
Abstract
Chronic stress response activation impairs cell survival and causes devastating degenerative diseases1,2,3. Organisms accordingly deploy silencing factors, such as the E3 ubiquitin ligase silencing factor of the integrated stress response (SIFI), to terminate stress response signalling and ensure cellular homeostasis4. How a silencing factor can sense stress across cellular scales to elicit timely stress response inactivation is poorly understood. Here we combine cryo-electron microscopy analysis of endogenous SIFI with AlphaFold modelling and biochemical studies to report the structural and mechanistic basis of the silencing of the integrated stress response. SIFI detects both stress indicators and stress response components through flexible domains within an easily accessible scaffold, before building linkage-specific ubiquitin chains at separate, sterically restricted elongation modules. Ubiquitin handover by a ubiquitin-like domain couples versatile substrate modification to linkage-specific ubiquitin polymer formation. Stress response silencing therefore exploits a catalytic mechanism that is geared towards processing many diverse proteins and therefore allows a single enzyme to monitor and, if needed, modulate a complex cellular state.
Similar content being viewed by others


Main
The resilience of metazoan development relies on stress response pathways that mitigate mutational, physiological or environmental challenges to cellular processes3,5,6. Defective mitochondrial protein import activates the stress response kinase HRI, which phosphorylates the translation initiation factor eIF2α to stall the synthesis of mitochondrial proteins7,8,9. In this manner, HRI reduces the cytoplasmic load of aggregation-prone proteins and provides cells with time to restore mitochondrial integrity. HRI is one of four kinases of the integrated stress response that enables cells to overcome a wide range of deleterious conditions1,3,10,11.
As stress responses put core processes on hold, their prolonged activation can trigger cell death and tissue degeneration1,2. To prevent this from happening, cells turn off stress responses using dedicated silencing factors4. After restoration of mitochondrial import, the E3 ligase SIFI marks HRI and its activator DELE1 for proteasomal degradation to terminate eIF2α phosphorylation and restart protein synthesis4. Deletion of the SIFI subunit UBR4 disrupts heart, brain and yolk-sac development, resulting in embryonic lethality12,13, and mutations in UBR4 cause ataxia and early onset dementia14,15,16. UBR4-deficient cells could be rescued by genetic or pharmacological inactivation of HRI4, which showed that stress response silencing is critical for cellular homeostasis.
To ensure timely silencing, SIFI must sense whether cells still experience stress. It accomplishes this task by recognizing mitochondrial precursors that accumulate in the cytoplasm only during stress4,17,18,19, and it also detects cleaved proteins that can be released from stressed mitochondria4,7,8,20. These proteins compete with HRI for access to SIFI, thereby delaying silencing of the stress response until stress has been resolved4. How SIFI can process hundreds of proteins that differ in size from around 50 to 2,500 residues and adopt different conformations is unclear. Indeed, how a single E3 ligase can monitor a global cellular state remains to be elucidated.
Here we combined cryo-electron microscopy (cryo-EM) analyses of SIFI with AlphaFold (AF) modelling and biochemical studies to reveal the molecular basis of mitochondrial stress response silencing. SIFI processes its many substrates using flexible domains within an easily accessible scaffold, while it builds Lys48-linked ubiquitin (Ub) polymers at separate modules centred on a sterically restricted E2. SIFI’s ubiquitylation centres communicate through ubiquitin handover orchestrated by a ubiquitin-like (UBL) domain. Silencing of the integrated stress response therefore relies on a mechanism that couples versatile substrate modification to linkage-specific chain elongation, which allows a single enzyme to process many diverse proteins for timely stress response silencing.
Structure of endogenous SIFI
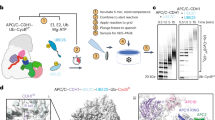
For structure determination, we purified endogenous SIFI by immunoprecipitating its largest subunit, UBR4, and then performing size-exclusion chromatography (Extended Data Fig. 1a). Having ensured that SIFI was active (Extended Data Fig. 1b), we used single-particle cryo-EM to resolve a partial C-terminal map of UBR4 and associated proteins to an overall resolution of 3.1 Å (Fig. 1a, Extended Data Fig. 1c and Supplementary Fig. 3). Purification of SIFI through its subunit KCMF1 led to a complementary map of the N-terminal half of UBR4 at an overall resolution of 3.4 Å (Fig. 1a and Extended Data Fig. 1d). The C-terminal region of UBR4, which includes its hemi-RING21, was not resolved until the SIFI E2 enzyme UBE2A was supplemented before cryo-EM analysis (Extended Data Fig. 1e). We integrated AF models of specific domains into less-well-resolved regions and combined all of the maps to build a near-complete structural model of human SIFI (Fig. 1b,c, Extended Data Table 1, Supplementary Video 1 and Supplementary Fig. 3).

a, Cryo-EM maps of SIFI, highlighting its C-terminal partial map (endogenous SIFI complex; top left; Electron Microscopy Data Bank (EMDB): EMD-46686, contour level 0.12) and N-terminal partial map (immunoprecipitation (IP) of KCMF1; bottom left; EMDB: EMD-46688, contour level 0.07). (A) and (B) denote the two heterotrimers. b, Composite structure of human SIFI built from partial maps (Protein Data Bank (PDB): 9D9Z), including two subunits each of UBR4 (blue), KCMF1 (purple), calmodulin (green) and UBE2A (red). c, Surface representations of the SIFI complex viewed from the N- and C-terminal dimerization regions, as well as views from above and the side of ring’s plane. All components are colour coded as in a and b.
SIFI forms an antiparallel dimer of heterotrimers that comprises two copies each of the E3 ligase UBR4, the E3 ligase KCMF1 and calmodulin22 (Fig. 1b,c and Extended Data Fig. 1f). UBE2A was captured by the C-terminal region of each UBR4 subunit that became ordered in its presence. Together, these proteins assemble into an approximately 1.3 MDa structure that is built around an open twisted-ring scaffold (292 Å × 229 Å) and two arms that extend to the side and span about 356 Å. SIFI is of comparable size to 80S ribosomes and 26S proteasomes (Extended Data Fig. 1g).
SIFI’s scaffold is primarily composed of α-helical armadillo repeats of UBR4 and relies on antiparallel dimer interfaces at its N- and C-terminal regions with buried surface areas of around 2,160 Å2 and 4,212 Å2, respectively (Fig. 1b,c). The N-terminal dimer interface is mediated by a domain-swapped helix from one protomer that bundles with three helices of the second, as well as two Arg residues that interlock the dimer through polar interactions (Extended Data Fig. 2a). The C-terminal interface is centred on extended interactions between helical repeats from each protomer that are cemented by horizontal coiled-coils at the inner side of the scaffold (Extended Data Fig. 2b). Revealing flexibility of the scaffold, the dimerization interfaces show ~10° rotations towards each other, which occur around a hinge close to UBR4 Ala2581, which is mutated in ataxia, and Arg2584, which is altered in cancer23,24 (Extended Data Fig. 2c and Supplementary Video 2).
Calmodulin docks onto the outer rim of the scaffold, close to a hinge around UBR4 Gly4301 that connects the C-terminal dimer interface to the catalytic module of UBR4 (Fig. 2a–c). The C-lobe of calmodulin, which is bound to calcium, captures an exposed UBR4 helix that fits the calmodulin-binding consensus and contains Arg residues mutated in cancer and ataxia24,25,26 (Fig. 2a,b). Hydrophobic calmodulin residues surround Trp4105 of UBR4 in a manner consistent with other calcium-activated structures26,27, and mutation of calcium-binding residues in the C-lobe28 disrupted UBR4 binding (Extended Data Fig. 2d). Calmodulin’s N-lobe forms a bundle with two helices of the UBR4 scaffold (Fig. 2b,c). The N-lobe is not bound to calcium, and calcium-binding residues are not required for UBR4 recognition (Extended Data Fig. 2d). By engaging two distinct regions of UBR4, calmodulin creates an intramolecular bridge that stabilizes the UBR4 lid to anchor KCMF1 to the scaffold, as described below. These findings may explain why deletion of UBR4 residues that include its exposed helix blocked calmodulin integration into SIFI and prevented stress response silencing4,29.

a, Calmodulin (green; CaM) binds to the outer rim of the SIFI scaffold above the C-terminal UBR4 dimerization interface. b, The N-terminal lobe of calmodulin (green) forms a helix bundle with UBR4 (light grey), while the Ca2+-bound C-terminal lobe of calmodulin engages a calmodulin-binding motif of UBR4 (gold), situated atop the UBR4 lid (blue grey) that encircles the C-terminal α-helix of KCMF1 (plum). Conserved residues involved in the interaction are highlighted within dashed boxes. c, Calmodulin stabilizes the base of the UBR4 C-terminal catalytic arm, including hemi-RING, UBE2A (coral) and UBL (blue), allowing flexible movement of the arm around residue Gly4301 (red dot) of UBR4. d, Two copies each of SIFI’s protein interaction modules (coloured) are located at the centre of the scaffold (left). Right, magnified view of the DOC2 (brown) and WD40 (teal) domains, and the KCMF1N138–DOC1–UBR subcomplex (plum, blue and yellow). e, The N-terminal ZZ domain of KCMF1 is required for ubiquitylation activity. WT or ΔZZ KCMF1 were purified from ΔUBR4 cells to prevent UBR4-dependent ubiquitylation and incubated with E1, E2 (UBE2D3, UBE2A) and ubiquitin. Activity was detected by formation of high-molecular-mass conjugates using anti-ubiquitin western blotting. Ubiquitylated species (UBI). Similar results were observed in n = 2 experiments. f, C-terminal helix of KCMF1 (plum) is anchored within α-helical bundles of UBR4’s armadillo repeats (light grey) through the UBR4 lid (blue grey). A conserved Phe319 of KCMF1 (plum), surrounded by three Trp residues of UBR4, is highlighted. g, Deletion or mutation (R316E/F319E/L323E/L325E) of the C-terminal helix (CTH mutant) impedes KCMF1 integration into SIFI, as shown by KCMF1–Flag affinity purification and detection of UBR4. A Q226/228/229/231/233E mutation in an unresolved linker of KCMF1 (mutLINK) did not affect integration into SIFI. ABHD10 binds to the ZZ domain of KCMF1 and was used as a positive control. Similar results were observed in n = 2 experiments. Gel source data are provided in Supplementary Fig. 1.
Tethered to the scaffold’s interior are the UBR box, the WD40 repeat and two DOC-homologous β-sandwich domains of each UBR4 subunit (Figs. 1b and 2d). Their lower local resolution indicates that these modules are flexibly attached to the scaffold (Extended Data Fig. 2e). The WD40 repeats reside symmetrically on either side of the ring, where they are supported by a helix protruding from the scaffold. A second short helix is predicted to plug each β-propeller using loops that often recognize binding partners of WD40 repeats (Extended Data Fig. 2f). The DOC1 domains are positioned near each β-propeller and engage the UBR box of UBR4 and the N-terminal region of KCMF1 to form a three-component subcomplex, referred to as the KCMF1 module, that fits well into the local ~16 Å map (Fig. 2d). Indicative of its inherent flexibility, 3D classification analyses indicated that this subcomplex can adopt distinct positions on SIFI (Supplementary Video 1). The DOC2 domains reside next to the KCMF1 module, where they combine a β-sandwich characteristic of DOC domains with two exposed Zn2+-coordinating loops to form a potential protein-binding interface (Extended Data Fig. 2g). Cross-linking mass spectrometry (MS) confirmed the position of these interaction modules within the centre of SIFI (Extended Data Fig. 2h).
SIFI possesses two subunits, KCMF1 and UBR4, with unconventional E3 ligase motifs. In KCMF1, we found that the N-terminal ZZ domain, which shares structural similarity to RING domains, supports ubiquitylation (Fig. 2e). This domain and two predicted C2H2-type zinc fingers are near the centre of the scaffold, where they bind to the DOC1 domain of UBR4, which has been shown to be required for KCMF1 binding4 (Fig. 2d). Following an unresolved linker that is predicted to adopt a flexible conformation and of which mutation impacted neither SIFI binding nor substrate degradation (Fig. 2g and Extended Data Fig. 3a,b), KCMF1 is further anchored above the C-terminal UBR4 dimer interface through a helix that is enclosed by the UBR4 lid (Fig. 2f). The UBR4 lid engages the KCMF1 helix through extensive van der Waals and polar interactions, including a group of Trp residues that encircle the conserved Phe319 of KCMF1 (Fig. 2f and Extended Data Fig. 3c). Underscoring the importance of these features, mutation or deletion of the N- or C-terminal regions of KCMF1 impaired its retention in SIFI (Fig. 2g and Extended Data Fig. 3d).
The ubiquitylation modules of UBR4 are found within the peripheral SIFI arms that make an approximately 90° turn to extend about 166 Å above the scaffold21 (Fig. 1b,c). These catalytic centres include a ten-turn α-helix, a hemi-RING and a UZI domain in which several ataxia mutations are found16,24. On the basis of X-ray structures and AF models21, the α-helix binds to the backside of the E2 and, together with the hemi-RING, captures UBE2A in a tight embrace that probably explains why this region was only visible when SIFI was incubated with its E2. Close to this module is a UBL domain that is tethered to the UBR4 scaffold through long linkers rich in Asp and Glu residues (Fig. 2c). As described below, the UBL domain has an important role in ubiquitin chain formation.
Together, our structural analyses reveal that SIFI contains several protein interaction modules within a flexible and easily accessible scaffold. The N-terminal, RING-like domains of its two KCMF1 subunits are located at the centre of this structure, while two additional ubiquitylation modules reside within the peripheral UBR4 arms. With four catalytic modules and a dynamic architecture, SIFI appears to be optimally configured to process many substrates for efficient stress response silencing.
Substrates bind at the SIFI centre
To prevent premature stress response silencing, SIFI must survey an entire cell to detect whether stress is still present. To this end, SIFI processes unimported or cleaved proteins that only accumulate in the cytoplasm during stress4, but how it engages its diverse substrates is unclear. In the cryo-EM map of endogenous SIFI, we noticed a low-resolution density that was attached to KCMF1 and could not be ascribed to SIFI components (Fig. 3a). To identify this factor, we compared SIFI immunoprecipitates from wild-type (WT) and ΔKCMF1 cells using MS and found that two mitochondrial proteins, ABHD10 and NIPSNAP3A, were lost in the absence of KCMF1 (Extended Data Fig. 4a and Supplementary Table 1). The same proteins were found to bind to KCMF1 in ΔUBR4 cells (Extended Data Fig. 4b and Supplementary Table 1). AF models showed that ABHD10 dimers fit well into the central density of the cryo-EM map (Fig. 3a). Both ABHD10 and NIPSNAP3A were ubiquitylated by SIFI (Extended Data Fig. 4c,d), showing that these proteins were substrates, rather than subunits, of the E3 ligase.

a, Overview of SIFI, highlighting a low-resolution density ascribed to ABHD10 dimers, with an ABHD10 AF2 model fitted in the map (Gaussian filter width 2.0, contour 0.052). b, AF2 model of ABHD10’s cleaved N terminus (cABHD10) fitting into the N-degron-binding pocket within the ZZ domain of KCMF1. c, ZZ-domain deletion or Asp29/Asp31 mutation in KCMF1 ablates recognition of ABHD10–HA, as seen by KCMF1–Flag immunoprecipitation from ΔUBR4 HEK293T cells. The experiment was performed once. d, Diagram of cross-linking MS analysis of the SIFI–HRI–Ub complex, highlighting cross-linked residues from UBR4, KCMF1 and HRI–Ub (left). Right, model of dimerized HRI–Ub placed at SIFI’s central cavity, with HRI located near to the DOC2 and KCMF1N138 domains, and fused ubiquitin near its cross-linked regions (red, salmon: cross-linked residues determined by MS). e, Conformational change of the KCMF1N138–DOC1–UBR subcomplex after SIFI binding to HRINT. f, SIFI was purified from WT or ΔDOC2 UBR4 cells and incubated with a fluorescently labelled MTS peptide, E1, E2 (UBE2D3 and UBE2A) and ubiquitin. Similar results were observed in n = 3 experiments. g, Deletion of the DOC2 domain in endogenous UBR4 stabilizes HRI to a similar extent to complete UBR4 inactivation. HRI stability was assessed by flow cytometry after HRI-GFP::mCherry expression. Similar results were observed in n = 3 experiments. h, DOC2 deletion leads to integrated stress response hyperactivation after mitochondrial stress induced by sodium arsenite (SA), as seen by induction of ATF4 using western blotting. Similar results were observed in n = 2 experiments. i, UBR4ΔDOC2 cells are hypersensitive to sodium-arsenite-induced stress, dependent on the integrated stress response. As indicated, the integrated stress response was inhibited using ISRIB. The fitness of GFP-labelled WT and mCherry-labelled UBR4ΔDOC2 cells was assessed through cell competition. n = 3 biologically independent replicates are presented together with the median of these experiments. Gel source data are provided in Supplementary Fig. 1.
SIFI recognized only ABHD10 variants that could be imported into mitochondria (Extended Data Fig. 4e). Instead of detecting the presequence, SIFI bound to ABHD10 that was released from mitochondria during cell lysis and possessed a new N terminus resulting from cleavage by mitochondrial presequence peptidase (Extended Data Fig. 4f). MS analyses of SIFI-bound ABHD10 revealed an N-terminal lysine (Extended Data Fig. 4g and Supplementary Table 1), a residue known to act as an N-degron30. SIFI recognized cleaved ABHD10 through the ZZ domain of KCMF1 (Fig. 3a,b and Extended Data Fig. 3d), which shows structural similarity to a domain in p62 that binds to N-degrons31 (Extended Data Fig. 4h). AF models showed that the processed ABHD10 N-termini fit well into the N-degron pocket of KCMF1 (Fig. 3b), which was validated by mutation of Asp residues in KCMF1 that ablated ABHD10 recognition (Fig. 3c). The fortuitous co-purification of ABHD10 dimers, which can access both ZZ domains at the centre of SIFI and therefore stably interact with the E3 ligase, showed how SIFI detects a set of N-degron targets. Note that DELE1 contains an N-degron that is produced by stress-induced cleavage through OMA14 and, as indicated by AF, fits well into the N-degron pocket of KCMF1 (Extended Data Fig. 4i).
To understand the recognition of HRI and mitochondrial presequences, we incubated SIFI with the degron-containing N-terminal domain of HRI (HRINT)4. To increase substrate solubility and provide reactive Lys residues for cross-linking, we fused HRINT to ubiquitin (HRINT–Ub). Cross-linking MS indicated that SIFI recognizes HRINT–Ub at the centre of its scaffold, close to the DOC2 domain and the C-terminal dimer interface of UBR4 (Fig. 3d). The binding of HRINT–Ub caused a substantial shift in the position of the KCMF1 module, while the density ascribed to ABHD10 was strongly reduced (Fig. 3e and Extended Data Fig. 5a). Reflecting the converging degrons in HRI and unimported mitochondrial proteins4, we observed similar conformational changes if SIFI was cross-linked to full-length HRI immunoprecipitated from cells or degron peptides derived from either HRI or mitochondrial presequences (Extended Data Fig. 5a). The architectural rearrangements caused by HRI or presequence binding underscore the flexibility of the central substrate-binding scaffold of SIFI (Supplementary Video 3). Although the resolution of these structures did not permit us to visualize the respective degrons, these results suggested that HRI and mitochondrial presequences bind close to UBR4’s DOC2 domain, and therefore at a different site compared with ABHD10.
To assess the role of UBR4’s DOC2 domain in stress response silencing, we excised it from all UBR4 loci. Affinity purification coupled to MS revealed that SIFI(ΔDOC2) was unable to engage mitochondrial proteins, including those with an MTS, during stress (Extended Data Fig. 5b and Supplementary Table 1). The binding of UBR4 to KCMF1 or calmodulin was not affected by the DOC2 deletion. SIFI(ΔDOC2) was strongly impaired in catalysing the ubiquitylation of mitochondrial presequences (Fig. 3f), while it retained its ability to modify the N-degron substrate ABHD10 (Extended Data Fig. 5c). Accordingly, the DOC2 deletion stabilized HRI, DELE1 and mitochondrial precursors to a similar extent to UBR4 inactivation (Fig. 3g and Extended Data Fig. 5d), resulting in increased stress signalling (Fig. 3h and Extended Data Fig. 5e). SIFI(ΔDOC2) cells also showed a strong fitness defect when experiencing mitochondrial stress, which was rescued by HRI deletion or treatment with the stress response inhibitor ISRIB (Fig. 3i and Extended Data Fig. 5f). The DOC2 domains at the centre of SIFI therefore have an important role in degrading HRI and mitochondrial precursors to ensure stress response silencing.
SIFI also contains a UBR box that interacts with N-degron peptides, albeit with comparably low affinity32,33,34,35. The UBR box in UBR4 is not required for HRI or DELE1 degradation, and it does not contribute to stress response silencing4. While the UBR box was not occupied in our structure (Extended Data Fig. 6a), its peptide-binding pocket is accessible (Extended Data Fig. 6b) and would place targets close to those bound through the ZZ and DOC2 domains (Extended Data Fig. 6c). While further studies are needed to investigate proteins processed through SIFI’s UBR box, our analyses suggest that SIFI recognizes all known targets at the centre of its flexible scaffold, where they occupy overlapping space and could therefore compete with DELE1 and HRI to ensure timely stress response silencing.
Distinct modules for chain formation
As substrates bind next to the N-terminal domains of KCMF1 (Fig. 4a), KCMF1 might help to transfer the first ubiquitin, a reaction that must accommodate the diverse sequence context of Lys residues in SIFI’s many targets. Loss of KCMF1 indeed impaired attachment of the first ubiquitin (Fig. 4b), which was mitigated when we bypassed chain initiation by fusing ubiquitin to a target (Fig. 4c). In cases in which initiation could occur, SIFI(ΔKCMF1) was able to build short ubiquitin chains that were connected through the correct Lys48 linkage (Extended Data Fig. 7a). KCMF1 therefore supports substrate modification but does not determine the linkage specificity of chain formation. Affinity purifications from ΔUBR4 cells showed that KCMF1 can modify ubiquitin independently of a particular lysine (Extended Data Fig. 7b). When encountering an internal subunit of a growing chain, KCMF1 can therefore branch off new conjugates (Extended Data Fig. 7c), which is equivalent to initiating a new chain off an existing polymer4,17.

a, Surface representation of SIFI with DOC2 (orange), KCMF1N138 (plum), UBE2A (coral) and the hemi-RING domain (blue grey) highlighted. b, A time-course ubiquitylation assay of a fluorescently labelled presequence peptide by SIFI or SIFI(ΔKCMF1) indicates that KCMF1 is required for the transfer of the first ubiquitin. Reactions were performed in the presence of ubiquitin(K48R) to prevent chain elongation. Similar results were observed in n = 2 experiments. c, Fusion of one or two ubiquitin molecules to HRINT (HRI-UB; HRI-UB2) overcomes the defect of SIFI(ΔKCMF1) in catalysing ubiquitin chain formation. 35S-labelled HRINT–SUMO (the SUMO tag was used for solubility) was incubated with WT SIFI or SIFI(ΔKCMF1) and ubiquitylation was followed by autoradiography. Similar results were observed in n = 2 experiments. d, AF2 model of UBE2A bound by the C-terminal region of UBR4 through three interfaces (middle). Mutations introduced to the interfaces are highlighted. Left, the interaction network between UBE2A and the backside binding helix of UBR4. Right, interactions near the N-terminal helix of UBE2A. e, Mutations in UBE2A that disrupt binding to the backside helix or the lock loop of UBR4, respectively, prevent stable integration of UBE2A into SIFI, as revealed by UBR4 affinity purification and western blotting using specific antibodies. The experiment was performed once and validated by in vitro ubiquitylation. f, UBE2A mutations impair MTS peptide ubiquitylation. Chain initiation occurred through an E2 that co-purified with SIFI. The experiment was performed once and validated with co-immunoprecipitation. Gel source data are provided in Supplementary Fig. 1.
With KCMF1 supporting initiation, we hypothesized that chain elongation relies on the catalytic modules provided by UBR436. UBR4 acts with only UBE2A or UBE2B21, and co-depletion of these E2 proteins stabilized HRI and increased stress signalling (Extended Data Fig. 7d,e). To test whether UBR4 drives chain elongation, we assessed the role of E2 enzymes in SIFI-dependent ubiquitylation, making use of the observation that SIFI co-purified with an E2 that could initiate some chains if supplied with E1 and ubiquitin. While addition of UBE2D3 increased the abundance of short chains, indicative of a role in initiation, supplementing SIFI with the UBR4-specific UBE2A resulted in selective chain extension (Extended Data Fig. 7f).
UBR4 recruits UBE2A through a three-sided embrace that to our knowledge has not been seen for other E3 ligases (Fig. 4d). While the hemi-RING of UBR4 binds to the N-terminal α-helix of UBE2A, as noted previously21, a long UBR4 helix and a short β-sheet together engage the backside of the E2. The helix connects to a UBR4 loop that contacts the C-terminal α-helix of UBE2A (Fig. 4d). Mutation of UBE2A residues at each interface prevented integration into SIFI and impaired chain elongation (Fig. 4e,f), showing that the E2 embrace is required for SIFI activity.
UBE2A mutations that cause the neurodevelopmental Nascimento syndrome map to each part of the E2 embrace37,38,39 (Fig. 4d): Arg7 of UBE2A faces the UBR4 hemi-RING; Arg11 binds to the loop that connects the hemi-RING to the backside helix; Gly23 is next to the backside β-sheet of UBR4; and the C-terminal helix of UBE2A, of which the deletion was first noted in patients with Nascimento syndrome39, engages the UBR4 loop. Mutants of Arg7, Arg11 or Gly23 were impaired in binding to SIFI and catalysing chain extension (Extended Data Fig. 8a,b), highlighting the physiological importance of the E2 embrace and implying that defective stress response silencing contributes to symptoms of patients with Nascimento syndrome.
Ubiquitin handover
As UBE2A lacks linkage specificity21,40,41, additional factors must encode SIFI’s preference for ubiquitin Lys48. In between initiation and elongation modules, SIFI contains a UBL domain (Fig. 5a). 1H-15N heteronuclear single quantum coherence (HSQC) nuclear magnetic resonance (NMR) spectroscopy showed that this domain binds to ubiquitin with a KD of 124 μM, an affinity sufficient to capture substrate-attached ubiquitin while still enabling processive chain formation (Fig. 5b). This interaction centres on UBR4 Met4444, Leu4447 and Arg residues mutated in cancer cells, which engage a surface centred on Leu8 of ubiquitin (Fig. 5b,c and Extended Data Fig. 8c,d). Notably, AF models showed that the UBL domain orients ubiquitin so that a small movement of the peripheral SIFI arms, as implicated by the flexibility of the UBE2A module (Supplementary Video 4), is sufficient to present Lys48 to the active site of UBE2A (Fig. 5d). The ε-amino group of Lys48 in UBL-bound ubiquitin is guided towards the catalytic Cys88 of UBE2A by Tyr82 and Ser120 of the E2, which are essential for chain elongation without affecting SIFI-binding (Extended Data Fig. 8e,f). These results explain observations that Ser120 of UBE2A is required for substrate ubiquitylation42, and strongly suggest that the UBL domain hands substrate-attached ubiquitin over to UBE2A for Lys48-specific chain formation.

a, SIFI’s UBL domain (blue) is located in between KCMF1 (pink) and the UBR4–UBE2A chain elongation centre (coral/blue grey). b, Overlaid 1H-15N HSQC NMR spectra of 15N-labelled ubiquitin titrating unlabelled UBL domain (left). Right, 1H-15N NMR spectroscopy titration analysis of unlabelled UBL domain and 15N-labelled ubiquitin. CSP, chemical shift perturbations. c, Model of the UBL–ubiquitin interaction, highlighting interface residues affected in NMR binding experiments. d, AF3 model illustrating the C-terminal conformational change of UBR4 that brings UBE2A (coral) and donor Ub close to UBL (blue) and acceptor Ub (left). Right, magnified view that highlights Gly76 of donor Ub (light pink), Lys48 of acceptor Ub (dark pink), and Ser120 and Tyr82 of UBE2A. e, In vitro ubiquitylation of a fluorescently labelled MTS peptide shows that SIFI(ΔUBL) is deficient in ubiquitin chain elongation, but not in initiation. Similar results were observed in n = 2 experiments. f, In vitro ubiquitylation of 35S-labelled HRINT–SUMO shows that the UBL domain is required for chain elongation. Similar results were observed in n = 3 experiments. g, HRI stability was analysed by flow cytometry using HRI-GFP::mCherry. Similar results were observed in n = 3 experiments. h, Endogenous mutation of key UBL residues stabilizes HRI to the same extent as complete UBR4 inactivation. Similar results were observed in n = 2 experiments. i, WT or UBR4ΔUBL cells were exposed to sodium arsenite, and ATF4 levels were monitored by western blotting. Similar results were observed in n = 2 experiments. j, Cell competition assay with sodium arsenite and ISRIB, performed as described above. n = 3 independent replicates shown with the median of these experiments. k, Model of SIFI-dependent substrate ubiquitylation. Substrates bind within the central cavity of SIFI. Ubiquitin chain initiation is mediated dependent on the ZZ-type domain of KCMF1, before a UBL domain hands substrate-attached ubiquitin over to UBE2A for Lys48-linkage specific chain elongation. Gel source data are provided in Supplementary Fig. 1.
We excised the entire UBL domain or mutated two residues at the interface with ubiquitin (Met4444/Leu4447) at all UBR4 loci. Affinity purifications confirmed that SIFI remained intact (Extended Data Fig. 9a,b and Supplementary Table 1). Notably, SIFI complexes containing UBR4(M4444A/L4447E) or UBR4(ΔUBL) were strongly compromised in chain elongation, while transfer of the first ubiquitin was not affected (Fig. 5e,f and Extended Data Fig. 9c,d). Deletion of the UBL domain also prevented chain elongation if chain initiation had been accomplished before SIFI addition (Extended Data Fig. 9e). HRI, DELE1 and mitochondrial precursors were stabilized by UBL mutation or deletion to the same degree as in ΔUBR4 cells (Fig. 5g,h and Extended Data Fig. 9f,g), leading to increased stress signalling (Fig. 5i and Extended Data Fig. 9h–j). Cells expressing SIFI without an intact UBL domain accordingly revealed a strong fitness defect when experiencing import stress, which was rescued by HRI loss or ISRIB (Fig. 5j and Extended Data Fig. 9k,l). We conclude that the UBL domain hands substrate-attached ubiquitin over to UBE2A for chain elongation. Coordination between flexible initiation and linkage-specific elongation modules allows SIFI to decorate many diverse proteins with a specific ubiquitin tag, a prerequisite for timely and efficient stress response silencing.
Discussion
Silencing of the integrated stress response requires that a single enzyme, SIFI, can sense stress across cellular scales. SIFI indeed processes a large array of mislocalized or cleaved proteins that accumulate in the cytoplasm only during stress and competitively inhibit ubiquitylation of the stress response kinase HRI4,14,17,20,32,43. Our study shows that SIFI manages such diverse proteins by combining flexible substrate-modification modules with ubiquitin handover to sterically restricted elongation centres (Fig. 5k).
SIFI binds to its targets within an easily accessible scaffold that also contains modules required for ubiquitin chain initiation. These SIFI domains show high degrees of flexibility that suggest that that they can adapt to targets ranging in size from around 5 to 250 kDa. Even if substrates are recognized through distinct degrons, they occupy overlapping space on SIFI, which provides a rationale for their competition with HRI as required for timely stress response silencing.
Elongation of Lys48-linkage specific polymers occurs at separate sites in the peripheral SIFI arms. Contrasting the flexibility of its initiation modules, SIFI’s elongation centres are built around an E2, UBE2A, that is captured by SIFI on three sides. UBE2A residues at each interface are required for chain extension and mutated in Nascimento syndrome, showing that the E2 embrace is critical for ubiquitylation. The elongation module is therefore sterically restricted. Given that E1 and E3 bind to overlapping sites on E2s44,45, it will be interesting to see how UBE2A is recharged with ubiquitin for efficient chain extension.
Although it was known that ubiquitin chain initiation and elongation can be executed by distinct enzymes46,47, SIFI illustrates how these activities are coordinated with each other by ubiquitin handover through a UBL domain that is tethered to the scaffold by long linkers. The UBL domain orients substrate-attached ubiquitin towards UBE2A so that only Lys48 is available for chain extension, a feature that requires some movement of the SIFI arms around a hinge close to the UBL domain. While distinct from previously described mechanisms of chain elongation48,49, ubiquitin handover is an efficient solution for enzymes that need to modify many substrates with a specific ubiquitin tag. Ubiquitin handover might therefore be a general feature of machines that ubiquitylate diverse targets and, indeed, ubiquitin-binding domains are often found in quality-control E3 ligases50,51,52,53,54.
Its ability to decorate conformationally diverse proteins with a linkage-specific ubiquitin tag suggests that SIFI should be harnessed for the emerging modality of targeted protein degradation. Our structures point to the ZZ, DOC2 and UBR domains of SIFI as potential binding sites for compounds. As SIFI already counteracts protein aggregation and is highly expressed in the brain17,55, it should be particularly tested for applications against neurodegenerative diseases. Our work therefore not only reveals the molecular basis of SIFI activity in the integrated stress response, but also points to strategies to unlock targeted protein degradation to disease areas of high unmet need.
Methods
Data reporting
No statistical methods were used to predetermine sample size. The experiments were not randomized and the investigators were not blinded to allocation during experiments and outcome assessment.
Mammalian cell culture
HEK293T cells were maintained in DMEM + GlutaMax (Gibco, 10566-016) plus 10% fetal bovine serum (VWR, 89510-186). HEK293T cells that were adapted to growth in suspension were cultured in FreeStyle 293 Expression medium (Gibco, 12338026) with 1% FBS. Expi293F cells used for transient transfection for protein purification were grown in Expi293 Expression medium (A1435101, Gibco). All cell lines were purchased directly from the UC Berkeley Cell Culture Facility, authenticated by short-tandem-repeat analysis before freezing stocks (9 February 2022) and were routinely tested for mycoplasma contamination using the Mycoplasma PCR Detection Kit (abmGood, G238). All of the cell lines tested negative for mycoplasma.
Plasmid transfections were performed using polyethylenimine (PEI, Polysciences 239661) at a 1:6 ratio of DNA (in μg) to PEI (in μl at a 1 mg ml−1 stock concentration), Lipofectamine 3000 transfection reagent (Thermo Fisher Scientific, L3000008) or Expifectamine according to the manufacturer’s instructions or ExpiFectamine 293 (Thermo Fisher Scientific, A14524). Lentiviruses were produced in HEK293T cells by co-transfection of lentiviral and packaging plasmids using PEI. Virus-containing supernatants were collected 48 h and 72 h after transfection, and the supernatants were spun down, aliquoted and stored at −80 °C for later use. For lentiviral transduction, 105 cells were seeded into 24-well plates and centrifuged for 45 min at 1,000g after addition of lentiviral particles and 6 μg ml−1 polybrene (Sigma-Aldrich, TR-1003). HEK293T transduced cells were drug-selected 24 h after infection with the following drug concentrations when applicable: puromycin (1 μg ml−1, Sigma-Aldrich, P8833), blasticidin (7.5 μg ml−1, Thermo Fisher Scientific, A1113903).
Plasmids
The list of all constructs used in this study is provided in Supplementary Table 2. Most cloning was performed using Gibson assembly with the HIFI DNA Assembly master mix (NEB, E2621L).
Generation of CRISPR–Cas9 genome-edited cell lines
All CRISPR–Cas9 edited cell lines used in this study were generated from HEK293T cells. sgRNA sequences were designed using the online resource provided by IDT. DNA oligonucleotides for sgRNA and their complementary sequence were phosphorylated (NEB, M0201), annealed and ligated (NEB, M0202) into pX330 (Addgene, 42230). HEK293T cells were cultured in a six-well plate and transfected at 50% confluence with 2 μg of px330 plasmids (and 1 μl of 10 μM single stranded donor oligo when applicable) using Mirus TransIT-293 Transfection reagent (Mirus, MIR2705). At 48 h after transfection, individual clones were expanded in 96-well plates. Homozygous clones were screened by PCR and DNA sequencing and confirmed by western blotting when applicable.
HEK293T Flag–UBR4, ΔUBR4, Flag–UBR4 ΔKCMF1 cells were generated previously4. For the generation of N-terminally Twin-Strep-tagged UBR4 cells (Twin-Strep–UBR4), we used the following sgRNA: 5′-GCGGAAGATGGCGACGAGCGG-3′ and ssODN 5′-CCGGTGGCAAGCCCCCGGAGGGAGCCGCAGTAGTACGACGGAAGATGAGCGCCTGGAGTCACCCTCAGTTTGAGAAAGGCGGAGGTAGCGGAGGTGGCTCTGGCGGAAGCGCCTGGTCACACCCACAGTTCGAGAAGGGCGGAGGTAGCGCGACGAGCGGCGGCGAAGAGGCGGCGGCAGCGGCTCCGGCGCCGGG-3′.
UBR4(ΔUBL) (Δ4342–4434), UBR4(ΔDOC2) (Δ3538–3721) were generated in the Flag–UBR4 background, with the following protospacer sequences that created in-frame deletions: UBR4(ΔUBL): 5′-CCTTGTTCCATGGACACTCG-3′ and 5′-CATTAGTCAGATGCCTCCAA-3′; UBR4(ΔDOC2): 5′-CGGGTTATTACACACCAGGC-3′ and 5′-ATGGGATCCACTGCACAGCA-3′ and the following sODN to repair: 5′-CATGCTAAGACTGGTTTCTTCCTTAGCACTTTGTCTGGCTTAGTGGAGTTTGATGGCTATTACCTGGAGAGCGATCCCTGCCTCGTGTGTAATAACGGCAGCAGCGCAGTGGATCCTATTGAGAATGAAGAAGACCGGAAGAAGGTGAGGCCAGATCTGGCCTAGACTCAGGGCTGTGGCCTTGATCTGGACTTTGGGCA-3′.
UBR4(M4444A/L4447E) cells were generated by nucleofecting Cas9–RNP complexes (sgRNA: 5′-GGAUUGUUUAUCGUGCCCGG-3′) and the ssODN template (5′-AGTCCAGGGACTCAATGAACTCCTCTGTGGCATCGCCCAGCTCCCCCCGGGCACGATAAACAATCCTCATGGGCTCTCCCTGAGCAGAGAAA-3′) into Flag–UBR4 cells using the Lonza 4D-Nucleofector X Unit (program CM130) and the SF Cell Line 4D-Nucleofector X Kit S. For each reaction, 2.5 µl of recombinant Cas9 (40 µM) was incubated with 1.3 µl of sgRNA (100 µM) and incubated for 15 min at room temperature with 4.7 µl of SF solution, after which 1.5 µl of ssODN (100 µM) and 2 × 105 cells in 10 µl of SF solution were added to the RNP complex. Immediately after nucleofection, cells were plated into 12-well plates. The editing efficiency was increased by adding Alt-R HDR Enhancer V2 from IDT for 16 h. Then, at 72 h after nucleofection, the bulk editing efficiency was determined by PCR and DNA sequencing and individual clones were expanded in 96-well plates. Homozygous clones were screened and confirmed multiple times.
Protein expression and purification
To purify the endogenous SIFI complex, HEK293T cells with the UBR4 gene endogenously tagged with a Flag or Strep tag were collected from 150 mm culture dishes or adapted suspension cells. To purify SIFI complex through affinity-tagged KCMF1, Twin-Strep-II-tagged KCMF1 was transiently expressed in Expi293F cells for 48 h before collection. Cells were lysed in cell lysis buffer containing 40 mM HEPES pH 7.5, 150 mM NaCl, 1 mM DTT, 0.1% Nonidet P-40 (NP-40), benzonase nuclease (Millipore-Sigma, 70746-4), proteasome inhibitor carfilzomib and Roche cOmplete protease inhibitor cocktail (Sigma-Aldrich, 11836145001). Cells were lysed for 20 min and homogenized using a Dounce homogenizer. Cell lysates were cleared by centrifugation at 4,000g for 10 min followed by centrifuging at 36,000g for 40 min. The supernatant solution was subsequently collected and incubated with M2 Flag resin (Sigma-Aldrich, A2220) or Strep-Tactin XT 4Flow resin (IBA Lifesciences, 16674714) for 4–6 h at 4 °C. Affinity resin was washed thoroughly with wash buffer (150 mM HEPES pH 7.5, 150 mM NaCl, 1 mM DTT), then eluted with elution buffer (500 μg ml−1 3×Flag peptide (Sigma-Aldrich, F4799) for M2 Flag resin, 50 mM biotin (IBA Lifesciences, 21016005) for Strep-Tactin XT 4Flow resin). Eluted proteins further purified using the Superose 6 10/300 size-exclusion chromatography (SEC) column (Cytiva, 17517201).
Cryo-EM sample preparation
Purified SIFI complex, SIFI–substrate mixture or cross-linked SIFI-substrate mixture samples were concentrated to 2–4 mg ml−1 concentration for cryo-preservation. The concentrated samples were mixed with a final concentration of 0.02% (w/v) fluorinated octylmaltoside (Anatrace) immediately before cryo-freezing to prevent protein denaturation at the air–water interface. Then, 2.6 μl of the sample was applied to a glow-discharged 300-mesh Quantifoil R1.2/1.3 grid and incubated for 15 s before being blotted and plunge-vitrified in liquid ethane, which was cool-protected by liquid nitrogen. Grid freezing was performed using a Mark IV Vitrobot (Thermo Fisher Scientific) system operating at 12 °C and 100% humidity.
Cryo-EM data collection and processing
Cryo-EM data were collected using the 300 kV Titan Krios G3i or G2 electron microscope (Thermo Fisher Scientific) equipped with a BIO Quantum energy filter (slit width 20 eV). Data were collected using SerialEM software56 at a nominal ×105,000 magnification with a pixel size of 1.05 Å per pixel (G3i) or 0.83 Å per pixel (G2). Videos were recorded using a 6k x 4k Gatan K3 Direct Electron Detector operating in super-resolution CDS mode. Each video was composed of 40 subframes with a total dose of 60 e− Å−2, resulting in a dose rate of 1.5 e− Å−2. A total of more than 6,000 videos was recorded for each sample. Data processing, including motion correction, CTF estimation, particle picking, 2D class averaging and 3D refinement, was performed using cryoSPARC v.4.3 workflow57. All videos were 2× binned and patch motion-corrected. After particle picking and several iterations of 2D class averaging, the initial 3D volume was calculated using ab initio 3D reconstruction. Selected particles were then used for non-uniform 3D refinements58. Protein motion and flexibility was calculated using cryoSPARC 3D Flexible Refinement59 (3DFlex).
Model building and structural analysis
Coordinates for the N- and C-terminal halves of SIFI complex were built separately into their respective cryo-EM maps. Coordinates for regions of the EM maps with higher local resolutions, such as the UBR4 α-helical armadillo repeats, were manually built into the map, whereas, for regions with lower local resolutions (~5–8 Å), AF260 models were used and protein secondary structures, including α-helices and β-strands, were fitted into the EM density. For regions with very low resolutions (<8 Å), such as the UBR4 WD40 domain, UBR, DOC1, N-terminal domain of KCMF1, UBL domain and hemi-RING/UBE2A, AF2 models were fitted into the low-resolution density in ChimeraX. Minimal adjustments were done at the secondary-structure level. Helices were adjusted to fit into the map, and amino acid side chains were refined to their preferred rotamers and clashes were minimized. Certain low-resolution models, such as the UBL domain of UBR4, the UBE2A-binding domains and the N-terminal domain of KCMF1, were validated through other biochemical assays such as NMR, binding assays and in vitro ubiquitylation assays. Coordinates of the medium-resolution to high-resolution regions were refined with multiple iterations of PHENIX real-space refinement61 and manual refinement in Coot62. Buried surface area of the protein–protein interface was calculated using PDBePISA63 with a 1.4 Å probe. Potential hydrogen bonds were assigned using the geometry criteria of <3.5 Å separation distance and >90° acceptor–donor–hydrogen (A-D-H) angle. A maximum distance of 4.0 Å was allowed for a potential van der Waals interaction. Structures were further analysed, and structural presentations were prepared using PyMOL v.2 (Schrödinger) and ChimeraX64,65.
MS analysis
MS was performed on immunoprecipitates prepared from forty 15-cm plates of endogenously Flag-UBR4 and Flag-UBR4 ΔKCMF1 HEK293T cell lines or twenty 15-cm plates of WT or ΔUBR4 HEK293T cells transfected with KCMF1-3×Flag DNA. Cells were lysed in lysis buffer (40 mM HEPES, pH 7.5, 150 mM NaCl, 0.2% NP-40, benzonase (Sigma-Aldrich, E1014) and 1× cOmplete protease inhibitor cocktail (Roche, 11836170001) and 1× PMSF), lysed extracts were clarified by centrifugation at 21,000g and bound to anti-Flag M2 affinity resin (Sigma-Aldrich, A2220) for 2 h with rotation at 4 °C. Immunoprecipitates were then washed four times and eluted three times at 30 °C with 0.5 mg ml−1 of 3×Flag peptide (Sigma-Aldrich, F4799) buffered in 1× PBS plus 0.1% Triton X-100. Elutions were pooled and precipitated overnight at 4 °C with 20% trichloroacetic acid. Centrifuged pellets were washed three times with an ice-cold acetone/0.1 N HCl solution, dried, resolubilized in 8 M urea buffered in 100 mM Tris pH 8.5, reduced with TCEP, at a final concentration of 5 mM, (Sigma-Aldrich, C4706) for 20 min, alkylated with iodoacetamide, at a final concentration of 10 mM (Thermo Fisher Scientific, A39271) for 15 min, diluted fourfold with 100 mM Tris-HCl pH 8.5 and digested with 0.5 mg ml−1 trypsin (Promega, v5111) supplemented with CaCl2 (at a final concentration of 1 mM) overnight at 37 °C. Trypsin-digested samples were submitted to the UC Berkeley Vincent J. Coates Proteomics/Mass Spectrometry Laboratory for analysis. Peptides were processed using multidimensional protein identification technology and run on the LTQ XL linear ion-trap mass spectrometer. To identify high-confidence interactors, CompPASS analysis was performed against MS results from unrelated Flag immunoprecipitates performed in our laboratory. KCMF1, ABHD10 and NIPSNAP3A were the only three proteins with more than 10 spectral counts and showing a tenfold or more reduction in Flag–UBR4(ΔKCMF1) compared with Flag–UBR4.
Flag–UBR4, Flag–UBR4(ΔUBL) and UBR4(M4444A/L4447E) MS experiments (Extended Data Fig. 9a,b) were performed from twenty 15-cm plates lysed in lysis buffer (40 mM HEPES, pH 7.5, 150 mM NaCl, 0.2% NP-40, benzonase (Sigma-Aldrich, E1014) and 1× cOmplete protease inhibitor cocktail (Roche, 11836170001), 1× PMSF), clarified by centrifugation at 21,000g, and bound to anti-Flag M2 affinity resin (Sigma-Aldrich, A2220) for 2 h with rotation at 4 °C. Immunoprecipitates were then washed four times with (40 mM HEPES, pH 7.5, 150 mM NaCl, 0.2% NP-40) followed by three washes in PBS, all of the remaining liquid was removed with a crushed gel tip and the beads were flash-frozen in liquid nitrogen.
To monitor mitochondrial proteins bound to SIFI under mitochondrial import stress, fifteen 15-cm plates of subconfluent cells per condition were treated with 10 µM arsenite for 16 h. Cells were co-treated with 200 nM ISRIB to eliminate differences in ISR-activation-induced translational changes. To reduce SIFI occupancy with ABHD10, NIPSNAP3A and other mitochondrial interactors present after lysing mitochondria, cells were collected without freezing cell pellets and lysed in digitonin lysis buffer (40 mM HEPES 7.5, 150 mM NaCl, 10 μg ml−1 digitonin (GoldBio, D-180-2.5), with Roche cOmplete Protease Inhibitor Cocktail (Sigma-Aldrich, 11873580001), carfilzomib (2 μM, Selleckchem, S2853) for 10 min with rotation at 4 °C. Lysed cells were spun down at 2,000g for 5 min at 4 °C and the supernatant was collected. The supernatant was added to equilibrated anti-Flag-M2 Affinity Agarose Gel slurry (Sigma-Aldrich, A2220) and rotated for 2 h at 4 °C. The following wash and elution steps were the same as described above for NP-40 lysis.
Further sample processing was performed at the UC San Diego Proteomics Facility, where the protein samples were diluted in TNE (50 mM Tris pH 8.0, 100 mM NaCl, 1 mM EDTA) buffer. RapiGest SF reagent (Waters) was added to the mix to a final concentration of 0.1%, and the samples were boiled for 5 min. TCEP was added to 1 mM (final concentration) and the samples were incubated at 37 °C for 30 min. Subsequently, the samples were carboxymethylated with 0.5 mg ml−1 of iodoacetamide for 30 min at 37 °C followed by neutralization with 2 mM TCEP (final concentration). The proteins samples were then digested with trypsin (trypsin:protein ratio, 1:50) overnight at 37 °C. RapiGest was degraded and removed by treating the samples with 250 mM HCl at 37 °C for 1 h followed by centrifugation at 14,000 rpm for 30 min at 4 °C. The soluble fraction was then added to a new tube and the peptides were extracted and desalted using C18 desalting columns (Thermo Fisher Scientific, PI-87782). Peptides were quantified using BCA assay and a total of 1 μg of peptides were injected for LC–MS analysis. Trypsin-digested peptides were analysed by ultra-high-pressure liquid chromatography (UPLC) coupled with tandem mass spectroscopy (LC–MS/MS) using nano-spray ionization. The nanospray ionization experiments were performed using a TimsTOF 2 pro hybrid mass spectrometer (Bruker) interfaced with nanoscale reversed-phase UPLC (EVOSEP ONE). The Evosep method of 30 samples per day was performed using a 10 cm × 150 μm reversed-phase column packed with 1.5 μm C18-beads (PepSep, Bruker) at 58 °C. The analytical columns were connected with a fused silica ID emitter (10 μm inner diameter, Bruker Daltonics) inside a nanoelectrospray ion source (captive spray source, Bruker). The mobile phases comprised 0.1% formic acid as solution A and 0.1% formic acid/99.9% acetonitrile as solution B. The MS settings for the TimsTOF Pro 2 were as follows: the dia-PASEF method for proteomics. The values for mobility-dependent collision energy were set to 10 eV. No merging of TIMS scans was performed. The ion mobility (IM) was set between 0.85 (1/k0) and 1.3 (1/k0) with a ramp time of 100 ms. Each method includes one IM window per dia-PASEF scan with variable isolation window at 20 amu segments; 34 PASEF MS/MS scans were triggered per cycle (1.38 s) with a maximum of 7 precursors per mobilogram. Precursor ions in an m/z range of between 100 and 1,700 with charge states ≥3+ and ≤8+ were selected for fragmentation. Protein identification and label-free quantification were performed using Spectronaut 18.0 (Biognosys).
The values obtained with DIA analysis for the Flag–UBR4, Flag–UBR4(ΔUBL) and UBR4(M4444A/L4447E) values are represented for a subset of interactors that were found significantly enriched over the HEK293T background control samples and were previously validated.
For analysis of mitochondrial proteins bound to SIFI under mitochondrial import stress, after DIA analysis, contaminant proteins were excluded from the analysis if they appeared in ≥25% of datasets in the CRAPOME database66. Mitochondrial proteins were defined as such if annotated in MitoCarta3.067. Identified peptides of mitochondrial proteins were run through the MTS prediction tool iMLP: iMTS-L predictor service68 and classified as a potential (i)MTS if there was a clear predicted iMTS-L propensity profile with a score >1. Detected intensities were normalized to bait (UBR4) and log2-transformed fold change values relative to the average DIA quantity of untreated WT samples are displayed as a heat map for all of the mitochondrial proteins and the top 19 abundant non-mitochondrial proteins. Gene-Ontology-term enrichment analysis was performed using Spectronaut 18.0 (Biognosys).
Protein cross-linking
Cross-linking reactions were performed in 40 mM HEPES, pH 7.4, 150 mM NaCl, 1 mM DTT buffer containing 3 mg ml−1 total protein. BS3 (bis(sulfosuccinimidyl)suberate) (Thermo Fisher Scientific, A39266) dissolved in water was added to a final concentration of 0.5 mM to initiate cross-linking. Cross-linking was performed at room temperature for 30 min before quenching by addition of 100 mM final concentration of ammonium bicarbonate. Cross-linked samples were reduced with 10 mM final concentration of TCEP for 1 h at 37 °C, alkylated with 15 mM final concentration of iodoacetamide at room temperature in the dark for 30 min followed by addition of 15 mM final concentration of DTT. Sequencing-grade modified trypsin (Promega, V5111) was added at an enzyme:substrate ratio of 1:15. Tryptic digestion was performed for 6 h at 37 °C before acidification by addition of HCl to a final concentration of 250 mM. Digested samples were centrifuged in a microfuge at 21,000g for 10 min, and the resulting supernatant was transferred to an autosampler vial and stored at −80 °C before analysis using LC–MS/MS.
MS analysis of cross-linked samples
MS and data analysis were based on previously described methods69. Sample digest (5 µl) was loaded onto a PepMap Neo Trap Cartridge (Thermo Fisher Scientific, 174500). The trap was brought online with a Bruker PepSep C18 15 cm × 150 µm, 1.9 µm column (Thermo Fisher Scientific, 1893471) connected to a 5 cm × 20 µm inner diameter Sharp Singularity Fossil Ion Tech tapered tip mounted in a custom constructed microspray source. Peptides were eluted from the column at 0.8 µl min−1 using a 90 min acetonitrile gradient. An Orbitrap Exploris 480 (Thermo Fisher Scientific) was used to perform MS in data-dependent acquisition (DDA) mode using the following parameters. Three methods were used, differing only by MS/MS resolution. The resolution at m/z 200 for MS was 60,000. MS/MS resolution was 15,000, 30,000 or 60,000. Cycle time was 2 s between MS scans. MS scan range was m/z 400–1,600. The automatic gain control was set to standard for MS and MS/MS, and the maximum injection times were set to auto. MS/MS spectra were acquired using an isolation width of 2 m/z and a normalized collision energy of 27. MS/MS acquisitions included +3 to +6 precursor ions and undefined precursor charge states were excluded. Dynamic exclusion was set at 10 s. All spectra were collected in centroid mode.
Acquired spectra were converted to mzML format using ProteoWizard’s msConvert (v.3.0.22335)70. Cross-links were identified from DDA data using the Kojak (v.2.0.3)71 search algorithm with post-processing using Percolator (v.2.08)72 and cross-link data visualization using the ProXL web application73. All data were filtered at a false-discovery rate (FDR) of 5% at the peptide level unless otherwise stated.
Growth competition assays
HEK293T, ΔUBR4, ΔDOC2, UBR4(M4444A/L4447E) and ΔUBL cells were transduced to express either GFP or mCherry, respectively using the lentiviral pLVX-GFP-P2A-Blasticidin or pLVX-mCherry-P2A-blasticidin vector as described previously4.
For drug competition assays, 5 × 104 WT GFP and 5 × 104 ΔUBR4 mCherry or UBR4 domain mutants were mixed in six-well plates. The next day, indicated concentrations of sodium arsenite (Ricca Chemical, 714216), were added for 72 h. The ratio of mCherry+/GFP+ cells was determined on the BD LSRFortessa instrument, analysed using FlowJo v.10.8.1 and normalized to the untreated sample. Gating strategies for flow cytometry analysis are shown in Supplementary Fig. 2.
Drug treatments
For 3 day competition experiments with drug-treated cells, we used the following drug concentrations: 0.5–2 μM sodium arsenite (Ricca Chemical, 714216). For overnight drug treatments we used 5 μM sodium arsenite unless otherwise indicated in the figure legends. To inhibit the proteasome, we used 2 μM carfilzomib (Selleck Chemicals, S2853) for 6 h. ISRIB (Sigma-Aldrich, SML0843) was used at a concentration of 200 nM.
Protein-stability reporter assay
The pCS2+-degron-GFP-IRES-mCherry reporter constructs (ISR, HRI and DELE1) were gene-rated as described previously4 and are listed Supplementary Table 2. Protein stability reporter assays were performed as described previously4. Cells were analysed on either the BD Bioscience LSR Fortessa or LSR Fortessa X20 system and the GFP/mCherry ratio was analysed using FlowJo. Gating strategies for flow cytometry analysis are shown in Supplementary Fig. 2.
Western blotting
For western blot analysis of whole-cell lysates, cells were collected at the indicated timepoints by washing in PBS, pelleting and snap-freezing. Cells were lysed in lysis buffer (150 mM NaCl, 50 mM HEPES pH 7.5, 1% NP-40 substitute) supplemented with Roche cOmplete Protease Inhibitor Cocktail (Sigma-Aldrich, 11836145001), PhosSTOP Phosphatase Inhibitor Cocktail (Roche, 4906837001), carfilzomib (2 μM) and benzonase (EMD Millipore, 70746-4) on ice. The samples were then normalized to protein concentration using the Pierce 660 nm Protein Assay Reagent (Thermo Fisher Scientific, 22660). Then, 2× urea sample buffer (120 mM Tris, pH 6.8, 4% SDS, 4 M urea, 20% glycerol, bromophenol blue) was added to the samples. SDS–PAGE and immunoblotting was performed using the indicated antibodies. Images were captured on the ProteinSimple FluorChem M device.
Small-scale immunoprecipitations
Cells were collected after washing in PBS, pelleted and snap-frozen. Frozen pellets were resuspended in lysis buffer (40 mM HEPES 7.5, 150 mM NaCl, 0.1% NP-40, with Roche cOmplete Protease Inhibitor Cocktail (Sigma-Aldrich, 11873580001), carfilzomib (2 μM, Selleckchem, S2853) and benzonase (EMD Millipore, 70746-4)). The lysates were incubated for 30 min on ice and cleared by centrifugation for 20 min at 21,000g at 4 °C. The supernatants were normalized to volume and protein concentration. Then, 5% of the sample was removed as an input and the sample was added to equilibrated anti-Flag-M2 Affinity Agarose Gel slurry (Sigma-Aldrich, A2220) and rotated for 1–2 h at 4 °C. The beads were washed three times in wash buffer (40 mM HEPES pH 7.5, 150 mM NaCl, 0.1% NP-40) and eluted with 2× urea sample buffer. SDS–PAGE and immunoblotting was performed using the indicated antibodies. Images were captured on a ProteinSimple FluorChem M device.
For immunoprecipitations performed from cells lysed in digitonin lysis buffer, cells were collected after washing in PBS and immediately lysed (never frozen) in digitonin lysis buffer (40 mM HEPES pH 7.5, 150 mM NaCl, 50 μg ml−1 digitonin (Sigma-Aldrich, D141)), with Roche cOmplete Protease Inhibitor Cocktail (Sigma-Aldrich, 11873580001), carfilzomib (2 μM, Selleckchem, S2853) for 10 min with rotation at 4 °C. Lysed cells were then spun down at 2,000g for 5 min at 4 °C and the supernatant was collected, and 5% input was removed. The supernatant was added to equilibrated anti-Flag-M2 Affinity Agarose Gel slurry (Sigma-Aldrich, A2220) and rotated for 1–2 h at 4 °C. The following wash and elution steps were the same as described above for NP-40 lysis. The pellet obtained after centrifugation of the digitonin-lysed cells was also subsequently lysed in NP-40 lysis buffer as described above to break open mitochondria and the sample was collected after centrifugation at 21,000g as the NP-40 fraction.
Antibodies
The following antibodies were used for immunoblot analyses: anti-Flag (mouse, clone M2, Sigma-Aldrich, F1804, 1:1,000), anti-Flag (rabbit, Cell Signaling Technology (CST), 2368, 1:1,000), anti-HA-tag (rabbit, C29F4, CST, 3724, 1:1,000), anti-Strep (strepMAB-Classic, 2-1507-001, Iba Lifesciences, 1:10,000), anti-GAPDH (rabbit, D16H11, CST, 5174, 1:1,000), anti-HSP90β (rabbit, D3F2, CST, 7411, 1:1,000), anti-α-tubulin (mouse, DM1A, Calbiochem, CP06, 1:1,000), anti-UBR4/p600 (rabbit, A302, Bethyl, A302-279A, 1:1,000), anti-UBE2A/B (mouse, G-9, Santa Cruz, sc-365507, 1:150), anti-ATF4 (rabbit, D4B8, CST, 11815S, 1:1,000), anti-EIF2AK1 (rabbit, Proteintech, 20499-1-AP, 1:1,000), anti-KCMF1 (rabbit, Sigma-Aldrich, HPA030383, 1:1,000), anti-NIPSNAP3A (rabbit, Thermo Fisher Scientific, PA5-20657, 1:1,000), anti-ubiquitin (rabbit, CST, 43124, 1:1,000), anti-ABHD10 (rabbit, Thermo Fisher Scientific, PA5-103553, 1:1,000), goat anti-rabbit IgG (H+L) HRP (Vector Laboratories, PI-1000, 1:5,000), Sheep anti-mouse IgG (H+L) HRP (Sigma-Aldrich, A5906, 1:5,000), goat anti-mouse IgG-light-chain-specific HRP conjugated (Jackson Immunoresearch, 115-035-174, 1:5,000).
In vitro transcription/translation of substrates
In vitro synthesized substrates were all cloned into pCS2 vectors containing a SP6 promoter (Supplementary Table 2) and generated using Wheat Germ Extract (Promega, L3260) as previously described4.
In vitro ubiquitylation assays
For in vitro ubiquitylations, human SIFI complex was purified using an endogenous Flag-UBR4 HEK293T cell line. Each in vitro ubiquitylation reaction required material from 2.5 15-cm plates of Flag-UBR4, Flag-UBR4(ΔUBL), Flag-UBR4(M4444A/L4447E) or Flag-UBR4(ΔDOC2) cells. For Flag-UBR4(ΔKCMF1) cells, we used ten 15-cm plates per reaction. Frozen cell pellets were lysed at 4 °C for 30 min in 1 ml of lysis buffer per ten 15-cm plates (40 mM HEPES, pH 7.5, 5 mM KCl, 150 mM NaCl, 0.1% NP-40, 1 mM DTT, 1× cOmplete protease inhibitor cocktail, 2 μM carfilzomib and 4 μl of benzonase per ten 15-cm plates). Lysed extracts were pelleted at 21,000g to remove cellular debris and the clarified lysate was bound to anti-Flag M2 resin (20 μl of slurry per 2.5 15-cm plates of material) for 2 h with rotation at 4 °C. UBR4-coupled beads were washed twice with (40 mM HEPES, pH 7.5, 5 mM KCl, 150 mM NaCl, 0.1% NP-40, 1 mM DTT) and twice without (40 mM HEPES, pH 7.5, 5 mM KCl, 150 mM NaCl, 1 mM DTT) detergent, all liquid was removed from the beads using a crushed gel loading tip before addition of the in vitro ubiquitylation reaction.
In vitro ubiquitylation assays were performed in a 10 μl reaction volume. 0.5 μl of 10 μM E1 (250 nM final), 0.5 μl of 50 μM Ube2A (2.5 μM final), 0.5 μl of 50 μM Ube2D3 (2.5 μM final), 1 μl of 10 mg ml−1 ubiquitin (1 mg ml−1 final) (R&D Systems, U-100H), 0.5 μl of 200 mM DTT, 1.5 μl of energy mix (150 mM creatine phosphate (Sigma-Aldrich, 10621714001-5G), 20 mM ATP, 20 mM MgCl2, pH to 7.5 with NaHCO3), 1 μl of 10× ubiquitylation assay buffer (250 mM Tris, pH 7.5, 500 mM NaCl and 100 mM MgCl2) and 0.5 μl of 1 mg ml−1 tandem ubiquitin binding entities were pre-mixed and added to 10 l of UBR4-coupled bed resin. Then, 3 μl of in vitro translated substrate and 1 μl of 100 μM TAMRA-labelled peptide were added to the reactions. In the ABHD10 ubiquitylation experiment, where ABHD10–3×HA was immunoprecipitated with Flag–UBR4, and in the KCMF1 auto-ubiquitylation experiment, where KCMF1–3×Flag (and mutants) were immunoprecipitated from cells, no additional substrate was added. PBS was added to reach final volume of 10 μl. Peptide sequences used in this study are summarized in Supplementary Table 3. Reactions were performed at 30 °C with shaking for 2 h or as indicated in the respective time course. Reactions were stopped by adding 2× urea sample buffer and resolved on SDS–PAGE gels before autoradiography in the case of radiolabelled substrates. MTS-TAMRA peptide ubiquitylations were run on 4–20% gradient gels (Thermo Fisher Scientific, EC6026BOX) and imaged on the ProteinSimple Fluorchem M imager. ABHD10–3×HA or KCMF1–3×Flag ubiquitylations were visualized by western blotting with anti-HA or anti-ubiquitin antibodies, respectively. Lys11/48-branched ubiquitin chains were visualized using a bispecific antibody developed previously17. The following commercially available recombinant human ubiquitin mutants used in this study were used: R&D Systems, UM-K11R, UM-K27R, UM-K29R, UM-K33R, UM-K48R, UM-K480, UM-K63R, UM-NOK. L8A ubiquitin was purified as described previously48. E1 enzyme UBA1 was purified as described previously74. Ube2A and Ube2A mutants, Ube2D3 and Tube recombinant proteins were purified as previously described4.
Protein purification for NMR analysis
For NMR isotopic labelling, WT untagged ubiquitin was expressed in Escherichia coli BL21 (DE3) grown in M9 minimal medium supplemented with 15N-ammonium chloride (Cambridge Isotope Labs). Ubiquitin was purified by cation exchange followed by SEC using the Superdex 75 column (GE Healthcare) equilibrated in NMR buffer (25 mM NaPi, pH 7.0, 150 mM NaCl, 1 mM TCEP) as previously described75. 6×His-tagged UBR4-UBL (residues 4340–4463; UniProt: Q5T4S7-1) variants were expressed in E. coli BL21 (DE3) and purified by Ni2+-NTA affinity chromatography using the Cytiva HisTrap FF crude column according to the manufacturer’s instructions followed by SEC using the Superdex 75 column equilibrated in NMR buffer.
NMR
All NMR samples were assembled with 200 mM 15N-labelled ubiquitin and the indicated amounts of unlabelled UBR4-UBL in NMR buffer supplemented with 10% deuterium oxide. 1H-15N heteronuclear single quantum coherence (HSCQ) spectra were collected on the Bruker Ascend 500 mHz magnet equipped with a Bruker Avance IV NEO console and a 5 mm BBO Prodigy CryoProbe. Data were transformed and phased using Bruker TopSpin (4.3) and plotted in NMRViewJ. Combined amide (NH) chemical shift perturbations (CSP) were calculated using the following equation: ΔδNH (ppm) = sqrt[ΔδH2 + (ΔδN/5)2]. NMR CSP titration data were fitted to the standard equation for a 1:1 binding equilibrium to derive a dissociation constant (Kd) for the ubiquitin–UBL interaction: ΔδNH (ppm) = Δδmax (([P]t + [L]t + Kd) − [([P]t + [L]t + Kd)2 − 4[P]t[L]t]1/2)/2[P]t where [L]t and [P]t are the concentrations of ligand (UBR4-UBL) and protein (ubiquitin), respectively76. A subset of NMR signals with substantial and well-resolved chemical shifts was used for Kd calculation. Data were plotted and fit to the binding equilibrium equation using Python. Plotted curves were fit to individual residues, while the reported Kd represents a global fit to all plotted residues with the s.d. derived from the residual sum of squares.
Software and code for data analysis
The following freely/commercially available software/codes were used to analyse data: FlowJo (v.10.8.1), GraphPad Prism (v.9), NMRViewJ (v9.2.0-b27), Bruker TopSpin (v.4.3.0), cryoSPARC (v.4.3), SerialEM (v.4.1), 3DFlex, AlphaFold (v.2; v.3), PHENIX (v.1.21.1-5286), Coot (v.0.9.8.92), PDBePISA (v1.52), Chimera (v.1.17.1), ChimeraX (v.1.8), PyMOL (v.2.5.5), Spectronaut (18.0), ProteoWizard’s msConvert (v.3.0.22335), Kojak (v.2.0.3), Percolator (v.2.08) and ProXL web application.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The consensus atomic coordinate model and cryo-EM map of the human UBR4–KCMF1–CaM (SIFI) complex has been deposited in the Protein Data Bank (PDB) and Electron Microscopy Data Bank (EMDB) under accession codes 9D9Z and EMD-46686, respectively. Coordinates and cryo-EM maps of the endogenous SIFI complex (C-terminal partial map with improved local densities) has been deposited under accession codes PDB 9NWE and EMD-49876, and the SIFI complex purified through affinity-tagged KCMF1 (N-terminal partial map) has been deposited under accession codes PDB 9NWD and EMD-46688. The cryo-EM map of the SIFI complex supplemented with UBE2A has been deposited under EMDB accession code EMD-46742. Source data for immunoblots are provided in Supplementary Fig. 1. Gating strategies for flow cytometry experiments are provided in Supplementary Fig. 2. The workflow for cryo-EM structure generation is provided in Supplementary Fig. 3. Source data for the IP–MS datasets are provided in Supplementary Table 1. Cross‐linking MS data are available interactively on the ProXL web application73 (https://proxl.yeastrc.org/proxl/p/sifi-hri) along with the raw MS spectra and search parameters used. Moreover, the complete search algorithm configuration files, fasta search databases, raw search output and raw MS data files were deposited to the ProteomeXchange Consortium via the PRIDE partner repository (https://www.ebi.ac.uk/pride/archive) under dataset identifier PXD055759. The publicly available CRAPome and Mitocarta 3.0 datasets can be accessed at https://reprint-apms.org and https://www.broadinstitute.org, respectively. There are no restrictions on data availability.
References
Winter, J. M., Yadav, T. & Rutter, J. Stressed to death: mitochondrial stress responses connect respiration and apoptosis in cancer. Mol. Cell 82, 3321–3332 (2022).
Acosta-Alvear, D., Harnoss, J. M., Walter, P. & Ashkenazi, A. Homeostasis control in health and disease by the unfolded protein response. Nat. Rev. Mol. Cell Biol. https://doi.org/10.1038/s41580-024-00794-0 (2024).
Costa-Mattioli, M. & Walter, P. The integrated stress response: from mechanism to disease. Science https://doi.org/10.1126/science.aat5314 (2020).
Haakonsen, D. L. et al. Stress response silencing by an E3 ligase mutated in neurodegeneration. Nature 626, 874–880 (2024).
Harper, J. W. & Bennett, E. J. Proteome complexity and the forces that drive proteome imbalance. Nature 537, 328–338 (2016).
Hipp, M. S., Kasturi, P. & Hartl, F. U. The proteostasis network and its decline in ageing. Nat. Rev. Mol. Cell Biol. 20, 421–435 (2019).
Fessler, E. et al. A pathway coordinated by DELE1 relays mitochondrial stress to the cytosol. Nature 579, 433–437 (2020).
Guo, X. et al. Mitochondrial stress is relayed to the cytosol by an OMA1-DELE1-HRI pathway. Nature 579, 427–432 (2020).
McEwen, E. et al. Heme-regulated inhibitor kinase-mediated phosphorylation of eukaryotic translation initiation factor 2 inhibits translation, induces stress granule formation, and mediates survival upon arsenite exposure. J. Biol. Chem. 280, 16925–16933 (2005).
Anderson, N. S. & Haynes, C. M. Folding the mitochondrial UPR into the integrated stress response. Trends Cell Biol. 30, 428–439 (2020).
Inada, T. & Beckmann, R. Mechanisms of translation-coupled quality control. J. Mol. Biol. 436, 168496 (2024).
Nakaya, T. et al. p600 plays essential roles in fetal development. PLoS ONE 8, e66269 (2013).
Tasaki, T. et al. UBR box N-recognin-4 (UBR4), an N-recognin of the N-end rule pathway, and its role in yolk sac vascular development and autophagy. Proc. Natl Acad. Sci. USA 110, 3800–3805 (2013).
Hunt, L. C. et al. A key role for the ubiquitin ligase UBR4 in myofiber hypertrophy in Drosophila and mice. Cell Rep. 28, 1268–1281 (2019).
Hunt, L. C. et al. Antagonistic control of myofiber size and muscle protein quality control by the ubiquitin ligase UBR4 during aging. Nat. Commun. 12, 1418 (2021).
Conroy, J. et al. A novel locus for episodic ataxia:UBR4 the likely candidate. Eur. J. Hum. Genet. 22, 505–510 (2014).
Yau, R. G. et al. Assembly and function of heterotypic ubiquitin chains in cell-cycle and protein quality control. Cell 171, 918–933 (2017).
Abdel-Nour, M. et al. The heme-regulated inhibitor is a cytosolic sensor of protein misfolding that controls innate immune signaling. Science https://doi.org/10.1126/science.aaw4144 (2019).
Girardin, S. E., Cuziol, C., Philpott, D. J. & Arnoult, D. The eIF2α kinase HRI in innate immunity, proteostasis, and mitochondrial stress. FEBS J. 288, 3094–3107 (2021).
Yamano, K. & Youle, R. J. PINK1 is degraded through the N-end rule pathway. Autophagy 9, 1758–1769 (2013).
Barnsby-Greer, L. et al. UBE2A and UBE2B are recruited by an atypical E3 ligase module in UBR4. Nat. Struct. Mol. Biol. 31, 351–363 (2024).
Grabarczyk, D. B. et al. Architecture of the UBR4 complex, a giant E4 ligase central to eukaryotic protein quality control. Preprint at bioRxiv https://doi.org/10.1101/2024.12.18.629163 (2024).
Tate, J. G. et al. COSMIC: the catalogue of somatic mutations in cancer. Nucleic Acids Res. 47, D941–D947 (2019).
Choi, K. D. et al. Genetic variants associated with episodic ataxia in Korea. Sci. Rep. 7, 13855 (2017).
Meyers, R. M. et al. Computational correction of copy number effect improves specificity of CRISPR-Cas9 essentiality screens in cancer cells. Nat. Genet. 49, 1779–1784 (2017).
Villalobo, A., Ishida, H., Vogel, H. J. & Berchtold, M. W. Calmodulin as a protein linker and a regulator of adaptor/scaffold proteins. Biochim. Biophys. Acta Mol. Cell. Res. 1865, 507–521 (2018).
Bhattacharyya, M., Karandur, D. & Kuriyan, J. Structural insights into the regulation of Ca2+/calmodulin-dependent protein kinase II (CaMKII). Cold Spring Harb. Perspect. Biol. https://doi.org/10.1101/cshperspect.a035147 (2020).
Spratt, D. E., Taiakina, V. & Guillemette, J. G. Calcium-deficient calmodulin binding and activation of neuronal and inducible nitric oxide synthases. Biochim. Biophys. Acta 1774, 1351–1358 (2007).
Belzil, C. et al. A Ca2+-dependent mechanism of neuronal survival mediated by the microtubule-associated protein p600. J. Biol. Chem. 288, 24452–24464 (2013).
Varshavsky, A. N-degron and C-degron pathways of protein degradation. Proc. Natl Acad. Sci. USA 116, 358–366 (2019).
Cha-Molstad, H. et al. p62/SQSTM1/sequestosome-1 is an N-recognin of the N-end rule pathway which modulates autophagosome biogenesis. Nat. Commun. 8, 102 (2017).
Jeong, D. E. et al. Insights into the recognition mechanism in the UBR box of UBR4 for its specific substrates. Commun. Biol. 6, 1214 (2023).
Tasaki, T. et al. A family of mammalian E3 ubiquitin ligases that contain the UBR box motif and recognize N-degrons. Mol. Cell. Biol. 25, 7120–7136 (2005).
Heo, A. J. et al. The N-terminal cysteine is a dual sensor of oxygen and oxidative stress. Proc. Natl Acad. Sci. USA https://doi.org/10.1073/pnas.2107993118 (2021).
Choi, W. S. et al. Structural basis for the recognition of N-end rule substrates by the UBR box of ubiquitin ligases. Nat. Struct. Mol. Biol. 17, 1175–1181 (2010).
Carrillo Roas, S. et al. Convergence of orphan quality control pathways at a ubiquitin chain-elongating ligase. Mol. Cell 85, 815–828 (2025).
Budny, B. et al. Novel missense mutations in the ubiquitination-related gene UBE2A cause a recognizable X-linked mental retardation syndrome. Clin. Genet. 77, 541–551 (2010).
Cordeddu, V. et al. Refinement of the clinical and mutational spectrum of UBE2A deficiency syndrome. Clin. Genet. 98, 172–178 (2020).
Nascimento, R. M., Otto, P. A., de Brouwer, A. P. & Vianna-Morgante, A. M. UBE2A, which encodes a ubiquitin-conjugating enzyme, is mutated in a novel X-linked mental retardation syndrome. Am. J. Hum. Genet. 79, 549–555 (2006).
Deng, Z. et al. Mechanistic insights into nucleosomal H2B monoubiquitylation mediated by yeast Bre1-Rad6 and its human homolog RNF20/RNF40-hRAD6A. Mol. Cell 83, 3080–3094 (2023).
Hibbert, R. G., Huang, A., Boelens, R. & Sixma, T. K. E3 ligase Rad18 promotes monoubiquitination rather than ubiquitin chain formation by E2 enzyme Rad6. Proc. Natl Acad. Sci. USA 108, 5590–5595 (2011).
Kumar, B., Lecompte, K. G., Klein, J. M. & Haas, A. L. Ser120 of Ubc2/Rad6 regulates ubiquitin-dependent N-end rule targeting by E3α/Ubr1. J. Biol. Chem. 285, 41300–41309 (2010).
Leto, D. E. et al. Genome-wide CRISPR analysis identifies substrate-specific conjugation modules in ER-associated degradation. Mol. Cell 73, 377–389 (2019).
Eletr, Z. M., Huang, D. T., Duda, D. M., Schulman, B. A. & Kuhlman, B. E2 conjugating enzymes must disengage from their E1 enzymes before E3-dependent ubiquitin and ubiquitin-like transfer. Nat. Struct. Mol. Biol. 12, 933–934 (2005).
Kamadurai, H. B. et al. Insights into ubiquitin transfer cascades from a structure of a UbcH5B approximately ubiquitin-HECTNEDD4L complex. Mol. Cell 36, 1095–1102 (2009).
Koegl, M. et al. A novel ubiquitination factor, E4, is involved in multiubiquitin chain assembly. Cell 96, 635–644 (1999).
Kaiho-Soma, A. et al. TRIP12 promotes small-molecule-induced degradation through K29/K48-branched ubiquitin chains. Mol. Cell 81, 1411–1424 (2021).
Wickliffe, K. E., Lorenz, S., Wemmer, D. E., Kuriyan, J. & Rapé, M. The mechanism of linkage-specific ubiquitin chain elongation by a single-subunit e2. Cell 144, 769–781 (2011).
Eddins, M. J., Carlile, C. M., Gomez, K. M., Pickart, C. M. & Wolberger, C. Mms2-Ubc13 covalently bound to ubiquitin reveals the structural basis of linkage-specific polyubiquitin chain formation. Nat. Struct. Mol. Biol. 13, 915–920 (2006).
Hodakova, Z. et al. Cryo-EM structure of the chain-elongating E3 ubiquitin ligase UBR5. EMBO J. 42, e113348 (2023).
Hehl, L. A. et al. Structural snapshots along K48-linked ubiquitin chain formation by the HECT E3 UBR5. Nat. Chem. Biol. 20, 190–200 (2024).
Mark, K. G. et al. Orphan quality control shapes network dynamics and gene expression. Cell 186, 3460–3475 (2023).
Liu, C., Liu, W., Ye, Y. & Li, W. Ufd2p synthesizes branched ubiquitin chains to promote the degradation of substrates modified with atypical chains. Nat. Commun. 8, 14274 (2017).
Kolla, S., Ye, M., Mark, K. G. & Rapé, M. Assembly and function of branched ubiquitin chains. Trends Biochem. Sci 47, 759–771 (2022).
Jevtic, P., Haakonsen, D. L. & Rapé, M. An E3 ligase guide to the galaxy of small-molecule-induced protein degradation. Cell. Chem Biol. https://doi.org/10.1016/j.chembiol.2021.04.002 (2021).
Mastronarde, D. N. Automated electron microscope tomography using robust prediction of specimen movements. J. Struct. Biol. 152, 36–51 (2005).
Punjani, A., Rubinstein, J. L., Fleet, D. J. & Brubaker, M. A. cryoSPARC: algorithms for rapid unsupervised cryo-EM structure determination. Nat. Methods 14, 290–296 (2017).
Punjani, A., Zhang, H. & Fleet, D. J. Non-uniform refinement: adaptive regularization improves single-particle cryo-EM reconstruction. Nat. Methods 17, 1214–1221 (2020).
Punjani, A. & Fleet, D. J. 3DFlex: determining structure and motion of flexible proteins from cryo-EM. Nat. Methods 20, 860–870 (2023).
Jumper, J. et al. Highly accurate protein structure prediction with AlphaFold. Nature 596, 583–589 (2021).
Afonine, P. V. et al. Real-space refinement in PHENIX for cryo-EM and crystallography. Acta Crystallogr. D 74, 531–544 (2018).
Emsley, P., Lohkamp, B., Scott, W. G. & Cowtan, K. Features and development of Coot. Acta Crystallogr. D 66, 486–501 (2010).
Krissinel, E. & Henrick, K. Inference of macromolecular assemblies from crystalline state. J. Mol. Biol. 372, 774–797 (2007).
Pettersen, E. F. et al. UCSF ChimeraX: structure visualization for researchers, educators, and developers. Protein Sci. 30, 70–82 (2021).
Meng, E. C. et al. UCSF ChimeraX: tools for structure building and analysis. Protein Sci. 32, e4792 (2023).
Mellacheruvu, D. et al. The CRAPome: a contaminant repository for affinity purification-mass spectrometry data. Nat. Methods 10, 730–736 (2013).
Rath, S. et al. MitoCarta3.0: an updated mitochondrial proteome now with sub-organelle localization and pathway annotations. Nucleic Acids Res. 49, D1541–D1547 (2021).
Boos, F., Muhlhaus, T. & Herrmann, J. M. Detection of internal matrix targeting signal-like sequences (iMTS-Ls) in mitochondrial precursor proteins using the TargetP prediction tool. Bio Protoc. 8, e2474 (2018).
Zelter, A. et al. The molecular architecture of the Dam1 kinetochore complex is defined by cross-linking based structural modelling. Nat. Commun. 6, 8673 (2015).
Chambers, M. C. et al. A cross-platform toolkit for mass spectrometry and proteomics. Nat. Biotechnol. 30, 918–920 (2012).
Hoopmann, M. R. et al. Kojak: efficient analysis of chemically cross-linked protein complexes. J. Proteome Res. 14, 2190–2198 (2015).
Kall, L., Canterbury, J. D., Weston, J., Noble, W. S. & MacCoss, M. J. Semi-supervised learning for peptide identification from shotgun proteomics datasets. Nat. Methods 4, 923–925 (2007).
Riffle, M., Jaschob, D., Zelter, A. & Davis, T. N. Proxl (Protein Cross-Linking Database): a public server, QC tools, and other major updates. J. Proteome Res. 18, 759–764 (2019).
Meyer, H. J. & Rapé, M. Enhanced protein degradation by branched ubiquitin chains. Cell 157, 910–921 (2014).
Lazar, G. A., Desjarlais, J. R. & Handel, T. M. De novo design of the hydrophobic core of ubiquitin. Protein Sci. 6, 1167–1178 (1997).
Williamson, M. P. Using chemical shift perturbation to characterise ligand binding. Prog. Nucl. Magn. Reson. Spectrosc. 73, 1–16 (2013).
Acknowledgements
We thank all of the members of our laboratory for enthusiastic support and many suggestions for this work; D. Toso, R. Thakkar, P. Tobias and the members of the Cal-Cryo cryo-EM facility at QB3-Berkeley for assistance on cryo-EM data collection; H. Celik, R. Giovine and the members of the Pines Magnetic Resonance Centre’s Core NMR Facility at University of Washington for spectroscopy assistance; P. Brzovic for sharing ubiquitin NMR chemical shifts; and M. Ghassemian and the members of the UCSD Biomolecular and Proteomics Mass Spectrometry facility for assistance in running the MS samples. The instruments used in this work were supported by the PMRC Core. D.L.H. was funded by an HHMI Helen Hay Whitney Fellowship; and M.H. by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation; HE 9330/1-1). This work was supported by the G. Harold and Leila Y. Mathers Charitable Foundation and the Howard Hughes Medical Institute. M.R. is an Investigator of the Howard Hughes Medical Institute.
Author information
Authors and Affiliations
Contributions
Z.Y., D.L.H. and M.H. performed most of the experiments, including SIFI purification, cryo-EM analysis, substrate ubiquitylation and mutant analyses. S.R.W. performed NMR analyses. A.Z. and M.J.M. performed and analysed cross-linking MS. T.B. supported biochemical analyses of SIFI. M.R. helped to plan and interpret experiments. Z.Y., D.L.H., M.H., S.R.W. and M.R. wrote the manuscript.
Corresponding author
Ethics declarations
Competing interests
M.R. is a co-founder and scientific advisory board member of Nurix Therapeutics; co-founder, consultant and scientific advisory board member of Lyterian Therapeutics; co-founder and consultant of Zenith Therapeutics; co-founder of Reina Therapeutics; and iPartner at The Column Group. The other authors declare no competing interests.
Peer review
Peer review information
Nature thanks Richard Youle and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data figures and tables
Extended Data Fig. 1 Structural characterization of human SIFI.
a. Size exclusion chromatography of affinity-purified endogenous UBR4FLAG. Red arrow denotes the SIFI peak, confirmed by Western blotting and negative stain EM (data not shown). b. In vitro ubiquitylation of a fluorescently labelled presequence peptide shows that SEC purified endogenous SIFI used for cryo-EM is active in catalysing ubiquitylation. Similar results in n = 2 independent experiments. c. Fourier shell correlation (FSC) plot of the cryo-EM map of endogenous SIFI complexes. Resolution cutoff for gold-standard FSC0.143 is shown. d. FSC plot of the cryo-EM map of SIFI complexes purified by affinity-purification of overexpressed KCMF1Strep. e. Comparison of the cryo-EM maps of SIFI (left. EMD-46686, Gaussian filter Width=2.0, contour level 0.04) and SIFI supplemented with recombinant UBE2A (right. EMD-46742, Gaussian filter Width=2.0 contour level 0.014) reveals stabilization of the C-terminal region of UBR4, including hemi-RING/UZI domain. f. Schematic representation of the SIFI complex. g. Comparison between SIFI, 26S proteasome (PDB 6MSB), and 80S ribosome (PDB 6QZP) reveals their similar size dimensions. For gel source data, see Supplementary Fig. 1.
Extended Data Fig. 2 Structural domains of SIFI.
a. Structural insights into the N-terminal dimerization region of SIFI. Left: two Lys residues at the tips of interlocking loops from each protomer engage in polar interactions with their paired counterpart. Right: α7 helix of UBR4 undergoes a helix swap with the paired protomer to form a helical bundle. b. C-terminal dimerization region. 180° rotation reveals the stabilizing helical pair at the inside of the SIFI scaffold. c. The SIFI scaffold shows flexibility around a central hinge region, leading to ~10° rotations of the N- and C-terminal dimerization domains relative to each other. Two disease mutations in UBR4, A2581 (ataxia) and R2584 (cancer) are located close to the hinge region. d. Calcium-binding to the C-lobe, but not the N-lobe of calmodulin is required for binding of UBR4, as seen by immunoprecipitation of calmodulin mutants that disrupt calcium-binding in the N- or C-lobe, respectively (N-lobe mutant: D21A, D23A, D57A, D59A; C-lobe mutant: D94A, D96A, D130A, D132A)28. Similar results in n = 2 independent experiments. e. Cryo-EM map of central SIFI showing docked WD40, DOC and UBR domains. These internal protein interaction modules are flexible and hence show lower overall resolution. The central density (ABHD10) is described below. f. The WD40 domain of UBR4 features a central plug helix at a surface commonly involved in protein interactions characteristic of WD40 repeats. g. The DOC2 domain of UBR4 contains two Zinc-binding loops that collectively form a potential protein interaction interface. h. Diagram of cross-linking mass spectrometry validates domain arrangement at the centre of SIFI. For gel source data, see Supplementary Fig. 1.
Extended Data Fig. 3 KCMF1 recruitment and function in SIFI.
a. top: Schematic of domain arrangement in KCMF1. bottom: KCMF1 AlphaFold model suggests that the KCMF1 linker not observed in SIFI structures (orange) does not adopt a compact fold. b. The ZZ-domain of KCMF1, but not its linker, is required for substrate degradation. Stability of HRI or cleaved DELE1 was measured in ΔKCMF1 cells using flow cytometry. As indicated, WT-KCMF1, KCMF1ΔZZ, or KCMF1mutLink (containing 5 Q/E mutations in conserved residues), were expressed. Similar results in n = 2 independent experiments. c. The central Phe residue in the C-terminal KCMF1 helix is highly conserved. d. Deletion of the N-terminal ZZ domain or mutation of R20E, E38R, L61E, Y64E, R68E in the ZZ domain strongly impaired the interaction between KCMF1FLAG and endogenous UBR4, as shown by affinity-purification and Western blotting. The ZZ deletion also interfered with recognition of the N-degron substrate ABHD10, as described below. Similar results in n = 2 independent experiments. For gel source data, see Supplementary Fig. 1.
Extended Data Fig. 4 ABHD10 binds SIFI as an N-degron substrate.
a. Immunoprecipitation of SIFI in WT or ΔKCMF1 cells coupled to mass spectrometry shows that two mitochondrial proteins, ABHD10 and NIPSNAP3A, are lost from SIFI purifications in the absence of KCMF1. b. Immunoprecipitation of KCMF1 coupled to mass spectrometry from WT or ΔUBR4 cells shows that KCMF1 binds to ABHD10 and NIPSNAP3A independently of UBR4. c. SIFI-bound ABHD10 is ubiquitylated if SIFI is incubated with E1, UBE2D3/UBE2A, and ubiquitin, as shown by Western blotting. Ubiquitin lacking Lys residues and hence unable to support chain formation (K0) is used as control. Similar results in n = 2 independent experiments. d. SIFI-bound NIPSNAP3A is ubiquitylated by SIFI in a similar manner as ABHD10. Experiment performed once. e. ABHD10 only binds KCMF1 if it contains an N-terminal presequence that is cleaved off upon entry into mitochondria. ΔMTS-ABHD10 does not contain its presequence and is not imported into mitochondria, thus initiating with a Met residue. Binding of ABHD10HA to KCMF1FLAG was analysed by αFLAG-affinity-purification and Western blotting. Similar results in n = 2 independent experiments. f. ABHD10 only binds KCMF1 after being released from mitochondria. Cells were either lysed with digitonin, which preserves mitochondrial integrity, or NP40, which disrupts mitochondrial integrity and results in release of mitochondrial proteins into the lysate. UBR4FLAG was affinity-purified and bound ABHD10HA was detected by Western blotting. CS: citrate synthase. Similar results in n = 2 independent experiments. g. Cleaved ABHD10 likely starts with a Lys residue, as determined by surveying all ABDH10-peptides retrieved in ABHD10 immunoprecipitations by mass spectrometry. h. Left: Structure of the ZZ-domain of p62, which is known to bind to N-degrons as determined by X-ray crystallography (PDB 5YPS). Right: overlay of p62 and KCMF1 N-domains bound to their respective N-degrons. i. AlphaFold model of cleaved DELE1 bound by the ZZ-domain of KCMF1. For gel source data, see Supplementary Fig. 1.
Extended Data Fig. 5 The DOC2 domain of UBR4 is important for SIFI function.
a. Crosslinking of HRI~Ub or a presequence peptide to SIFI results in similar conformational rearrangements of the KCMF1 module composed of the N-terminal domains of KCMF1 and the DOC1 and UBR domains of UBR4, as determined by cryo-EM. fl: full-length; MTS: mitochondrial targeting sequence. All maps were Gaussian filtered with width sdev=2.0. b. SIFI binds endogenous mitochondrial proteins, when import has been disrupted by arsenite treatment. When SIFI was purified from cells expressing endogenous UBR4ΔDOC2, the binding of mitochondrial proteins was lost. Binding partners were determined after endogenous SIFI affinity-purification using mass spectrometry. Mitochondrial proteins, for which predicted presequence peptides could be identified, are shown in red. IP-MS samples were run as technical triplicates. c. The DOC2 domain of SIFI is not required for ABHD10 ubiquitylation. SIFI was purified from either WT or UBR4ΔDOC2 cells that also expressed ABHD10HA and incubated with E1, UBE2D3/UBE2A, and ubiquitin. Ubiquitylation of bound ABDH10 was detected by Western blotting. Experiment performed once. d. The DOC2 domain of SIFI is required for degradation of DELE1 or unimported mitochondrial proteins. The stability of DELE1 or the presequence of COX8A was determined in WT or UBR4ΔDOC2 cells by flow cytometry, as described before. Similar results in n = 2 independent experiments. e. Deletion of the DOC2 domain in UBR4 results in stronger activation of the integrated stress response. A reporter for integrated stress response activation based on uORFs of ATF4 was expressed in either WT, ΔUBR4, or UBR4ΔDOC2 cells, which were treated with sodium arsenite and analysed by flow cytometry. Similar results in n = 2 independent experiments. f. The DOC2 domain of UBR4 is essential for cell survival upon mitochondrial import stress. WT and UBR4ΔDOC2 cells were labelled with GFP and mCherry, respectively, mixed, and depleted of either TIMM8A or TIMM8A/HRI, as indicated. After 12 d of co-culture, the ratio between cell types was determined by flow cytometry. n = 3 independent replicates shown with the median of these experiments. For gel source data, see Supplementary Fig. 1.
Extended Data Fig. 6 The UBR boxes of UBR4 present substrates towards the interior of SIFI.
a. The UBR box of UBR4 binds to DOC1 domain of UBR4 and the N-terminal domain of KCMF1, forming the KCMF1N138-DOC1-UBR subcomplex. X-ray structure of the UBR box of UBR1 (PDB 3NIH)35 reveals that its peptide binding groove is open. b. Modelling a N-degron peptide into the open substrate-binding groove of the UBR box of UBR4, based on the UBR1 X-ray structure (PDB 3NIH). c. Structure of SIFI showing the proximity of UBR box to other substrate-binding modules of SIFI, including the DOC2 domain of UBR4 and the ZZ domain of KCMF1.
Extended Data Fig. 7 UBE2A promotes ubiquitin chain elongation by SIFI.
a. KCMF1 does not determine linkage specificity of SIFI. SIFI was purified from WT or ΔKCMF1 cells and tested for its ability to catalyse ubiquitylation of a mitochondrial presequence. As indicated, either wt-ubiquitin or ubiquitin-K48R were used. Experiment performed once. b. KCMF1 can modify ubiquitin residues independently of a specific Lys residue. KCMF1 was affinity-purified from ΔUBR4 cells and incubated with E1, UBE2D3, and indicated ubiquitin variants. Ubiquitin polymers were detected by Western blotting. Similar results in n = 2 independent experiments. c. KCMF1 can introduce ubiquitin branches. KCMF1 was affinity-purified from ΔUBR4 cells and incubated with E1, UBE2D3, and indicated ubiquitin variants. Reaction products were visualized with either ubiquitin antibodies or with bispecific K11/K48-antibodies that preferentially detect K11/K48-branched ubiquitin molecules17. Experiment performed once. d. UBE2A and its close homologue UBE2B are required for HRI degradation. UBE2A and UBE2B were co-depleted from 293 T cells, and HRI stability was monitored using a HRI-GFP::mCherry reporter, as described before. Similar results in n = 2 independent experiments. e. UBE2A and UBE2B co-depletion results in increased activation of the integrated stress response, as determined by αATF4-Western blotting. Experiment performed once. f. UBE2A promotes ubiquitin chain elongation. SIFI was affinity-purified via UBR4FLAG and incubated with E1, ubiquitin and a mitochondrial presequence peptide (left) or HRI degron helix 2 peptide (right), resulting in short ubiquitin chains on the presequence substrate likely catalysed by an E2 that co-purified with SIFI. Addition of the E2 UBE2D3 results in an increase in short ubiquitin chains, indicative of a role of UBE2D3 in chain initiation. Addition of UBE2A leads to selective elongation of ubiquitin chains that had already been initiated. Similar results in n = 2 independent experiments. For gel source data, see Supplementary Fig. 1.
Extended Data Fig. 8 Structural insight into UBE2A binding and linkage specificity.
a. UBE2A mutants detected in patients of Nascimento Syndrome are impaired in binding SIFI in cells, as determined by UBR4 affinity-purification and Western blotting. Similar results in n = 2 independent experiments. b. UBE2A variants in Nascimento Syndrome are impaired, but not fully inhibited, in catalysing ubiquitin chain elongation on SIFI substrates. D114 of UBE2A is at the interface with donor ubiquitin. Similar results in n = 2 independent experiments. c. Chemical shift perturbation analysis of 15N-ubiquitin with 2.5 molar equivalents of UBR4 UBL domain. d. NMR analysis of UBR4 UBL mutants binding to 15N-ubiquitin. Lys11 of ubiquitin was chosen as a reporter of binding because it is far away from the interface directly affected by UBL mutations. e. Y82 and S120 of UBE2A are essential for ubiquitin chain elongation by SIFI, as detected in an in vitro ubiquitylation assay as described above. Experiment performed once. f. Mutation of Y82 or S120 of UBE2A does not prevent binding of the E2 to SIFI. Endogenous SIFI was purified from UBR4FLAG cells and co-purifying UBE2AHA variants were detected by Western blotting. Experiment performed once. For gel source data, see Supplementary Fig. 1.
Extended Data Fig. 9 The UBL domain of UBR4 is essential for ubiquitin chain elongation.
a. Mass spectrometry of SIFIΔUBL indicates that the UBL domain is not required for structural integrity of SIFI. Endogenous SIFI was affinity-purified from WT, UBR4ΔUBL or UBR4M4444A/L4447E cells and bound proteins were detected by mass spectrometry. Technical replicates are shown. b. Western blotting of SIFIΔUBL shows that the UBL domain is not required for structural integrity of SIFI. Endogenous SIFI was affinity-purified from WT or UBR4ΔUBL cells and bound proteins were detected by Western blotting. Experiment performed once and validated by mass spectrometry. c. Mutation of the UBL domain in UBR4 prevents ubiquitin chain elongation, as seen with a TAMRA-labelled presequence peptide as substrate. Similar results in n = 2 independent experiments. d. Mutation or deletion of the UBL domain in UBR4 both prevent ubiquitin chain elongation by SIFI, as seen with 35S-labelled HRINT ~ SUMO as a substrate. e. Deletion of the UBL prevents ubiquitin chain elongation, even if initiation was performed independently of SIFI by a 2 h preincubation with E1/E2. Similar results in n = 2 independent experiments. f. The UBL domain of UBR4 is required for degradation of DELE1 or unimported mitochondrial proteins. Protein stability was determined by flow cytometry, as described before. Similar results in n = 2 independent experiments. g. Mutation of the UBL domain stabilizes cDELE1 and unimported mitochondrial proteins, as seen by flow cytometry with two independent clones expressing endogenous UBR4M4444A/L4447E. Similar results in n = 2 independent experiments. h. Mutation of the UBL domain in UBR4 results in increased stress signalling, as seen by an increase in ATF4 levels after import was compromised with arsenite. Experiment validated with two independent clones. i. Deletion of the UBL domain in UBR4 increases stress response activation upon sodium arsenite treatment, as detected by uORF-ATF4 reporters in flow cytometry as described above. Similar results in n = 2 independent experiments. j. Mutation of the UBL domain in UBR4 increases stress response activation, as measured by flow cytometry. Experiment validated with two independent clones. k. Mutation of the UBL domain in UBR4 compromises cell survival upon arsenite-induced import stress, as seen in a competition experiment with WT cells. n = 3 independent replicates shown with median of these experiments. l. The UBL domain of UBR4 is essential for cell survival upon mitochondrial import stress. WT and UBR4ΔUBL cells were labelled with GFP and mCherry, respectively, mixed, and depleted of either TIMM8A or TIMM8A/HRI, as indicated. After 12 d of co-culture, the ratio between cell types was determined by flow cytometry. n = 3 independent replicates shown with median of these experiments.
Supplementary information
Supplementary Fig. 1
Source data for western Blots, fluorescence gels and autoradiographs.
Supplementary Fig. 2
Gating strategy for flow cytometry experiments.
Supplementary Fig. 3
Workflow of EM processing.
Supplementary Table 1
Source data for proteomics data.
Supplementary Tables 2 and 3
Supplementary Table 2: plasmids. Supplementary Table 3: synthetic peptide sequences.
Supplementary Video 1
Overview of SIFI structure and important domains. Structures shown in the video are based on PDB 9D9Z and AlphaFold models.
Supplementary Video 2
Movements of the SIFI scaffold around a central hinge region analysed by cryoSPARC 3D flexible refinement using particle images of EM map EMD-46742.
Supplementary Video 3
Flexibility of the substrate-binding cavity of SIFI. Data were analysed by cryoSPARC 3D flexible refinement using particle images of EM map EMD-46686.
Supplementary Video 4
Movements of the SIFI arms. Data were analysed by cryoSPARC 3D flexible refinement using particle images of EM map EMD-49876.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Yang, Z., Haakonsen, D.L., Heider, M. et al. Molecular basis of SIFI activity in the integrated stress response. Nature 643, 1117–1126 (2025). https://doi.org/10.1038/s41586-025-09074-z
Received:
Accepted:
Published:
Issue date:
DOI: https://doi.org/10.1038/s41586-025-09074-z
