-nucleotide polymorphisms on autosomes with vireo53. Chromosome Y and pre-characterized somatic single-nucleotide variants (SNVs) were used as controls (Supplementary Fig. 10 and see the section on ‘Bioinformatic analysis (human)’).
Single-cell DNA methylation profiling with scTAM-seq
Single-cell DNA methylation profiling
For profiling DNA methylation at single-cell resolution, we used scTAM-seq5, which leverages the Mission Bio Tapestri technology to investigate up to 1,000 CpGs in 1,000s of cells per experiment. In brief, we loaded 120,000–140,000 cells into the Tapestri machine and followed the default Mission Bio DNA+Protein protocol for V2/V3 chemistry for the experiments (v.2: https://missionbio.com/wp-content/uploads/2021/02/Tapestri-Single-Cell-DNA-Protein-Sequencing-V2-User-Guide-PN_3360A.pdf; v.3: https://missionbio.com/wp-content/uploads/2023/08/Tapestri-Single-Cell-DNA-Protein-Sequencing-v3-User-Guide_MB05-0018.pdf; see also Supplementary Table 1), but with the following modifications: (1) we added a DNA methylation-sensitive restriction enzyme (HhaI) to digest non-methylated targets (CpGs) before amplification; and (2) in the case of the mouse experiments, we used TotalSeq-B antibodies and different primers for the amplification of antibody oligonucleotide tags. The default Mission Bio protocol uses a different type of oligonucleotide tag, TotalSeq-D, which we used here for the experiments using human samples, but which are currently not available for mouse antigens.
For the mouse samples stained with TotalSeq-B, we added 5 μl of highly concentrated HhaI (150,000 U ml–1, NEB) enzyme and 2 µl of 30 µM of a custom antibody tag primer specific for the amplification of the oligonucleotide tags of TotalSeq-B antibodies (ACTCGCAGTAGTCTTGCTAGGACCGGCCTTAAAG) to the Tapestri barcoding mix reagent. An incubation at 37 °C for 30 min was added to the start of the targeted PCR thermal cycling program to allow for the restriction enzyme digest to take place before the PCR amplification step. The use of TotalSeq-B antibodies primarily affected the ‘Protein Library Cleanup I’ section of the protocol, for which we replaced the 2× binding and washing (B&W) buffer from the kit with the following buffer prepared with nuclease-free water: Tris-HCl (final concentration 10 mM, pH 7.5), EDTA (final concentration 1 mM) and NaCl (final concentration 2 M). We used 2 µl of 5 µM of our custom biotin oligonucleotide (/5Biosg/GTGACTGGAGTTCAGACGTGTG/3C6/) to isolate the antibody tags. Moreover, during the isolation of antibody tags, we performed the second wash of streptavidin beads with 1 ml nuclease-free water instead of 1× B&W buffer. Finally, each tube of streptavidin beads was resuspended in 45 µl of nuclease-free water then transferred and combined into a new tube for a total of 90 µl. To amplify the final protein target library, we used 5 µl of 4 µM of each custom indexed primers (forward: CAAGCAGAAGACGGCATACGAGAT[i7 index]GTGACTGGAGTTCAGACGTGTGCTCTTCCGATCT; reverse: AATGATACGGCGACCACCGAGATCTACAC[i5 index]TCGTCGGCAGCGTC). Typically, we performed twice as many reactions to amplify the DNA target library, but this may be increased to achieve sufficient yield. Last, we adjusted the AMPure XP reagent-to-sample ratio in the second size-selection step in the ‘DNA Library Cleanup II’ section from 0.72× to 0.65×.
For the human samples stained with TotalSeq-D, we followed the scTAM-seq protocol as previously described5.
Using the stained cells that we used as input to scTAM-seq, we also performed 10x Genomics Chromium Single Cell 3′ for transcriptomic profiling of the cells, following the standard protocol. This step was exclusively performed for experiment M.1 (Supplementary Table 1). For the transcriptomic data, LARRY barcodes were later amplified using a modified version of the protocol8 (see Supplementary Table 4 for an updated list of primers).
Mouse panel design for scTAM-seq
We aimed to design a panel with CpGs dynamically methylated in HSCs, as well as in more committed progenitors (MPPs). We collected bulk whole-genome bisulfite sequencing data from a previous publication22 profiling DNA methylation in three replicates of HSCs (LSK and CD135−CD48−CD150+CD34−), MPP1 (LSK and CD135−CD48−CD150+CD34+), MPP2 (LSK and CD135−CD48+CD150+CD34+), and a mixture of MPP3 (LSK and CD135−CD150−CD48+CD34+) and MPP4 (LSK and CD135+CD150−CD48+CD34+). Using these data, we selected CpGs that were variably methylated in HSPCs (Extended Data Fig. 1b,c) using three criteria: (1) CpGs differentially methylated between the HSCs and the different MPP populations (DMCs); (2) CpGs intermediately methylated within HSCs (IMCs); and (3) CpGs harbouring within-sample heterogeneity in HSCs (WSHs). The code for selecting CpGs is available from GitHub (https://github.com/veltenlab/EPI-CloneSelection).
For DMCs, we used RnBeads54 to determine CpGs that were specifically methylated in one of the HSPCs (that is, in HSC, MPP1, MPP2 or MPP3/MPP4) but not methylated in all the remaining HSPC populations. We only focused on CpGs that were covered by at least 10 sequencing reads in all samples and that had a methylation difference of at least 0.2 between the target cell type and the average of the remaining cell types.
IMCs had to be non-overlapping with DMCs and were then defined by a DNA methylation level in the bulk samples (HSCs) between 0.25 and 0.75. Such CpGs may be differentially methylated between two sub-cell types of HSCs. IMCs were required to have a low proportion of discordant reads (PDRs)55 together with a high quantitative fraction of discordant read pairs (qFDRPs)56. PDR and qFDRP are measures of WSH in bulk bisulfite sequencing data and quantify the concordance of methylation states on the same sequencing read (PDR) or of multiple CpGs across different sequencing reads (qFDRP).
CpGs with high WSH were non-overlapping with DMCs and IMCs. The CpGs were then identified on the basis of the high levels of both PDR and qFDRP. These CpGs were therefore located in regions showing variable methylation profiles in bulk sequencing data and might represent regions with stochastic methylation in HSCs.
After identifying all CpGs that fulfilled the above criteria, we excluded any CpGs that were not in the context of a HhaI cut site and enriched the selected CpGs for those located in the vicinity (100 bp) of at least one TFBS of an important haematopoietic transcription factor (Supplementary Table 5). We then selected 105 CpGs specifically methylated in HSCs, 70 in MPP1, 70 in MPP2, 75 in MPP3/MPP4, 210 IMCs and 80 WSH (Extended Data Fig. 1b,c). We also included the following control amplicons: 20 constitutively methylated, 20 constitutively unmethylated and 50 amplicons without a HhaI cut site. Control amplicons were required to identify cells from the data because the remaining amplicons were digested depending on their methylation state. We uploaded this list to the Mission Bio Designer tool (https://designer.missionbio.com/) to receive a final list of 663 amplicons and corresponding primer sequences (Supplementary Table 5). The CpGs were further annotated according to their location in the genome with respect to chromatin states as previously defined57. From the 573 non-control amplicons, a subset of 453 amplicons with low dropout rate in an experiment without HhaI digest was used for analysis.
For amplifying the LARRY barcodes, we spiked in an additional primer into the primer pool targeting the LARRY barcode sequence (forward: GCATCGGTTGCTAGGAGAGA; reverse: GGGAGTGAATTAGCCCTTCCA). We could therefore read out the LARRY barcode together with information about the DNA methylation state from the same single cell.
Human panel design for scTAM-seq
The design for the human panel closely followed the strategy applied for the mouse panel. Two previously published datasets21,58 were used to similarly profile DMCs, IMCs and, additionally, CpGs with interindividual heterogeneity (IIH). Sites were selected to not include single-nucleotide polymorphisms according to dbSNP v.151 and to be located in the HhaI cut sequence.
For DMCs, we considered peripheral blood and bone marrow samples from a previous study21. Samples with an average coverage across all CpGs below 1 were removed. DMCs between HSCs, MPPs, multilymphoid progenitors (MLPs; combining MLP0, MLP1, MLP2 and MLP3), common lymphoid progenitors (CLPs), common myeloid progenitors (CMPs) and GMPs were computed using RnBeads. CpGs with a mean methylation difference higher than 0.1 between the cell types were identified as DMCs.
We performed IMC detection on HSC-enriched lineage-negative (LIN−CD34+CD38−) samples from eight male donors using a previously published dataset58. To deal with data sparsity, we set the maximum quantile of missing values per site to 0.005 and removed any sites that exceeded this threshold. IMCs were defined as CpGs with a DNA methylation level between 0.25 and 0.75 in at least 5 samples. When checking for a HhaI cut site, we allowed for a maximum of 25 CpG sites in the extended region around the IMC.
For CpGs with IIH, we used the same dataset to identify CpGs with a variance higher than 0.1 across all individuals of the dataset from ref. 58.
We also created genotyping amplicons that cover mutations in ASXL1, DNMT3A, TET2, TP53, JAK2, IDH2, PPM1D, SF3B1, IDH1 and SRSF2. We used 62 amplicons covering these genes from the Tapestri single-cell DNA myeloid panel by Mission Bio (https://missionbio.com/products/panels/myeloid/) as a base panel, excluding amplicons with the HhaI restriction sequence GCGC. We designed further amplicons for exons in the aforementioned genes that had a coverage of less than 60% in the default myeloid panel. To prevent these amplicons from having a recognition site, we performed a virtual digestion of the exonic sequences using the HhaI cut sequence. We then uploaded a list containing the fragmented genomic regions to the Mission Bio Designer tool, which resulted in 82 additional amplicons. We also included 20 amplicons targeting chromosome Y and 50 control amplicons without a HhaI cut-sequence. We uploaded the CpG targets and readily designed genotyping, chromosome Y, and control amplicons using the Mission Bio Designer tool. The final list comprises 665 amplicons and corresponding primer sequences. The resulting 448 CpG targeting amplicons are divided into 215 DMC, 145 IMC and 88 IIH amplicons (Supplementary Table 6).
Sequencing
Libraries were sequenced on an Illumina NovaSeq 6000 with 2 × 100 bp (scTAM-seq mouse), 2 × 150 bp (scTAM-seq human), 2 × 50 bp (scRNA-seq) and 2 × 50 bp (protein libraries) reads. For an overview of the sequencing statistics, see Supplementary Table 7.
Combined profiling of DNA methylation and RNA in the same cell
To jointly profile DNA methylation and RNA in the same cell (scTAMARA-seq, experiment X.1), we took advantage of the recently published SDR-seq method45 and combined it with scTAM-seq. In total, we profiled 120 RNA and 367 gDNA (200 DNA methylation and 167 genotyping) targets (Supplementary Table 6). DNA methylation targets were a subset of the original set, excluding amplicons that were not identified as consensus static or dynamic CpGs in the total bone marrow original cohort and low-performing amplicons. RNA targets were selected from a RNA-seq reference map59 using LASSO regression to identify 120 RNAs most predictive of cell state in the CD34+ compartment. We followed the SDR-seq protocol45 using the glyoxal fixation condition. Once the cells had been fixed, permeabilized and reverse-transcribed, they were loaded onto the Mission Bio Tapestri platform and processed as for scTAM-seq. The final RNA and DNA sequencing libraries were individually generated as previously described45.
Combined profiling of DNA methylation and mitochondrial variants
For this experiment (scTAMito-seq, experiment X.2), we performed scTAM-seq using the same 367 DNA methylation and genotyping amplicons as for scTAMARA-seq. We spiked in a pre-designed mitochondrial panel (https://designer.missionbio.com/catalogpanels/Virtual-mtDNA) at a ratio of 1:20 as previously described60.
Bioinformatic analysis (mouse)
Data processing
For processing of raw data, we used a modified pipeline that was based on the originally described pipeline for scTAM-seq5 (https://github.com/veltenlab/scTAM-seq-scripts), which is available from GitHub (https://github.com/veltenlab/EPICloneProcessing). In brief, cellular barcodes were extracted from the raw sequencing files before alignment to the reference genome subset to the CpG panel. Reads mapping to each of the amplicons were quantified to generate a count matrix, and DNA methylation states were determined using a cut-off value of one sequencing read as in the original scTAM-seq publication5. We used those cellular barcodes that had more than 10 sequencing reads in at least 70% of the control (non-HhaI) amplicons. Doublets were removed using the DoubletDetection tool (https://zenodo.org/record/2678042).
At the single-cell level, we differentiated methylated from unmethylated CpGs through the presence of at least one sequencing read for the corresponding amplicon as in the original publication of scTAM-seq5. Sequencing reads can uniquely originate from amplicons with methylated CpGs, whereas the lack of sequencing reads from an amplicon originates either from an unmethylated CpG or from a dropout. To minimize the effect of dropout, we determined the primer combinations that reliably amplified in our panel using a single experiment without the restriction enzyme. For this experiment in mice, LIN−KIT+ cells from a young, wild-type mice (12 weeks) were used and we determined that 453 out of the 573 non-control amplicons (79%) amplified in more than 90% of the cells. These amplicons were used for subsequent analyses.
For the surface-protein data, the Mission Bio pipeline was used to extract sequencing reads for a particular cell-barcode–antibody-barcode combination. We restricted analyses of the protein data to those cellular barcodes identified in the DNA methylation library.
Processing of LARRY barcodes
LARRY barcodes could be directly identified from the scTAM-seq sequencing library because an additional primer pair capturing the LARRY barcode was included (see the section ‘Mouse panel design for scTAM-seq’). Sequencing reads mapping to the amplicon with the LARRY barcode were extracted from the raw sequencing reads using the fluorophore sequence GCTAGGAGAGACCATATGGGATCCGAT. The LARRY barcode was determined using the base pairs following the GFP sequence, given that the sequence matches the rules by which the LARRY barcode was constructed (see the section ‘Barcode lentivirus library generation and diversity estimation’). Barcode extraction was performed using a modified version of the scripts provided in the original LARRY publication8 (https://github.com/AllonKleinLab/LARRY). Barcodes supported by fewer than five sequencing reads were discarded, and LARRY barcodes with a Hamming distance lower than three were merged for each of the experimental batches individually.
Notably, each cell can have more than one unique LARRY barcode owing to multiple lentiviral infections. In these cases, groups of LARRY barcodes were jointly passed on to the progeny. To call clones in this setting, we computed for any pair of LARRY barcodes the extent to which these two barcodes were observed in an overlapping set of cells (formally a Jaccard index). LARRY barcodes were then clustered according to this distance metric. We used a permutation test to determine LARRY barcodes that are merged together to a clone. When LARRY barcodes were merged, cells were assigned to the merged clone if any constituent LARRY barcode was observed.
Data integration and annotation of cell states
We constructed Seurat61 objects for each of the scTAM-seq samples individually using the binary DNA methylation matrix. To integrate all the samples from experiments M.1–M.3, we used Seurat’s IntegrateData62 function. Then we used Seurat’s standard workflow without normalization to obtain a low-dimensional representation of our data using UMAP. We removed cells in low-density parts of the UMAP because we found that these cells were of lower quality using the non-digested control amplicons. To annotate the cell-type clusters we obtained as result of the Seurat workflow, we inspected the expression of surface proteins, the DNA methylation states of CpGs in bulk data and the DNA methylation states of important lineage-specific transcription factors. To compare single-cell to bulk DNA methylation we computed the relative methylation state by dividing the average methylation state of all CpGs in the given group of bulk data (for example, HSC-specific) by the mean methylation state of all CpGs. To that end, we performed differential analysis for each cell-type cluster individually and selected CpGs with a log fold change larger than 1 for each cluster. For those sites, we investigated whether they are in the vicinity (100 bp) of any of the 39 transcription factors in Supplementary Table 5 and computed enrichment P values with the Fisher exact test. A full vignette is available from GitHub (https://github.com/veltenlab/EPI-clone).
All remaining experiments (M.4–M.8) were analysed without batch correction, as the samples were processed as single batches. Annotation of the cell-type clusters was performed in dynamic CpG space (see below) using bulk methylation values, demethylation of TFBSs and surface-protein expression. In addition to this information, for experiments M.7 and M.8, we projected cell-type labels from the initial analysis (experiments M.1–M.3) using scmap63. EPI-Clone was then used with the standard parameters as described below.
For processing the protein data of scTAM-seq, we used the centred-log-ratio (CLR) normalization methods. To generate a low-dimensional representation of the protein data only, we opted to use SCTransform64, which produced an improved cell-state resolution.
For the scRNA-seq dataset, we used cellranger to generate transcriptomic and surface-protein count matrices, which were used as input to Seurat. Harmony65 was used for batch integration and the cell-type annotation was performed using known haematopoietic marker genes together with the expression of surface proteins.
EPI-Clone
The EPI-Clone algorithm is divided into three steps: (1) identification of static CpGs, (2) identification of cells from expanded clones and (3) clustering of cells from expanded clones. A detailed, step-by-step vignette is available from GitHub (https://github.com/veltenlab/EPI-clone; v.2.0 used in this article). A brief description is given below.
-
1)
To identify static CpGs, for each combination of CpG and surface protein, EPI-Clone performs a Kolmogorov–Smirnov test to investigate whether cells with methylated CpG differ in surface-antigen expression relative to cells with unmethylated CpG. CpGs with no significant antigen association (determined by the lowest P value for any of the surface proteins) according to a Bonferroni criterion were then selected if their average methylation across all cells was less than 90% but higher than 25% in mouse and higher than 5% in human. In the main LARRY experiment in Figs. 1 and 2, this resulted in the identification of 110 CpGs, which we annotated for enhancer–heterochromatic regions57 and early–late-replicating domains66.
-
2)
To identify cells from expanded clones, cells stemming from an expanded clone should be in a higher density region of the space defined by the static CpGs than cells stemming from non-expanded clones. A density estimate was therefore computed as follows. PCA was performed on all static CpGs from step (1). In the reduced dimensional space obtained by the first n = 100 principal components, the average Euclidean distance to the k = 5 nearest neighbours was determined. Effects of cell state, batch and sequencing depth on this measure of local density were then removed by linear regression. We observed that smoothing the resulting quantity locally over 20 nearest neighbours additionally improved performance. Optimal parameters n and k of this step, as well as the density threshold for a cell to be classified as stemming from an expanded clone, were identified through a systematic grid search on experiment M.1, using LARRY barcodes as a ground truth. Here clones >0.25% in size were defined as expanded.
-
3)
To cluster expanded clones, cells from expanded clones were clustered using the standard Seurat workflow, again in a space spanned by n = 100 principal components.
The parameters of steps (2) and (3) were established on the basis of the original LARRY experiment (M.1: LARRY main experiment) and used for all subsequent analyses of the mouse haematopoietic system (M.2–M.5, M.7 and M.8) without further adjustments. Static CpGs were defined in experiment M.1 and used for all remaining experiments. In particular, the performance on a replicate LARRY ground-truth experiment (M.2) is analysed in Extended Data Fig. 3.
In the native ageing experiment (M.7), we opted for a more conservative threshold for defining large, expanded clones (1%), as native haematopoiesis is more polyclonal than the transplantation setting. This threshold resembled what we found in human native haematopoiesis, using CH mutations and mitochondrial mutations as a partial ground truth (Fig. 4).
For when no or only a partial ground truth was available (mouse endothelia (M.6), see below for more details, and the human analysis), we opted instead for a parameter-free approach to identify expanded clones. We used a recently published clustering method, CHOIR44, which automatically determines clusters that have statistical support in the data. Unlike the density-based criterion, CHOIR does not have free parameters (for example, number of principal components, density threshold, number of nearest neighbours to consider). We confirmed that on the mouse LARRY experiment, CHOIR had a similar quantitative performance to the density-based criterion at optimal parameter values (Extended Data Fig. 3g).
EPI-Clone of endothelial data
As the first step of this experiment (M.6), all CpGs were used for dimensionality reduction and clustering. Consequently, we identified a cluster of contaminating non-endothelial (CD31–) cells that we removed. We then used the dynamic CpGs defined in experiment M.1 to construct a cell-state map of ECs. The CLR-transformed protein levels enabled us to annotate ECs as capillary, Car4 or lymphatic, in concordance with transcriptomic references. Finally, the 110 static CpGs defined in experiment M.1 were used to identify clones in these lung ECs. Binary data were used as input for CHOIR44 using false-discovery rate adjustment. Only clones with a relative clone size greater than 1% are highlighted in the figures. For comparison with transcriptomic data, the Mouse LungMAP31 was downloaded from CELLxGENE Datasets (Mus musculus + Lung + 10×3′ v.2 + Smart-seq2) and subset for adult samples. The lung EC atlas67 was also downloaded (https://endotheliomics.shinyapps.io/ec_atlas/).
EPI-Clone of transplantation
To understand whether EPI-Clone robustly identifies clones before and after transplantation, we investigated replicate 2 (old mouse) of the M.7 experiment together with the transplanted mouse (experiment M.8). Notably, the HSCs that were barcoded with LARRY and used for transplantation were obtained from replicate 2 (old mouse) of experiment M.7 (donor mouse). We then performed EPI-Clone on the combined Seurat object of the transplanted mouse with its donor using the static CpGs identified in experiment M.1 without further adjustments of the parameters. Moreover, and to estimate the false-positive rate of this approach, we performed the same analysis using replicate 1 (old mouse) of experiment M.7. This mouse had no relationship with the transplanted mouse and we would not expect clonal clusters to have cells from both samples to appear from a joint analysis.
Bioinformatic analysis (human)
Data processing, demultiplexing and mutation calling
Data processing followed the methods described for mice. For sample pairs that had been multiplexed into a single Tapestri lane (B.1 and B.5, B.2 and B.4, and A.2 and A.5), vireo53 was used for donor deconvolution based on germline SNVs. SNVs were called with cellsnp-lite using a minimum allele frequency of 0.05 and a count threshold of 5. Donor assignments were validated by detecting the presence of the Y chromosome in cases when male and female donors had been multiplexed, and/or the presence of previously known donor-specific CH mutations (Supplementary Fig. 9).
For samples with previously characterized CH mutations, the mutational status of each cell was determined using a custom script written in pysam. Any cell for which more than 5% of reads covering the relevant genomic site displayed the CH mutation were classified as mutant. Cells with a low number of reads covering the site were excluded, using a read threshold that was determined as a function of total site coverage.
Additional CH mutations were identified using SComatic68 based on the assumption that T cells and B cells are depleted from CH mutations. For that purpose, the BAM files of the TBM cohort were split by cell type. Base counts per cell type were calculated using BaseCellCounter with a minimum mapping quality of 30 and a maximum depth proportional to the number of cells in each group. Beta-binomial parameters were estimated across 35,000 genomic sites to model the distribution of reference and alternate alleles. Final mutation calling was performed using BaseCellCalling, considering all identified cell groups and estimated beta-binomial parameters. Mutations of interest were then identified by comparing T cells to myeloid cells. This strategy led to the identification of CH mutations in donors A.4 and A.7. Finally we repeated the same analysis for the TBM and CD34+ cohorts and comparing cells that EPI-Clone had annotated as expanded clones to cells annotated as stemming from non-expanded clones. This enabled us to identify the CH mutation in donor B.5, and the DNMT3A(C666Y) variant in donor A.4.
Data integration
Unlike in mouse data, data integration across all CpGs in the human dataset did not effectively remove interindividual differences (for example, large CH clones still clustered apart). However, a larger set of CITE-seq antibodies was included in the human cohort. We therefore identified surface-protein associated (‘dynamic’) CpGs across all cells in the TBM cohort and performed data integration using three approaches: CITE-seq data alone, dynamic CpG data alone or a combination of both modalities concatenated into a single feature matrix. All three strategies produced similar results (Extended Data Fig. 7c). Notably, the inclusion of both modalities provided more consistency across donors than dynamic CpGs alone, and was less susceptible to technical variation within the donors than CITE-seq data alone. Data integration was performed using scanorama69, as it offered a higher biological resolution of cell types or cell states compared with Seurat integration. In the TBM cohort, we identified a cluster of overstained cells (positive for all antibodies) that were removed before further analyses.
EPI-Clone applied to human samples
The same strategy was followed as for mouse; however, several adjustments were made as described below.
EPI-Clone analyses were performed while excluding mature T cells and B cells, unless denoted otherwise.
For identification of static CpGs, we proceeded as described above for each donor from cohort A (TBM) individually. We then defined consensus static CpGs as those CpGs that were identified as ‘static’ in at least five donors. Eventually, the same set of 94 consensus static CpGs was used for EPI-Clone analysis in all samples. The use of consensus static CpGs in some donors led to substantial improvements in the performance of EPI-Clone with respect to the ground-truth clonal labels (for example, CH mutations). Moreover, it eliminated the need for a static CpG identification step in future studies, as it established a reference set of static CpGs.
We used CHOIR44 with false-discovery rate adjustment for identifying expanded clones, see the section on EPI-Clone (above)
Analysis of scTAMARA-seq data
For this experiment (X.1), DNA methylation data were projected to the CD34+ reference (Fig. 4b) using scmap63. The RNA-seq reads were processed as previously described45, and data were analysed using default Seurat routines. EPI-Clone was used on the DNA methylation data with identical settings to all other human samples.
Analysis of scTAMito-seq data
For this experiment (X.2), cell types were identified by clustering on all surface antigens. EPI-Clone was then applied using consensus static CpGs. Heteroplasmies of mitochondrial mutations that had previously been identified for that sample using mt-scATACseq46 were called in single cells using pysam by dividing the number of reads supporting the mutant allele by the total number of reads covering the site. Cells with fewer than ten reads on the site were excluded as potential dropout.
Data visualization
Plots were generated using the R packages ggplot2 (ref. 70) and ComplexHeatmap71. Boxplots are defined as follows: the middle line corresponds to the median; the lower and upper hinges correspond to first and third quartiles, respectively; the upper whisker extends from the hinge to the largest value no further than 1.5× the interquartile range (or the distance between the first and third quartiles) from the hinge and the lower whisker extends from the hinge to the smallest value at most 1.5× the interquartile range of the hinge. Data beyond the end of the whiskers are called ‘outlying’ points and are plotted individually. For computing lineage-specific output as shown in Fig. 3, we defined output as the fraction of all HSC/MPP1 or myeloid cells per EPI-Clone cluster compared with all HSC/MPP1 or myeloid cells per experiment. In the bubble plots of Fig. 3 and Supplementary Fig. 7, the radius of the circles scales with the square of frequency.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The following single-cell DNA methylation datasets and scRNA-seq dataset are available as Seurat objects from Figshare: M.1–M.3 (https://doi.org/10.6084/m9.figshare.24204750)72; M.4 (https://doi.org/10.6084/m9.figshare.25472467.v1)73; M.5 (https://doi.org/10.6084/m9.figshare.27917427.v1)74; M.6 (https://doi.org/10.6084/m9.figshare.27960771.v1)75; M.7, replicate 1 (https://doi.org/10.6084/m9.figshare.25472434.v1)76 and replicate 2 (https://doi.org/10.6084/m9.figshare.27917454.v1)77; M.8 (https://doi.org/10.6084/m9.figshare.27917331.v1)78; A.1–A.7 (https://doi.org/10.6084/m9.figshare.25526899.v2)79; B.1–B.5 (https://doi.org/10.6084/m9.figshare.28082048.v1)80; X.1 (https://doi.org/10.6084/m9.figshare.27991574.v1)81; X.2 (https://doi.org/10.6084/m9.figshare.28082066.v1)82; and scRNA-seq (https://doi.org/10.6084/m9.figshare.24260743.v1)83. Count matrices are available from the Gene Expression Omnibus under accession number GSE282971. Raw reads for the mouse experiments are available from the NCBI Sequence Read Archive with BioProject number PRJNA1191391. Raw sequencing data for the human cohort has been deposited into the European Genome-phenome Archive (accession number EGAS00001008056). To address ethics board mandates and patient privacy concerns, access is restricted to research projects in haematology and development of bioinformatic methods, and excludes ancestry research, surname inference and other research. Requests for access need to be addressed to L.V. For comparison of our endothelial data with published data, we downloaded the following data from the CELLxGENE database: https://cellxgene.cziscience.com/collections/48d354f5-a5ca-4f35-a3bb-fa3687502252. The lung EC atlas was downloaded from https://endotheliomics.shinyapps.io/ec_atlas/. Source data are provided with this paper.
Code availability
The code used for processing scTAM-seq data, the EPI-Clone algorithm and generating the figures of the paper is available from GitHub (https://github.com/veltenlab/EPI-clone). Release v.2.0 was used for the work included in this article.
References
Sankaran, V. G., Weissman, J. S. & Zon, L. I. Cellular barcoding to decipher clonal dynamics in disease. Science 378, eabm5874 (2022).
Wagner, D. E. & Klein, A. M. Lineage tracing meets single-cell omics: opportunities and challenges. Nat. Rev. Genet. 21, 410–427 (2020).
Mitchell, E. et al. Clonal dynamics of haematopoiesis across the human lifespan. Nature 606, 343–350 (2022).
Ludwig, L. S. et al. Lineage tracing in humans enabled by mitochondrial mutations and single-cell genomics. Cell 176, 1325–1339 (2019).
Bianchi, A. et al. scTAM-seq enables targeted high-confidence analysis of DNA methylation in single cells. Genome Biol. 23, 229 (2022).
Kasbekar, M., Mitchell, C. A., Proven, M. A. & Passegué, E. Hematopoietic stem cells through the ages: a lifetime of adaptation to organismal demands. Cell Stem Cell 30, 1403–1420 (2023).
Jaiswal, S. & Ebert, B. L. Clonal hematopoiesis in human aging and disease. Science 366, eaan4673 (2019).
Weinreb, C., Rodriguez-Fraticelli, A., Camargo, F. D. & Klein, A. M. Lineage tracing on transcriptional landscapes links state to fate during differentiation. Science 367, eaaw3381 (2020).
Naik, S. H. et al. Diverse and heritable lineage imprinting of early haematopoietic progenitors. Nature 496, 229–232 (2013).
Chan, M. M. et al. Molecular recording of mammalian embryogenesis. Nature 570, 77–82 (2019).
Bowling, S. et al. An engineered CRISPR–Cas9 mouse line for simultaneous readout of lineage histories and gene expression profiles in single cells. Cell 181, 1410–1422 (2020).
Pei, W. et al. Resolving fates and single-cell transcriptomes of hematopoietic stem cell clones by PolyloxExpress barcoding. Cell Stem Cell 27, 383–395 (2020).
Lareau, C. A. et al. Massively parallel single-cell mitochondrial DNA genotyping and chromatin profiling. Nat. Biotechnol. 39, 451–461 (2021).
Weng, C. et al. Deciphering cell states and genealogies of human haematopoiesis. Nature 627, 389–398 (2024).
Chapman, M. S. et al. Mitochondrial mutation, drift and selection during human development and ageing. Preprint at ResearchSquare https://doi.org/10.21203/rs.3.rs-3083262/v1 (2023).
Lareau, C. A. et al. Artifacts in single-cell mitochondrial DNA mutation analyses misinform phylogenetic inference. Preprint at bioRxiv https://doi.org/10.1101/2024.07.28.605517 (2024).
Gabbutt, C. et al. Fluctuating methylation clocks for cell lineage tracing at high temporal resolution in human tissues. Nat. Biotechnol. 40, 720–730 (2022).
Li, L. et al. A mouse model with high clonal barcode diversity for joint lineage, transcriptomic, and epigenomic profiling in single cells. Cell 186, 5183–5199 (2023).
Gaiti, F. et al. Epigenetic evolution and lineage histories of chronic lymphocytic leukaemia. Nature 569, 576–580 (2019).
Chaligne, R. et al. Epigenetic encoding, heritability and plasticity of glioma transcriptional cell states. Nat. Genet. 53, 1469–1479 (2021).
Farlik, M. et al. DNA methylation dynamics of human hematopoietic stem cell differentiation. Cell Stem Cell 19, 808–822 (2016).
Cabezas-Wallscheid, N. et al. Identification of regulatory networks in HSCs and their immediate progeny via integrated proteome, transcriptome, and DNA methylome analysis. Cell Stem Cell 15, 507–522 (2014).
Liu, H. et al. DNA methylation atlas of the mouse brain at single-cell resolution. Nature 598, 120–128 (2021).
Nichols, R. V. et al. High-throughput robust single-cell DNA methylation profiling with sciMETv2. Nat. Commun. 13, 7627 (2022).
Verovskaya, E. et al. Heterogeneity of young and aged murine hematopoietic stem cells revealed by quantitative clonal analysis using cellular barcoding. Blood 122, 523–532 (2013).
Ashcroft, P., Manz, M. G. & Bonhoeffer, S. Clonal dominance and transplantation dynamics in hematopoietic stem cell compartments. PLoS Comput. Biol. 13, e1005803 (2017).
Zink, F. et al. Clonal hematopoiesis, with and without candidate driver mutations, is common in the elderly. Blood 130, 742–752 (2017).
Nam, A. S. et al. Single-cell multi-omics of human clonal hematopoiesis reveals that DNMT3A R882 mutations perturb early progenitor states through selective hypomethylation. Nat. Genet. 54, 1514–1526 (2022).
Hui, T. et al. High-resolution single-cell DNA methylation measurements reveal epigenetically distinct hematopoietic stem cell subpopulations. Stem Cell Reports 11, 578–592 (2018).
Dykstra, B. et al. Long-term propagation of distinct hematopoietic differentiation programs in vivo. Cell Stem Cell 1, 218–229 (2007).
Guo, M. et al. Guided construction of single cell reference for human and mouse lung. Nat. Commun. 14, 4566 (2023).
Pei, W. et al. Polylox barcoding reveals haematopoietic stem cell fates realized in vivo. Nature 548, 456–460 (2017).
Rodriguez-Fraticelli, A. E. et al. Clonal analysis of lineage fate in native haematopoiesis. Nature 553, 212–216 (2018).
Beerman, I. et al. Functionally distinct hematopoietic stem cells modulate hematopoietic lineage potential during aging by a mechanism of clonal expansion. Proc. Natl Acad. Sci. USA 107, 5465–5470 (2010).
Sudo, K., Ema, H., Morita, Y. & Nakauchi, H. Age-associated characteristics of murine hematopoietic stem cells. J. Exp. Med. 192, 1273–1280 (2000).
Rossi, D. J. et al. Cell intrinsic alterations underlie hematopoietic stem cell aging. Proc. Natl Acad. Sci. USA 102, 9194–9199 (2005).
Yamamoto, R. et al. Large-scale clonal analysis resolves aging of the mouse hematopoietic stem cell compartment. Cell Stem Cell 22, 600–607 (2018).
Dykstra, B., Olthof, S., Schreuder, J., Ritsema, M. & de Haan, G. Clonal analysis reveals multiple functional defects of aged murine hematopoietic stem cells. J. Exp. Med. 208, 2691–2703 (2011).
Rodriguez-Fraticelli, A. E. et al. Single-cell lineage tracing unveils a role for TCF15 in haematopoiesis. Nature 583, 585–589 (2020).
Kuribayashi, W. et al. Limited rejuvenation of aged hematopoietic stem cells in young bone marrow niche. J. Exp. Med. 218, e20192283 (2021).
Konturek-Ciesla, A. et al. Temporal multimodal single-cell profiling of native hematopoiesis illuminates altered differentiation trajectories with age. Cell Rep. 42, 112304 (2023).
Säwen, P. et al. Murine HSCs contribute actively to native hematopoiesis but with reduced differentiation capacity upon aging. eLife 7, e41258 (2018).
Jakobsen, N. A. et al. Selective advantage of mutant stem cells in human clonal hematopoiesis is associated with attenuated response to inflammation and aging. Cell Stem Cell 31, 1127–1144 (2024).
Mucke, L. & Ryan Corces, M. CHOIR improves significance-based detection of cell types and states from single-cell data. Nat. Genet. https://doi.org/10.1038/s41588-025-02148-8 (2025).
Lindenhofer, D. et al. Functional phenotyping of genomic variants using multiomic scDNA-scRNA-seq. Preprint at bioRxiv https://doi.org/10.1101/2024.05.31.596895 (2024).
Lareau, C. et al. Codon affinity in mitochondrial DNA shapes evolutionary and somatic fitness. Preprint at bioRxiv https://doi.org/10.1101/2023.04.23.537997 (2023).
Li, X. et al. The comprehensive DNA methylation landscape of hematopoietic stem cell development. Cell Discov. 7, 86 (2021).
Kreger, J., Mooney, J. A., Shibata, D. & MacLean, A. L. Developmental hematopoietic stem cell variation explains clonal hematopoiesis later in life. Nat. Commun. 15, 10268 (2024).
Chen, M., Fu, R., Chen, Y., Li, L. & Wang, S.-W. High-resolution, noninvasive single-cell lineage tracing in mice and humans based on DNA methylation epimutations. Nat. Methods 22, 488–498 (2025).
Aksöz, M. et al. Hematopoietic stem cell heterogeneity and age-associated platelet bias are evolutionarily conserved. Sci. Immunol. 9, eadk3469 (2024).
Scherer, M. et al. EPI-Clone protocol. protocols.io https://doi.org/10.17504/protocols.io.4r3l29dzjv1y/v1 (2025).
Wilkinson, A. C. et al. Long-term ex vivo haematopoietic-stem-cell expansion allows nonconditioned transplantation. Nature 571, 117–121 (2019).
Huang, Y., McCarthy, D. J. & Stegle, O. Vireo: Bayesian demultiplexing of pooled single-cell RNA-seq data without genotype reference. Genome Biol. 20, 273 (2019).
Müller, F. et al. RnBeads 2.0: comprehensive analysis of DNA methylation data. Genome Biol. 20, 55 (2019).
Landau, D. A. et al. Locally disordered methylation forms the basis of intratumor methylome variation in chronic lymphocytic leukemia. Cancer Cell 26, 813–825 (2014).
Scherer, M. et al. Quantitative comparison of within-sample heterogeneity scores for DNA methylation data. Nucleic Acids Res. 48, e46 (2020).
Vu, H. & Ernst, J. Universal chromatin state annotation of the mouse genome. Genome Biol. 24, 153 (2023).
Adelman, E. R. et al. Aging human hematopoietic stem cells manifest profound epigenetic reprogramming of enhancers that may predispose to leukemia. Cancer Discov. 9, 1080–1101 (2019).
Triana, S. et al. Single-cell proteo-genomic reference maps of the hematopoietic system enable the purification and massive profiling of precisely defined cell states. Nat. Immunol. 22, 1577–1589 (2021).
Heimlich, J. B. et al. Multiomic profiling of human clonal hematopoiesis reveals genotype and cell-specific inflammatory pathway activation. Blood Adv. 8, 3665–3678 (2024).
Stuart, T. et al. Comprehensive integration of single-cell data. Cell 177, 1888–1902 (2019).
Hao, Y. et al. Integrated analysis of multimodal single-cell data. Cell 184, 3573–3587 (2021).
Kiselev, V. Y., Yiu, A. & Hemberg, M. scmap: projection of single-cell RNA-seq data across data sets. Nat. Methods 15, 359–362 (2018).
Hafemeister, C. & Satija, R. Normalization and variance stabilization of single-cell RNA-seq data using regularized negative binomial regression. Genome Biol. 20, 296 (2019).
Korsunsky, I. et al. Fast, sensitive and accurate integration of single-cell data with Harmony. Nat. Methods 16, 1289–1296 (2019).
Marchal, C. et al. Genome-wide analysis of replication timing by next-generation sequencing with E/L Repli-seq. Nat. Protoc. 13, 819–839 (2018).
Kalucka, J. et al. Single-cell transcriptome atlas of murine endothelial cells. Cell 180, 764–779 (2020).
Muyas, F. et al. De novo detection of somatic mutations in high-throughput single-cell profiling data sets. Nat. Biotechnol. 42, 758–767 (2024).
Hie, B., Bryson, B. & Berger, B. Efficient integration of heterogeneous single-cell transcriptomes using Scanorama. Nat. Biotechnol. 37, 685–691 (2019).
Wickham, H. ggplot2: Elegant Graphics for Data Analysis (Springer, 2009).
Gu, Z., Eils, R. & Schlesner, M. Complex heatmaps reveal patterns and correlations in multidimensional genomic data. Bioinformatics 32, 2847–2849 (2016).
Velten, L. et al. EPI-Clone datasets M.1-M.3: Single cell targeted DNA methylation profiling of hematopoietic stem and progenitor cells. Figshare https://doi.org/10.6084/m9.figshare.24204750.v2 (2023).
Velten, L. EPI-Clone dataset: Mature cell experiment. Figshare https://doi.org/10.6084/m9.figshare.25472467.v1 (2024).
Scherer, M. et al. EPI-Clone dataset M.5: LARRY mature immune cells. Figshare https://doi.org/10.6084/m9.figshare.27917427.v1 (2024).
Scherer, M. et al. EPI-Clone dataset M.6: endothelial cells (Lung). Figshare https://doi.org/10.6084/m9.figshare.27960771.v1 (2024).
Velten, L. EPI-Clone dataset: Native hematopoiesis, old and young mouse. Figshare https://doi.org/10.6084/m9.figshare.25472434.v1 (2024).
Scherer, M. et al. EPI-Clone dataset M.7: Native hematopoiesis: old and young mouse (replicate 2). Figshare https://doi.org/10.6084/m9.figshare.27917454.v1 (2024).
Scherer, M. et al. EPI-Clone dataset M.8: Transplantation experiment. Figshare https://doi.org/10.6084/m9.figshare.27917331.v1 (2024).
Velten, L. EPI-Clone dataset: Human total bone marrow(A.1-A.7). Figshare https://doi.org/10.6084/m9.figshare.25526899.v2 (2024).
Velten, L. EPI-Clone dataset: Human CD34+ cells (A.1-A.7,B.1-B.5). Figshare https://doi.org/10.6084/m9.figshare.28082048.v1 (2025).
Velten, L. EPI-Clone dataset X.1 : Targeted DNAm+DNA+RNA-seqfrom CD34+ BM cells of a healthy donor. Figshare https://doi.org/10.6084/m9.figshare.27991574.v1 (2025).
Velten, L. EPI-Clone dataset X.2: EPI-Clone + Mitochondrial mutation profiling from PBMCs of a healthy donor. Figshare https://doi.org/10.6084/m9.figshare.28082066.v1 (2025).
Velten, L. et al. EPI-Clone supplementary dataset: Single cell RNA-seq of clonally barcoded hematopoietic progenitors. Figshare https://doi.org/10.6084/m9.figshare.24260743.v1 (2023).
Acknowledgements
We thank staff at Mission Bio for support and at the CRG Core Technologies Programme, specifically to the CRG Genomics Unit for assistance with sequencing and the CRG/UPF Flow Cytometry Unit for flow sorting. Funding for this project was provided to L.V. by an EHA Research Grant award granted by the European Hematology Association, by the Fundación Asociación Española Contra el Cáncer (AECC laboratory grant) and by the the Ministry of Science and Innovation (PID2023-146699NB-I00 funded by MCIN / AEI / 10.13039/501100011033 / FEDER, UE). M.S. was supported through the Walter Benjamin Fellowship funded by Deutsche Forschungsgemeinschaft (DFG, German Research Foundation, reference 493935791) and a postdoctoral fellowship provided by the Dr. Rurainski Foundation for Cancer Research. I.S. was supported through the European Union’s Horizon 2020 research and innovation programme under the Marie Skłodowska-Curie grant agreement no 945352. The project that gave rise to these results received the support of a fellowship to M.M.B. from “la Caixa” Foundation (ID 100010434). The fellowship code is LCF/BQ/DI24/12070016. L.V. acknowledges support of the Spanish Ministry of Science and Innovation through the Centro de Excelencia Severo Ochoa (CEX2020-001049-S, MCIN/AEI /10.13039/501100011033), the Generalitat de Catalunya through the CERCA programme and to the EMBL partnership. A.R.-F has been supported by the Cris Foundation Excellence Award (PR_EX_2020-24), the ERC Starting Grant MemOriStem (101042992), the Spanish National Research Agency (PID2020-114638RA-I00), the Agencia de Gestio d’Ajuts Universitaris i de Recerca (AGAUR, 2017 SGR 1322), and the CERCA Program/Generalitat de Catalunya. A.R.-F. acknowledges support from the Institut Catalá de Recerca i Estudis Avançats (ICREA), the American Society of Hematology (ASH) Scholar Award, the Leukemia Lymphoma Society Special Fellow Career Development Program Award (3391–19), the NIH NHLBI K99/R00 transition to independence award (K99 HL146983), the Ministry of Science Ramon y Cajal Fellowship, and the LaCaixa Junior Fellows Incoming Fellowship. C.A.L. is supported by NIH grants P30CA008748 and R00HG012579. L.S.L. acknowledges supported by grants by the German Research Foundation (DFG), including an Emmy Noether fellowship (LU 2336/2-1), LU 2336/3-1, LU 2336/6-1, STA 1586/5-1, TRR241, SFB1588, and the Heinz Maier-Leibnitz Award. N.A.J. was supported by a Medical Research Council and Leukaemia UK Clinical Research Training Fellowship (MR/R002258/1) and MRC DTP Supplementary Funding 2021. P.V. acknowledges funding from the Medical Research Council Molecular Haematology Unit Programme Grant (MC_UU_00029/8), Blood Cancer UK Programme Continuity Grant 13008, NIHR Senior Fellowship, and the Oxford BRC Haematology Theme.
Author information
Authors and Affiliations
Contributions
M.S., I.S., A.R.-F. and L.V. conceptualized the study. I.S. performed the mouse experiments. C.S.-T. and I.S. generated all sequencing libraries. D.L. and L.M.S. supported the SDR-seq experiment. M.S. analysed the mouse data and developed EPI-Clone. M.M.B. analysed the human data with support from R.B., A.B. and L.C. P.S.S. and A.R.-F. analysed the mouse aged endothelial data. L.V. supervised data analyses, with conceptual input from I.S. and A.R.-F. M.S. and M.B. created the targeting panels with support from R.F. J.R. and S.B.-C. analysed the scRNA-seq data. M.K., N.A.J., V.K., C.A.L., A.T.S., L.S.L., L.N., P.K., S.R. and P.V. provided and characterized the human samples. M.S., I.S., A.R.-F. and L.V. wrote the manuscript with input from all co-authors. A.R.-F. and L.V. supervised all aspects of this work.
Corresponding authors
Ethics declarations
Competing interests
A.R.-F. serves as an advisor for Retro Bio. Parts of this study have been supported with reagents donated by Mission Bio. The other authors declare no competing interests.
Peer review
Peer review information
Nature thanks Elisa Laurenti, Shalin Naik and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Peer reviewer reports are available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data figures and tables
Extended Data Fig. 1 Overview of experimental design and CpG panel.
a. Experimental design of the mouse experiments M.1-M.8. See also Supplementary Table 1. LSK: LIN−SCA1+KIT+, LK: LIN−KIT+. b. Distribution of the CpGs covered by all 663 amplicons in our panel. From this set of amplicons, 453 WSH/DMC/IMR CpGs were selected based on a low dropout in a control experiment, see methods. c. Schematic overview of the CpG selection for scTAM-seq. Bulk DNA methylation data was collected from Cabezas-Wallscheid et al.22. We identified three classes of CpGs, which we included in the final panel design shown in Fig. 1b: DMCs, IMCs, and WSH. DMCs are defined by comparisons between cell types, IMCs are regions with intermediate methylation in HSCs, and WSHs are regions with intermediate methylation in HSCs and a high degree of intra-molecule variability. The lines represent sequencing reads, where filled circles stand for methylated and unfilled circles for unmethylated CpGs, respectively.
Extended Data Fig. 2 Comparison of different data modalities for the identification of cell state (experiment M.1-M.3).
a. UMAP of transcriptomic data from the same cell pool as for DNAm for experiment M.1. b. Confusion matrices between scRNA-seq celltypes and scTAM-seq celltypes (Fig. 1c vs. panel A). To compute the confusion matrix, a random forest classifier was trained to predict cell type from surface antigen expression data, using the scRNA-seq modality. The confusion matrix for that classifier during 10-fold cross validation is shown in the plot on the left. The same classifier was then applied to predict cell type in the scTAM-seq experiment, where the same surface antigens were measured using the same TotalSeq-B cocktail. Label transfer accuracy is shown. c. Integrated UMAP of the LARRY main experiment, replicate, and native haematopoiesis (experiments M.1-M.3) as in Fig. 1c, highlighting the LARRY barcodes and donor mouse. d. UMAP defined only on the dynamic CpGs. The plot shows all 13,885 cells from the experiment M.1 (LARRY main experiment). Indicated in colors are the cell types defined in Fig. 1c. e. Surface protein UMAP of experiment M.1 (13,885 cells) with the cell type labels obtained from the DNA methylation UMAP as shown in Fig. 1c. Protein data was normalized using SCTransform64 prior to generating a low-dimensional representation with PCA and UMAP. f. Expression of selected surface proteins in the protein UMAP. g. Bar chart depicting the percentage of static and dynamic CpGs within early/late replicating domains66, respectively.
Extended Data Fig. 3 Validation of EPI-clone’s capability on a biological replicate (experiment M.2).
a,b,c. Clonal UMAP based on static CpGs as in Fig. 2b, computed for experiment M.2: LARRY replicate experiment. Indicated are the cell state (A) and the LARRY barcode (B). C highlights cells that were selected as part of expanded clones, based on local density in PCA space. d. Receiver-Operating Characteristics Curve characterizing the performance of the local density criterion in selecting expanded clones for the biological replicate. e. Overlap between clones defined using EPI-clone and ground truth labels for the biological replicate. The remark ‘small clones’ indicates all LARRY clones with a relative size less than 0.25%. f. Same UMAP as in Fig. 2b highlighting the LARRY donor labeled by two unique fluorophore sequences. For experiment M.1, two donor mice were sacrificed and HSCs were labeled with LARRY constructs containing a GFP label in one case, and LARRY constructs containing a Sapphire label in the other case. Subsequently, labeled cells from each donor were transplanted into two recipient mice each. Accordingly, the data set contains cells from four mice that contain two sets of clones, labeled with GFP and Sapphire, respectively, see also methods. g. Comparison between the performance of the density-based clustering of EPI-Clone with the performance of CHOIR44, a parameter-free clustering method. Precision and recall were calculated for the identification of cells from expanded (>0.25%) clones. ARI: Adjusted rand index. The results are shown for experiment M.1: LARRY main experiment.
Extended Data Fig. 4 EPI-Clone’s performance in mature myeloid cells (experiment M.4).
a. Overview of the sorting scheme for experiment M.4: Mature myeloid cells. b. UMAP based on dynamic CpGs (defined from experiment M.1) showing the differentiation state of mature myeloid cells and their progenitors. c. Enrichment of CpGs specifically unmethylated in a cell-type cluster according to the vicinity to the annotated TFBS, see also main Fig. 1e. d. Expression of surface proteins in the different cell type clusters for stem-cell-specific markers (KIT, SCA1, CD201) and markers of mature myeloid cells (CD9, CD44). e. UMAP as in B, highlighting relative methylation state of cells across all CpGs that are methylated in HSCs or MPP3/4 in bulk data. See also main Fig. 1d. f. UMAP computed on static CpGs (defined from experiment M.1) with the LARRY barcodes indicated. g. Same UMAP as in F, with the cell states as defined in B indicated. h. UMAP representation as in F visualizing the different cellular compartments including progenitors (LSK, LK) and mature cells from lung and BM/PB. i. Overlap between clones defined using EPI-clone and ground truth clonal labels for the mature myeloid experiment. j. Receiver-Operating Characteristics Curve characterizing the performance of the local density criterion in selecting expanded clones for the mature myeloid experiment. k. Adjusted rand indices quantifying the overlap between EPI-clone clusters and LARRY barcodes stratified by the different cell types identified in B. l. Cell type distribution and clone sizes in different clones identified by EPI-Clone and stratified by cellular compartment m. Number of unique LARRY barcodes per cell type cluster. The elevated number of LARRY barcodes per cell in the macrophage cluster suggests the presence of contaminant DNA from doublets or phagocytosis in this cluster.
Extended Data Fig. 5 Cell type mapping and clonality of lung endothelial cells by scTAM-seq and EPI-clone (experiment M.6).
a. Lung cells were isolated from an old mouse, then purified and sorted to filter out CD45+ cells and enrich for CD31 + , before profiling with scTAM-seq. b. UMAP embedding and low-resolution clustering of endothelial cells using the dynamic CpGs identified in experiment M.1. c. Differential expression analysis of surface markers in the different clusters from panel B. d. CLR-normalized expression values of surface markers across the different clusters. e. Normalized expression of the corresponding genes (scRNA-seq) for endothelial cells from the Mouse LungMAP, only for adult samples31. f. Normalized expression of the corresponding genes (scRNA-seq) for endothelial cells from the lung EC atlas67. g. UMAP computed on static CpGs (identified in experiment M.1). Colors highlight clones identified by EPI-Clone with a relative clone size greater than 1%. h. Barplot of endothelial cell types contributions across clones; again, only EPI-clones with a relative clone size greater than 1% are visualized; numbers in the top of the bars represent the absolute clone size, i.e. number of cells. i. Mutual information between methylation status of all CpGs and the EPI-clones for endothelial and haematopoietic cells.
Extended Data Fig. 6 Transplantation experiment profiling EPI-clones before and after transplantation (experiment M.8).
a. Overview of the experimental design for experiment M.8: transplantation experiment. HSCs from an old donor mouse (100 weeks) were either LARRY-barcoded and transplanted into a recipient mouse or directly used for processing with scTAM-seq/EPI-clone. In the negative control, we performed EPI-clone analysis on a set of unrelated HSCs from an old mouse (100 weeks) and the transplanted mouse. b. Joint EPI-clone clustering of the donor and the transplanted mouse. Highlighted in red are HSCs from the donor mouse. c,d. Same EPI-clone UMAP as in B highlighting the sample origin (C) and the LARRY barcode (D). e. Quantification of the fraction of EPI-clone clones that have at least one HSC from the donor mouse. This would indicate that a progenitor cell of this HSC gave rise to this clone. If a HSC successfully engrafts, it should keep its clonal DNA methylation pattern (i.e., EPI-clone identity) and pass it to all of its progeny. Since all blood progeny in the transplantation setting comes from the transplanted HSCs, the donor HSC giving rise to the blood cells should also be part of the same EPI-clone cluster. We observe that this is the case for the transplantation experiment, but not for clustering together the transplanted mouse with an unrelated, aged mouse (negative control). f. Correlation between the clone sizes observed in the Donor and in the transplanted mouse for the shared EPI-clones. The values indicate the Pearson correlation coefficient and corresponding p-values from a Correlation test. g. Spearman correlation between the clonal output of each clone towards the three main blood lineages compared between the donor mouse and the transplanted mouse. The asterisk indicated p-values below 0.1 from a correlation test.
Extended Data Fig. 7 Application of EPI-Clone to human bone marrow samples.
a. Scheme illustrating selection of target CpGs from bulk whole genome bisulfite sequencing data, see also Methods. DMCs are differentially methylated between cell types, IMCs display intermediate methylation levels in HSCs and IIH are variably methylated across individuals in HSCs. b. Bar chart illustrating the composition of the panel. c. Cell state clustering for the TBM cohort using antibodies, DNA methylation or both modalities. Colors correspond to clustering on the DNA methylation (DNAm)+AB data, see main Fig. 4b for color scheme. UMAPs were computed using data integration by scanorama across donors from the TBM cohort, using the indicated modality. d. Average protein expression levels in the different clusters, for the TBM cohort. e. UMAPs of the CD34+ cohort highlighting the surface expression of various antigens. f. Selection of static and dynamic CpGs for donor A.6, see also main Fig. 1i. g. Scatter plot depicting for all CpGs the average methylation across myeloid cells per donor, as well as the classification of the CpG as static or dynamic. h. CpGs that were classified as static in at least five donors were selected as consensus static CpG and used for the EPI-clone analysis.
Extended Data Fig. 8 Characterization of human EPI-Clones.
a. Static CpG UMAPs and EPI-clone clustering result for donor B.5. Left panel highlights a CH mutation identified in this donor, right panel highlights EPI-clone clusters. b. Scatter plot displaying the percentage of cells from each EPI-Clone displaying CH mutations, for the CD34+ cohort. Dots in colors correspond to EPI-clones dominated by a CH mutation, see Fig. 4c for a color scheme. All donors from the CD34+ cohort with a detected CH mutation are shown. c. Scatter plot displaying the percentage of cells from each EPI-Clone displaying CH mutations, for NK and immature B cells. EPI-Clone was run on all cells except T and mature B cells, but the overlap was computed on NK and immature B cells only. See main Fig. 4c for color scheme. d. Static CpG UMAPs as in main Fig. 4c,d, highlighting NK and immature B cells classified according to CH status. e. Static CpG UMAP computed for all cells (including mature B and T cells) for patient A.4, highlighting T cells classified according to CH status. Mature and immature B cells are also highlighted to demonstrate that mature B and T cells mostly cluster in lymphoid clusters. Barchart depicts precision and recall for the task of classifying T cells as CH or non-CH based on EPI-Clone labels. f. Scatter plot depicting the fraction of the different cell types observed per clone, relative to the fraction of the same cell type observed in non-expanded clones from the same patient. Grey dots correspond to EPI-clones with no known driver mutation. Dots in colors correspond to EPI-clones dominated by a CH mutation, see Fig. 4c for a color scheme. g. Same as F, for cell states within the CD34+ compartment.
Extended Data Fig. 9 scTAMARA-seq enables multiplexed readout out RNA, DNA methylation and genotyping amplicons from the same single cell.
a. Scheme of the method, adapted from45. b. Composition of the panel used, see Supplementary Table 6. RNA-seq amplicons were selected using a scRNA-seq reference59 to identify the set of 120 genes with highest information on cell states in the CD34+ compartment by LASSO regression. c. Scatter plot depicting the number of RNA, DNA methylation (DNAm) and genotyping amplicons observed per cell. d. Boxplot comparing the number of features (RNA species) observed per cell in scTAMARA-seq to the number of features observed in whole transcriptome analysis (WTA) on CD34+ cells for the same 120 genes59. See methods, section Data visualization for a definition of boxplot elements. e. Heatmap depicting correlation in DNA methylation profiles between sample X.1 and the other CD34+ BM donors. f. UMAPs computed on the RNA information from scTAMARA-seq highlighting cell state annotation based on RNA (left) and based on DNAm (right). g. Heatmap depicting scaled expression of marker genes for the different RNA-based cell states.
Extended Data Fig. 10 Comparison of EPI-Clone and mitochondrial lineage tracing by scTAMito-seq.
a. Static CpG UMAP computed on all cells from the patient, highlighting cell types identified using surface antigen expression levels. b. Average coverage in reads per cell for the mitochondrial variants previously described for donor X.246. c. Scatter plot comparing average heteroplasmies for these mutations, as determined by mt-scATAC-seq (reference46) or scTAMito-seq (this study). d. Scatter plot depicting, for all mitochondrial variants, the average heteroplasmy and the statistical association with EPI-Clone. Specifically, a linear model was trained on EPI-Clone clusters to predict heteroplasmy at the single cell level, and the p value from an F-test is shown. e. Heatmap relating the single-cell heteroplasmies of mitochondrial variants to EPI-Clones, for T cells only. The columns correspond to different T cells and the rows comprise mitochondrial mutations measured by scTAMito-seq.
Supplementary information
Supplementary Figures
This file contains Supplementary Figs. 1–10.
Supplementary Table 1
Overview of the experiments and cell pools used as input to scTAM-seq for the different experiments. This table details which cell types and pools of cell types were used as input to scTAM-seq across the different experiments, for both human and mouse samples.
Supplementary Table 2
TotalSeq-B antibodies used for generating surface-protein-expression libraries. This table includes the 20 antibodies used for staining for the expression of surface proteins for the mouse experiments.
Supplementary Table 3
Characterization of mitochondrial variants by mt-scATAC–seq. This table contains information on mitochondrial variants derived from ref. 46.
Supplementary Table 4
List of primers used for constructing and amplifying LARRY barcodes. These primers are used for amplifying LARRY barcodes from a cDNA library and enables the association between a lineage barcode and gene expression.
Supplementary Table 5
List of all amplicons and CpGs covered by the scTAM-seq mouse panel with further annotations. This table comprises 663 amplicons comprising CpGs, their genomic location and further annotation such as vicinity to TFBSs and DNA methylation values in bulk data.
Supplementary Table 6
List of all amplicons and CpGs covered by the scTAM-seq human panel with further annotations. This table comprises (1) a list of primers and corresponding genomic locations for the human scTAM-seq experiments, (2) a list of antibodies for assessing the expression of surface proteins for the human experiments, and (3) the gene-specific primers used for scTAMARA-seq.
Supplementary Table 7
Sequencing statistics for scTAM-seq and scRNA-seq libraries. Shown are the number of sequencing reads generated, the number of reads in cells, as well as the number of cells for all of the experiments performed for both the human and mouse experiments.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Scherer, M., Singh, I., Braun, M.M. et al. Clonal tracing with somatic epimutations reveals dynamics of blood ageing. Nature 643, 478–487 (2025). https://doi.org/10.1038/s41586-025-09041-8
Received:
Accepted:
Published:
Issue date:
DOI: https://doi.org/10.1038/s41586-025-09041-8
This article is cited by
-
Epigenetic clues from cancer’s past foretell its future
Nature (2025)
