
Abstract
Prokaryotes create adaptive immune memories by acquiring foreign DNA snippets, known as spacers, into the CRISPR array1. In type II CRISPR–Cas systems, the RNA-guided effector Cas9 also assists the acquisition machinery by selecting spacers from the protospacer adjacent motif-flanked DNA2,3. Here we uncovered the first biological role of Cas9 that is independent of its dual RNA partners. Following depletion of CRISPR RNA (crRNA) and/or trans-activating CRISPR RNA, Neisseria apoCas9 stimulates spacer acquisition efficiency. Physiologically, Cas9 senses low concentrations of crRNA in cells with short CRISPR arrays, such as those undergoing array neogenesis or natural array contractions, and dynamically upregulates acquisition to quickly expand the small immune memory banks. As the CRISPR array expands, rising crRNA abundance in turn reduces apoCas9 availability, thereby dampening acquisition to mitigate autoimmunity risks associated with elevated acquisition. Although the nuclease lobe of apoCas9 alone suffices to stimulate acquisition, only full-length Cas9 responds to crRNA concentrations to boost acquisition in cells with low immunity depth. Finally, we showed that this activity is evolutionarily conserved across several type II-C Cas9 orthologues. Altogether, we established an auto-replenishing feedback mechanism in which apoCas9 safeguards CRISPR immunity depth by acting as both a crRNA sensor and a regulator of spacer acquisition.
Similar content being viewed by others


Main
A cornerstone of CRISPR–Cas prokaryotic adaptive defence is adaptation, in which snippets of invader DNA are incorporated into the host CRISPR loci as new immune memories called spacers1. Following transcription of the CRISPR locus, the spacer-derived segment of mature CRISPR RNA (crRNA) guides Cas nucleases to destroy complementary foreign targets during interference4,5. DNA-targeting CRISPR–Cas systems also require a protospacer adjacent motif (PAM) next to the target site to distinguish self from non-self6,7,8. Therefore, functional spacers must be acquired from PAM-flanked sequences to ensure immunity.
Diverse CRISPR–Cas systems use the conserved Cas1–Cas2 integrase complex to catalyse spacer insertion at the leader–proximal end of the CRISPR array1,9,10,11. Numerous studies have shown that many CRISPR–Cas systems further require auxiliary Cas or host factors, such as Csn2, Cas4 and DnaQ, to faithfully create functional memories1,12,13,14,15,16,17,18. In the Streptococcus pyogenes (Spy) type II-A system, Cas9 has dual functions both as an interference effector enzyme19,20 and as the PAM selector for spacer acquisition2. When cleaving a DNA target, Cas9 must form a ternary complex with two RNAs (crRNA and trans-activating CRISPR RNA (tracrRNA)) to act as an RNA-guided endonuclease19,21. In II-A spacer acquisition, Cas9 specifies memory precursors from PAM-flanked viral sequences independently of its nuclease activity, although the precise molecular roles of both small RNAs are unclear2,3. The tracrRNA is genetically required for forming viral memories in vivo2, but tracrRNA itself is notably absent from Cas protein co-complexes detected in biochemical and structural studies of Spy II-A acquisition2,22. Moreover, how Cas9 coordinates with the Cas1–Cas2 integrase and Csn2 in acquisition remains to be elucidated. The mechanism and regulation of acquisition in the other main branch of Cas9-containing systems (type II-C23) remain largely unknown. Type II-C systems account for roughly 40% of all type II CRISPR–Cas and are notable for their compact Cas9 and a minimal cas1–cas2–cas9 gene composition, lacking the adaptation factors Csn2 or Cas4 (ref. 24).
Here we reveal that the type II-C Cas9 protein lacking its RNA partners (apoCas9) potently stimulates spacer acquisition to safeguard CRISPR immunity depth. This immunity depth is reflected by CRISPR array sizes, which scale with spacer diversity, and crRNA abundance, which influences the strength of interference25. Physiologically, Cas9 senses crRNA abundance to dynamically regulate acquisition efficiency, rapidly replenishing short CRISPR arrays while reducing autoimmunity risk when the CRISPR array is lengthened.
Cas9 enables PAM-compliant acquisition
The type II-C CRISPR–Cas system of Neisseria meningitidis (Nme) (Fig. 1a) is recognized as a key player in impacting gene transfer through natural transformation26, but its functional interplay with bacteriophages is unexplored. As a model system to study acquisition, we challenged N. meningitidis strain 8013 with meningococcal disease-associated phage (MDAΦ), a well-characterized Neisseria filamentous phage (Fig. 1b) associated with hypervirulent strains27,28. Because MDAΦ causes neither lysis nor detectable growth burden to infected hosts, we used MDAΦ marked with a kanamycin (Kan) resistance gene to track infection27. Neisseria cells were incubated with MDAΦ–KanR in liquid culture for 3 h to allow transduction and then plated on GCB–Kan plates to select for transductants. We found that MDAΦ lysogenized the wild-type (WT) 8013 strain with a transduction frequency of 10−4 (Fig. 1c). MDAΦ lysogenization depends on type IV pili but not on other host factors essential for natural transformation (Extended Data Fig. 1a,b). Real-time polymerase chain reaction (PCR) and nanopore sequencing revealed that MDAΦ exists primarily as an episome but can also integrate into the host genome as a prophage through dRS3 repeats (Extended Data Fig. 1c–i), consistent with previous knowledge of MDAΦ and related filamentous phages27,29. MDAΦ lysogenization did not occur in strains carrying a spacer targeting the sense or antisense of MDAΦ genome (Fig. 1a–c). This interference phenotype required the N4GATT PAM sequence26 of NmeCas9 next to the target sequence (Fig. 1c). In summary, we demonstrate that N. meningitidis CRISPR–Cas9 can restrict the spread of phages, thereby influencing the prophage content and pathogenicity of meningococci (Supplementary Discussion).

a, The II-C CRISPR–Cas locus of N. meningitidis. Coloured squares, spacers; white diamonds, repeats; arrowheads, repeat-embedded crRNA promoters. b, MDAΦ genome organization. White arrows, phage open reading frames; black triangles, inverted repeats; red lines, protospacers targeted. c, CRISPR–Cas9 blocked MDAΦ lysogenization. MDAΦ was marked by a Kan-resistant cassette to track infection. Data are log-scale of colony-forming units (CFU) per millilitre (mean ± s.d.) of total colonies (grey bars) versus transductants (white bars) of three independent experiments. NS (P ≥ 0.05), *0.005 ≤ P < 0.05 and **P < 0.005; P values calculated using two-tailed Welch’s t-tests. d, Detecting new spacers at the leader end of CRISPR from transductants. 0 and +1, PCR amplicons from WT and elongated CRISPRs. Top, PCR primer design. The leader (L) that flanks CRISPR and contains essential elements for acquisition is depicted. Bottom, representative adaptation PCR gel; ‘adapted %’, adaptation efficiency measured as the percentage of total amplicons with new spacers on the basis of band intensities. Cas1–cas2 OE is driven by a lac regulatory system and integrated at the iga–trpB genomic site. e, Motif analysis for 3′ flanks of viral protospacers for lane 4 of d. f, Δcas9 modestly reduced efficiency. Δcas9 and its derivatives with genomic complementation of WT or mutant cas9 were assayed. Top, representative adaptation PCR gel; bottom, quantification of efficiencies. Data are mean ± s.d., n = 3. NS (P ≥ 0.05), *0.005 ≤ P < 0.05 and **P < 0.005; P values calculated using two-tailed Welch’s t-tests. H1024A, cas9 mutant with PAM-contacting residue H1024 mutated; PID swap, cas9 chimaera with PID switched. g, 3′ Flanking motif for viral spacers of lanes 2–6 of f. Shown is a representative sequence logo of three independent experiments that give similar results. NS, nonsignificant; NT, non-targeting; OE, overexpressing.
We next asked whether new viral spacers are acquired during infection. Following MDAΦ transduction, we extracted genomic DNA from the resulting transductant pool and PCR amplified the leader end of the CRISPR array, where evolutionarily divergent (that is, new) spacers tend to exist in native arrays26 to detect acquisition events (Fig. 1d, top). Although no discernible +1 band indicating acquisition was observed (Fig. 1d, lane 1), we gel extracted the presumed +1 band position for deep sequencing and detected 183 unique new spacer sequences, of which 99 matched the MDAΦ genome, indicative of bona fide acquisition. This low acquisition efficiency is consistent with the previous notion that acquisition is a rare event in native CRISPR–Cas systems under laboratory conditions30. We therefore adopted a widely used method to enhance adaptation to PCR-detectable level by overexpressing the cas1–cas2 integrase genes2,3,9. Upon isopropyl β-d-1-thiogalactopyranoside (IPTG) induction of Cas1–2 (Extended Data Fig. 2a,b), acquisition was readily detectable in 6% of the CRISPR arrays of the transductants (Fig. 1d, lane 4). Deep sequencing of this +1 band yielded 2.8 million acquisition reads, including nearly 54,000 unique spacers, of which 78% were mappable to MDAΦ genome (Extended Data Fig. 2c–f and Supplementary Discussion). Alignment of the flanking sequences of the protospacers matched by viral spacers revealed a 3′-N4GATT motif (Fig. 1e and Extended Data Fig. 2g,h) identical to the interference PAM required by NmeCas9 for target cleavage26. Collectively, our system recapitulated the formation of PAM-compliant functional viral spacers.
Previous genetic studies have shown that the cas9 gene in both the Spy and Streptococcus thermophilus (Sth) type II-A systems are essential for acquisition, and that their PAM-interacting domains (PIDs) ensure selection of PAM-flanked spacers2,3. To test if NmeCas9 plays a similar role in the type II-C system of meningococci, we deleted cas9 in the cas1–2 overexpressing background, finding a modest reduction in acquisition efficiency and a complete absence of discernible PAM for new viral spacers (Fig. 1f,g). Genomic complementation by a WT cas9 or catalytically dead cas9, but not a cas9H1024A allele in which the key PAM-contacting residue (H1024) was mutated to disrupt DNA binding31, restored the N4GATT PAM of new spacers (Fig. 1f,g and Extended Data Fig. 2i–k). Complementation by a PID-swapped chimaera32 (cas9PID-swap), in which the wedge domain (WED)/PID of NmeCas9 is replaced by that of a closely related natural variant Nme2Cas9 that recognizes a distinct 3′ N4CC PAM32, altered the PAM specificity to 3′-N4CC (Fig. 1f,g and Extended Data Fig. 2i–k). These data demonstrate that the role of type II-C NmeCas9 in specifying the adaptation PAM relies on its PAM-binding specificity but not its nuclease activity, consistent with previous reports for type II-A SpyCas9 and SthCas9 (refs. 2,3).
Notably, deep sequencing of the +1 band for the Δcas9-derivative strain with complemented dcas9 revealed that approximately 70% of the acquisition reads were MDAΦ-derived (Extended Data Fig. 2d). Given the difference in genome size between MDAΦ (8 kb) and the host (2.2 Mb) and the ratio of approximately ten copies of MDAΦ per host genome (Extended Data Fig. 1f,g), this result reflects an approximately 60-fold preference of foreign-over-host DNA for N. meningitidis acquisition (Supplementary Discussion), which is independent of the nuclease activity of Cas9 or the CRISPR interference phenotype.
Loss of tracr or crRNA boosts adaptation
To define the RNA partner requirements for acquisition, we deleted the tracrRNA gene (that is, Δtracr) in the cas1–2 overexpressing background (Fig. 2a and Extended Data Fig. 3a,b) and measured acquisition by adaptation PCR in MDAΦ-infected meningococci. The loss of tracrRNA caused a marked increase in acquisition efficiency, from undetectable to 4% under the cas1–2 uninduced condition, or from 6% to 61% when cas1–2 was overexpressed (Fig. 2b). The induced cells also yielded +2, +3 and +4 adapted bands corresponding to arrays with two, three and four new spacers added, respectively. In all cases, the genomic complementation of tracrRNA reverted the enhanced acquisition phenotype to the respective baseline WT level (Fig. 2b). This ‘super-adaptation’ phenotype retained a strong bias for acquiring spacers from viral over the host genome (Extended Data Fig. 3c; 68% of spacer reads were MDAΦ-derived) and displayed the canonical 3′ N4GATT PAM (Fig. 2c), indicating that tracrRNA is not required for the acquisition of PAM-compliant spacers in this II-C system. The Δtracr strain did not alter the expression levels of Cas9 or Cas1 (Extended Data Fig. 3d,e). Moreover, the ΔtracrΔcas9 strain (Extended Data Fig. 3a,b) exhibited 3% basal adaptation with nullified PAM (Fig. 2b,c), suggesting that Δtracr exerts its effect by modulating NmeCas9.

a, Schematic of CRISPR–Cas9 loci of WT and Δtracr N. meningitidis strains. b, Δtracr and its derivatives complemented with tracrRNA or empty vector, and a ΔtracrΔcas9 strain was assayed for MDAΦ infection and acquisition, with or without induction of cas1–2. Top, a representative adaptation PCR gel. Bottom, quantification of adaptation efficiencies. Data are mean ± s.d., n = 3. NS (P ≥ 0.05), *0.005 ≤ P < 0.05 and **P < 0.005; P values calculated by two-tailed Welch’s t-tests. c, 3′ Flanking motif analysis for new viral spacers from b. Lx, lane number. d, Schematic of CRISPR–Cas9 loci of WT and crRNA null allele R26. e, R26 and its derivative strain lacking cas9 were assayed for MDAΦ infection and acquisition, with or without the IPTG induction of cas1–2. Top, a representative adaptation PCR gel. Bottom, quantification of adaptation efficiencies. Data are mean ± s.d., n = 3. NS (P ≥ 0.05), *0.005 ≤ P < 0.05 and **P < 0.005; P values calculated by two-tailed Welch’s t-tests. CR, CRISPR; FL, full length. f, 3′ Flanking motif analysis for new viral spacers from e. nt, nucleotide.
One possible explanation for these results is that the loss of CRISPR interference activity in Δtracr strains (Extended Data Fig. 3f) could abolish selection against newly acquired self-targeting spacers and thereby enriching for cells with expanded arrays, as observed previously in S. thermophilus with deactivated SthCas9 (ref. 3). To address this possibility, we measured acquisition in isogenic strains, in which WT cas9 was replaced by interference-deficient dcas9 at the endogenous locus. The WT cas9 and dcas9 strains exhibited similar acquisition phenotypes in response to tracrRNA deletion and complementation (Extended Data Fig. 3g,h), demonstrating that super-adaptation is not because of interference defects and is independent of the nuclease activity of Cas9. In summary, we conclude that tracrRNA is a negative regulator of acquisition efficiency in meningococci, in contrast to previous findings that tracrRNA is genetically required for acquisition in the Spy type II-A system2.
Any function or requirement for crRNA in CRISPR–Cas9 adaptation has remained elusive2,3,33 owing to the difficulty of blocking crRNA production without affecting the leader element essential for acquisition. In the distantly related Escherichia coli type I-E system, crRNA transcription from its leader-embedded promoter is dispensable for acquisition9. Given that crRNA and tracrRNA are co-processed by RNase III during maturation20 and that they must load into Cas9 as a pair19, we propose that crRNA would, like tracrRNA, play a regulatory role in N. meningitidis acquisition. We constructed a mutant strain R26 in which the endogenous full-length 1.7-kb CRISPR array was removed, except for the leader-adjacent repeat, R26 (Fig. 2d), which was expected to be sufficient for enabling acquisition2,9. We predicted and validated by northern blot (Extended Data Fig. 4a–c) that this R26 allele does not produce any crRNA or leader-derived crRNA mimic extraneous crRNA34, defining R26 as a proxy for a crRNA null allele.
In the cas1–2 overexpression background, R26 enhanced acquisition from 5% to a super-adaptation level of 44% (Fig. 2e). Deletion of cas9 from this context reduced the acquisition efficiency to 2% (Fig. 2e) and abolished PAM (Fig. 2f). A similar super-adaptation phenotype was observed in an isogenic R26 derivative that expresses dcas9 in place of WT cas9 (Extended Data Fig. 4d–f), indicating that this phenotype is dependent on Cas9 but independent of CRISPR interference. Moreover, R26-triggered super-adaptation was not because of changes in Cas9 or Cas1 protein concentrations (Extended Data Fig. 4g,h). We therefore conclude that both crRNA and tracrRNA are negative regulators of acquisition.
ApoCas9 stimulates adaptation efficiency
Because the loss of crRNA or tracrRNA resulted in similar super-adaptation phenotypes, we propose that these RNAs work together as a regulatory switch, implying that apoCas9 (that is, Cas9 in an RNA-free state) may be the driver of super-adaptation. Combining the Δtracr and R26 in cells carrying WT cas9 and cas1–2 overexpression cassette resulted in super-adaptation (Fig. 3a–d and Extended Data Fig. 5a), which was reversed to a basal level by simultaneous complementation by both tracrRNA and a WT CRISPR array, or by a single guide RNA (sgRNA), which is a chimaeric fusion of tracrRNA and crRNA19 but not by tracrRNA or the CRISPR array alone (Fig. 3a–d). The simultaneous loss of tracrRNA and crRNA did not alter Cas9 protein concentrations (Extended Data Fig. 5b). Together, our data showed that apoNmeCas9 is indeed driving highly efficient acquisition, and that the tracrRNA–crRNA pair is a regulatory switch for NmeCas9-stimulated adaptation (Extended Data Fig. 5c).

a, Schematic of the strains used. Blue box, cas9 complemented at the iga–trpB genomic locus, alone or with RNA partners. Cas9 was driven by leaky expression from the Tet promoter, and tracr and sgRNA were driven from a copy of the tracrRNA promoter. b, Northern blot confirming the absence or presence of small RNAs in accordance with strain genotypes. The orange stars denote sgRNA, detectable by both anti-repeat (top) and anti-tracr (middle) northern probes. 5S ribosomal RNA (rRNA), loading control (bottom). c, ApoNmeCas9 was the driver of super-adaptation and was suppressed by the crRNA–tracrRNA pair. Top, a representative adaptation PCR gel. Bottom, bar graph of adaptation efficiencies. Data are mean ± s.d., n = 3. NS (P ≥ 0.05), *0.005 ≤ P < 0.05 and **P < 0.005; P values calculated by two-tailed Welch’s t-tests. d, 3′ Flanking motif analysis for new viral spacers from c, showing successful PAM enrichment for super-adaptation and baseline adaptation conditions.
These findings are further supported by biochemical data confirming that the apo and RNA-loaded forms of NmeCas9 exist in distinct states. In an RNA electrophoretic mobility shift assay, apoCas9 only assembled stable ribonucleoprotein complexes with its crRNA–tracrRNA pair or sgRNA but not with crRNA alone, tracrRNA alone or control RNAs (Extended Data Fig. 5d). In a limited proteolysis assay, apoNmeCas9 was rapidly degraded by trypsin in conditions lacking RNAs or in the presence of crRNA or tracrRNA alone, whereas it became protected from degradation when the crRNA–tracrRNA pair or sgRNA was present (Extended Data Fig. 5e). This is consistent with reports of substantial conformational changes to NmeCas9 and other Cas9 orthologues upon sgRNA loading31,35. Together, our findings uncovered the first role of apoCas9, which is independent of its RNA partners and nuclease activity. We also established an important role of the crRNA–tracrRNA pair in minimizing the inherent risk of high acquisition activity, which can acquire self-targeting spacers from the host genome.
Domain requirements for super-adaptation
We next investigated the mechanism by which apoNmeCas9 stimulates super-adaptation by systematically removing each of its domains: RuvC (split into three subdomains), bridge helix, recognition lobe 1 (REC1), REC2, histidine–asparagine–histidine (HNH) and WED/PID31,36 (Fig. 4a). In the Δtracr + R26 and Cas1–2 overexpressing background that lacks both RNAs, we found that deletion of the RuvCII, HNH, RuvCIII or WED/PID domain abrogated super-adaptation, with acquisition efficiency dropping from 45% in the full-length Cas9 to 3–4% (Fig. 4b and Extended Data Fig. 5f,g) and abolishing PAM selection for new spacers (Fig. 4c), resembling the Δcas9 condition in Fig. 1. On the other hand, a cas9ΔREC1/2 allele lacking the entire REC lobe and a cas9ΔBHΔREC1/2 allele further lacking the bridge helix motif that connects the two lobes remained competent for super-adaptation (Fig. 4b,c). These two alleles elicited super-adaptation even in the presence of RNA partners, indicating that their adaptation-stimulating ability was no longer repressible by RNAs (Fig. 4d,e). Because it is highly unlikely that NmeCas9 lacking the REC lobe could still bind its RNA partners31, our results further support the notion that tracrRNA and crRNA act through NmeCas9 to regulate adaptation. Altogether, our results indicate that the NUC lobe of NmeCas9 (comprising the HNH, RuvC and WED/PID domains) is sufficient for stimulating adaptation, and the REC lobe mediates regulation by RNA partners. By contrast, all domains of NmeCas9 are essential for interference (Extended Data Fig. 5h), further validating that the apo and RNA-guided states are functionally distinct.

a, Schematics of NmeCas9 domain architecture and serial deletion mutants. aa, amino acid. b, NUC lobe of NmeCas9, which lacks bridge helix (BH) and REC1/2 domains, is sufficient for super-adaptation. Top, a representative adaptation PCR gel. Bottom, quantification of adaptation efficiencies. Data are mean ± s.d., n = 3. NS (P ≥ 0.05), *0.005 ≤ P < 0.05 and **P < 0.005; P values calculated using two-tailed Welch’s t-tests. c, 3′ Flanking motif analysis for new viral spacers of b. d,NmeCas9 mutants ΔREC1/2 and ΔBHΔREC1/2 became resistant to RNA regulation. Top, a representative adaptation PCR gel. Bottom, quantification of adaptation efficiencies. Data are mean ± s.d., n = 3. NS (P ≥ 0.05), *0.005 ≤ P < 0.05 and **P < 0.005. e, 3′ Flanking motif analysis for new viral spacers of d.
ApoCas9 assists CRISPR array neogenesis
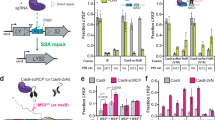
Although microbes may restrain acquisition to minimize the fitness cost associated with possible acquisition of self-targeting spacers, many physiological conditions could necessitate high rates of acquisition. We therefore explored three biological scenarios in which dynamic control of acquisition may be beneficial: (1) CRISPR neogenesis, in which a new CRISPR array arises from a single repeat; (2) array contraction, which would eliminate existing spacers to allow the uptake of beneficial traits previously targeted; and (3) array shortening driven by recombination between repeats. In the first scenario, to mimic CRISPR arrays at various stages of neogenesis, we created N. meningitidis 8013 derivatives in which the endogenous CRISPR array was shortened from its typical 25-spacer length to varying degrees (Fig. 5a), whereas the other cas and tracrRNA genes remained unmodified. Although NmeCas9 protein concentrations remained constant across strains (Extended Data Fig. 6a), the concentrations of mature crRNA and tracrRNA co-increased gradually, from undetectable levels in the single repeat strain to weak signals for short arrays with one to three spacers and then to intermediate and plateaued levels with mid-sized and longer arrays (Fig. 5b). By assaying acquisition following MDAΦ infection in these strains overexpressing Cas1–2, we observed a strong inverse correlation between acquisition efficiency and array length, with 31% adaptation efficiency in the single repeat array diminishing to 20–30% for arrays with one to three spacers, 8–10% for arrays with five to seven spacers and 4–6% for longer arrays (Fig. 5c and Extended Data Fig. 6b). CrRNA abundance quantified by northern blot (Fig. 5b) also inversely correlated with acquisition efficiency (Extended Data Fig. 6c,d). These results indicate that crRNA may be the limiting agent to enable such array-length-based regulation. We therefore proposed that NmeCas9 primarily exists in the apo state during early CRISPR neogenesis, a condition that unleashes super-adaptation to quickly build up an initial immune memory repertoire. This represents a dynamic feedback mechanism for regulating acquisition; array elongation increases crRNA abundance, which in turn reduces apoCas9 availability to dampen acquisition.

a, Schematic of serial arrays of varying lengths to imitate CRISPR evolution from a single lone repeat. The WT array in N. meningitidis strain 8013 has 25 spacers. b, Northern blot showing the trend of mature crRNA increase. Total RNAs extracted were probed for crRNA (top), tracrRNA (bottom) and 5S rRNA loading control (middle). The dashed grey line indicates non-adjacent lanes from the same gel. c, The efficiency of spacer acquisition inversely correlates with CRISPR array length. Top, a representative adaptation PCR gel; bottom, quantification of adaptation efficiencies. Data are mean ± s.d., n = 3. NS (P ≥ 0.05), *0.005 ≤ P < 0.05 and **P < 0.005; P values calculated using two-tailed Welch’s t-tests. d, Schematic for CRISPR interference of a beneficial horizontally transferred trait. The transforming DNA bears a chloramphenicol (Cm) resistance marker (green) and protospacer (ps) 6 (orange), together flanked by homology arms (black) for recombination into the host genome. e, Transformation interference assay using high amounts of DNA (3 μg) to obtain escapers. Y axis, log-scale of CFU per millilitre (mean ± s.d.; n = 3) for total cells (blue bars) and transformants (red bars). f, PCR diagnostic of CRISPR length in representative escapers (e). Array truncations were confirmed by Sanger sequencing and illustrated at the bottom. Red, spacer 6. g, Interference assay retesting escapers using different targets, protospacers 25 (left) versus 6 (right). Data plotted as in e. h, CRISPR array-collapsed escapers produced much less crRNA than WT strain. Northern blot was conducted and shown as in b. i, The array-collapsed escapers, assayed in a Cas1–2 overexpression context, elevated their spacer acquisition efficiencies. Data are shown as in c.
To further demonstrate that Cas9 senses the cellular crRNA concentration to govern acquisition, we examined array co-existing scenarios in R26 derivatives that carry a second leader-flanked array inserted at a separate chromosomal site (Extended Data Fig. 6e). Under Cas1–2 overexpressing conditions, the total acquisition efficiency was evenly distributed between the two co-existing arrays, regardless of their individual sizes. As the second array increased from 0 to 2, 3 and 25 spacers, the acquisition efficiency at both arrays in the same strain progressively co-declined (Extended Data Fig. 6f). These observations indicate that the overall acquisition efficiency is governed by the total cellular crRNA abundance, rather than the number of arrays or the size and crRNA output of individual arrays.
ApoCas9 safeguards CRISPR immunity depth
Microbes may benefit from super-adaptation following natural CRISPR array contraction to rapidly rebuild the memory repertoire. To illustrate a physiological context when natural array compaction occurs, we examined how Neisseria accommodates beneficial traits that are targeted by a resident spacer. In Staphylococcus epidermidis, CRISPR interference against a beneficial conjugative plasmid has been shown to select host escapees that resolve this conflict by deteriorating CRISPR–Cas, allowing plasmid retention37. We conducted a similar experiment in meningococci by transforming WT 8013 strain with DNA containing a target sequence for spacer 6 and a chloramphenicol resistance marker (CmR–ps6), together flanked by homology arms for recombination into the genomic capsule locus (Fig. 5d). Host cells must obtain the CmR trait to survive on chloramphenicol plates, but a genomically integrated CmR–ps6 cassette would trigger autoimmunity through the native spacer 6. As expected, we obtained more than 6,000-fold fewer CmR transformants relative to the non-target control (Fig. 5e), indicating strong CRISPR interference as seen before26. The high amounts of DNA used here allowed us to obtain more than 100 CmR transformants as escapers (escape rate of 1.2 × 10−4; Fig. 5e). We randomly picked and sequenced about half of the escapers at their CRISPR–Cas and capsule loci, revealing Cas9 deactivation and CRISPR array collapse as the two most common mechanisms by which cells escape the targeting conundrum (Extended Data Fig. 6g,h). Array-contracted escapers lost continuous blocks of spacer–repeat units, including the targeting spacer 6 (Fig. 5f) while remaining competent for interference through the remaining spacers (Fig. 5g).
These array-contracted escapers form the basis of our second scenario to study acquisition regulation. Because CRISPR compaction logically comes at the cost of immunity depth, reflected in the loss of spacer diversity and crRNA abundance25, elevated acquisition can compensate by replenishing the array more quickly. Although Cas9 protein concentrations remained constant over various array lengths (Extended Data Fig. 6i), crRNAs are much less abundant in cells with contracted arrays (Fig. 5h). This NmeCas9–crRNA imbalance augmented acquisition, for example, escaper colonies e10 and e5 lost 16 and 22 spacer–repeat units, respectively, elevating their acquisition efficiencies under cas1–cas2 induction from a baseline 5% to 9% and 15%, respectively (Fig. 5f,i and Extended Data Fig. 6j). We assayed native acquisition in these escapers without introducing the Cas1–2 overexpression cassette or any other modifications to the endogenous CRISPR–Cas locus (Extended Data Fig. 7a,b). Deep sequencing of all CRISPR amplicons, without gel extraction to enrich for adapted arrays, revealed that native acquisition occurs at a low efficiency of 0.6–1 × 10−5, which was stimulated by up to 5-fold in natural escapers (Extended Data Fig. 7a,b) and approximately 2.5-fold to 7.7-fold in isogenic strains carrying either an evolutionarily short-CRISPR mimic or a natural short array from other Neisseria isolates (Extended Data Fig. 7c,d). These results align well with the findings under Cas1–2 overexpressing conditions (Fig. 5c,i), supporting that the stimulatory effect of Cas9 on acquisition is a genuine physiological response, rather than a rogue phenotype arising from the removal of key CRISPR–Cas components.
For the third scenario, we examined array collapse caused by the homologous recombination between repeats25,38. We transformed WT cells with a DNA fragment containing a repeat and a selectable marker and recovered transformants that have undergone recombination between the incoming repeat and resident genomic CRISPR (Extended Data Fig. 8a). We detected array shortening in 82% (180 of 220) of the transformants (Extended Data Fig. 8b,c), and the resulting collapsed arrays exhibited elevated adaptation when assayed under Cas1–2 overexpressing conditions (Extended Data Fig. 8d,e). The natural competence of Neisseria allows frequent uptake of genomic DNA from neighbouring cells39. Consequently, repeat-mediated recombination between incoming and resident CRISPRs, along with array collapse, are likely common events, highlighting the importance of a dynamic array-replenishing mechanism in naturally competent bacteria.
Collectively, our data indicate that array contraction reduces crRNA concentrations and in turn shifts a portion of NmeCas9 from the RNA-loaded to the apo state, thereby stimulating acquisition to quickly replenish the memory repertoire. ApoNmeCas9, acting both as a sensor to gauge low crRNA abundance (that is, shallow immunity depth) and an actuator to upregulate acquisition, represents an elegant feedback mechanism that dynamically replenishes contracted arrays in response to crRNA concentration (Fig. 6). Finally, among N. meningitidis 8013 derivatives that all lack MDAΦ-targeting spacers to begin with, strains with short arrays exhibited stronger MDAΦ resistance upon acquisition induction than those with longer arrays (Extended Data Fig. 9), linking the array compaction-stimulated acquisition to enhanced phage defence.

Top, with high concentration of crRNAs produced from a regular-sized CRISPR array, spacer acquisition is in check to balance the need to defend new threats and avoid autoimmunity. Bottom, memory purging and array contraction led to low crRNA abundance, which was sensed by apoCas9 to stimulate acquisition efficiency to rapidly build up immunity depth.
Role of apoCas9 across II-C systems
Finally, we investigated whether other type II-C systems regulate acquisition through a similar mechanism. Using meningococci lacking NmeCas9 and dual RNAs as surrogate hosts, we assessed seven II-C Cas9 orthologues, with 31–96% protein sequence identity to NmeCas9 (Extended Data Fig. 10a–c), for their ability to confer super-adaptation in collaboration with N. meningitidis Cas1–Cas2 and R26. All orthologues tested, except for Campylobacter jejuni (Cje) Cas9 (ref. 40) (Extended Data Fig. 10d,e), elicited apoCas9-stimulated super-adaptation (Extended Data Fig. 10f, left). Cas9 orthologues from Haemophilus parainfluenzae (Hpa), Actinobacillus succinogenes (Asu), Pasteurella pneumotropica (Ppn) and Pasteurella multocida (Pmu)40,41,42, which share 62–65% sequence identity with NmeCas9 and 67–85% pairwise identity with each other, stimulated adaptation to 20–47% efficiencies without RNA partners. Nme2Cas9 and Nme3Cas9, two natural variants related to NmeCas9 but possessing divergent PID and PAM preferences32, also elicited super-adaptation with approximately 40% efficiency. In all cases, new spacers were primarily drawn from viral sequences flanked by a prominent PAM (Extended Data Fig. 10g, left), consistent with the known interference PAM of each Cas9 (refs. 40,41,42) (Extended Data Fig. 10h). By contrast, isogenic strains co-expressing the cognate sgRNA for each II-C Cas9 (Extended Data Fig. 10a,c) exhibited 4–6% basal acquisition efficiency (Extended Data Fig. 10f, right) while maintaining the cognate PAM specificity (Extended Data Fig. 10g, right). Altogether, our data highlight the conserved roles of apoCas9 and crRNA–tracrRNA in acquisition regulation across type II-C CRISPR–Cas.
Discussion
Although Cas9 is primarily known as an RNA-guided nuclease for prokaryotic adaptive immunity, more roles of Cas9 in endogenous gene regulation have been unveiled43,44. A natural sgRNA variant in S. pyogenes (tracr-L) directs Cas9 to bind and auto-repress the promoter of the CRISPR–Cas operon to dampen autoimmunity45. In Francisella novicida (Fno), Cas9 is guided by an atypical CRISPR-associated RNA called small CRISPR–Cas-associated RNA to repress the transcription of an endogenous lipoprotein to control bacterial pathogenicity46. Beyond these RNA-guided roles, type II-A Cas9 also acts as the PAM selector for new viral memories2,3 in a manner that requires its tracrRNA2.
Given the small RNA requirements for target interference and spacer acquisition in type II-A system, apoCas9 has been perceived as non-functional. This study reports the first biological function of apoCas9 in bacterial immunity, which is independent of but regulatable by its RNA partners. We showed that apoCas9 boosts the acquisition efficiency during filamentous phage infection in meningococci (Figs. 2–4 and Extended Data Figs. 3–5 and 10). This acquisition-stimulatory role requires the nuclease lobe of Cas9 but not its catalytic activity and is regulated by the crRNA–tracrRNA pair through its REC lobe (Fig. 4 and Extended Data Fig. 5). As such, the two RNA partners are integral to CRISPR biology, both in directing foreign DNA targeting and in reining in super-adaptation to reduce the likelihood of acquiring autoimmunity spacers from the host genome. Our findings fundamentally expand the roles of Cas9, crRNA and tracrRNA in bacterial immunity.
The size of the CRISPR array and the concentration of crRNA produced from it reflect the depth of the adaptive immunity25. Although acquisition allows for constant memory updates, most bacterial CRISPR arrays contain fewer than 50 spacers and do not grow infinitely, with occasional cases of arrays reaching 100 or 200 spacer counts, meaning that older spacers become obsolete and get lost over time25,47. Memory purging often occurs through array rearrangement that deletes a continuous block of spacer–repeat units, presumably through recombination between repeats25. In S. epidermidis, experiments and mathematical modelling indicate that host cells bear spontaneous CRISPR–Cas mutations at the rate of approximately 10−4 per cell per generation; these mutations, including CRISPR array contraction as a common type, will be selected for when the host is under pressure to acquire laterally transferred beneficial traits targeted by a resident spacer37. We showed that in meningococci, immunity escapees also arise through the host losing a block of spacer–repeat units, including the targeting spacer (Fig. 5d–g). The resultant reduced crRNA concentration was read out by Cas9, which stimulated acquisition to swiftly replenish the memory bank (Fig. 5h,i). This auto-replenishing feedback mechanism helped to reduce the vulnerability window caused by short arrays that did not provide a sufficiently diverse repertoire of spacers to ensure robust protection (Fig. 6). Our findings demonstrated the physiological role of Cas9 in safeguarding immunity depth as a crRNA sensor and acquisition regulator.
Our findings opened new possibilities for regulating bacterial adaptation that merit further investigation. For example, are there environmental cues or physiological conditions that tap into this II-C acquisition regulatory network by affecting the expression of crRNA, tracrRNA or Cas9? Although tracr-L homologues have not been detected in previous small RNA sequencing studies in Neisseria26,48, other unknown mechanisms may regulate CRISPR–Cas expression. Some phages use crRNA decoys to hijack CRISPR–Cas enzymes, forming interference-incompetent complexes and blocking immunity49. It will be interesting to investigate whether phages use similar crRNA or tracrRNA mimicry strategies or anti-CRISPRs that occlude crRNA binding to Cas9 to disrupt the activity or regulation of II-C Cas9-regulated acquisition. On a more practical note, we envision that researchers can take advantage of the super-adaptation phenotype to unveil the mechanistic role of Cas9 in acquisition, the molecular details of which have remained unclear. From a biotechnology perspective, the apoCas9-based II-C acquisition machineries offer new avenues to develop robust molecular recording, barcoding and lineage-tracing tools50,51,52.
Methods
Bacterial strains and growth conditions
The N. meningitidis strains MC58, 8013 and 8013 mutant derivatives used in this study are listed in Supplementary Table 3. These strains were grown on Gonococcal Medium Base (GCB) (BD; 228950) plates with appropriate antibiotics and Kellogg’s supplements I and II (22.2 mM glucose (Thermo Fisher Scientific; D16-1), 0.68 mM glutamine (Sigma; G3126), 0.45 mM co-carboxylase (also known as thiamine pyrophosphate; Sigma; C8754) and 1.23 mM Fe(NO3)3 (Sigma; F8508)). The antibiotic concentrations used were 2.5 μg ml−1 for erythromycin (Erm), 100 μg ml−1 for Kan, 100 μg ml−1 for streptomycin, 2.5 μg ml−1 for chloramphenicol and 100 μg ml−1 for spectinomycin. The plates were incubated at 37 °C in a 5% CO2 humidified atmosphere.
JM109 (Promega; L2005) and Mach1 (Invitrogen; C862003) chemically competent E. coli cells were used for plasmid cloning. E. coli BL21(DE3) (Novagen; 69450-3) cells were used for protein purification. The E. coli cells were grown at 37 °C in lysogeny broth Lennox or on lysogeny broth agar plates supplemented, when needed, with 50 μg ml−1 of Kan, 50 μg ml−1 of carbenicillin, 20 μg ml−1 of chloramphenicol or 100 μg ml−1 of spectinomycin.
Plasmid construction
A complete list of all the plasmids and their detailed construction strategies are provided in Supplementary Table 2. All oligonucleotides and gBlocks used in this study were ordered from Integrated DNA Technologies, Eurofins Genomics and Twist Bioscience (see Supplementary Tables 1 and 4 for their sequences). PCR amplifications to generate insert or vector backbone for plasmid cloning were performed using Q5 High-Fidelity DNA Polymerase (New England Biolabs (NEB); M0493L). Cloning reactions were performed using one of the following methods: Q5 site-directed mutagenesis, Gibson assembly or DNA ligation, using the NEB products E0554S, E2621L and M0202L, respectively. All plasmid constructs were verified by means of colony PCR using 2X GoTaq Green Master Mix (Promega; M7122), restriction digestion and Sanger sequencing (Eurofins Genomics).
Phage preparation
MDAΦ prepared from strain Z5463–MDAΦ–orf6::aph3′ was a kind gift from E. Bille and X. Nassif. MDAΦ for transduction assay in this study was prepared from 8013–MDAΦ–founder strain, as previously described27 with minor modifications. The founder strain was grown overnight on GCB plate and subsequently resuspended in GCB liquid medium (1.5% proteose peptone no. 3 (Gibco; 211693), 0.1% NaCl (Fisher; S271-10), 0.4% K2HPO4 (Sigma; P3786) and 0.1% KH2PO4 (Sigma; P5655)) containing 10 μg ml−1 of Kan, 2.5 mM MgCl2, 2.5 mM MgSO4, 10 mM NaHCO3 and Kellogg’s supplements I and II. This suspension was diluted to optical density at 600 nm (OD600) of 0.1 and incubated at 37 °C with 200 rpm shaking for 2–3 h until OD600 reached 0.4–0.5. Mitomycin C (RPI; M92010) was then added to a final concentration of 0.015 μg ml−1, and the bacteria continued to be cultured overnight. The bacteria were pelleted, and the supernatant was collected and filtered through a 0.45-μm filter (CELLTREAT; 229763), followed by an enzymatic treatment step at room temperature for 3 h with 25 μg ml−1 of DNase I (Roche; 10104159001) and RNase A (QIAGEN; 19101) each. To precipitate phage particles, NaCl and polyethylene glycol 6000 (Sigma; 8074911000) were added to the supernatant to final concentrations of 0.5 mM and 10% (w/v), respectively. This mixture was incubated at 4 °C overnight, centrifuged at 11,000g at 4 °C for 45 min and resuspended in sodium chloride–magnesium sulfate buffer (50 mM Tris (pH 7.5), 100 mM NaCl, 8 mM MgSO4 and 0.002% (w/v) gelatin (Sigma; G1393)) with 5% glycerol and stored at −80 °C for up to 1 month.
To quantify the concentration of phage DNA/particles, a 20-fold diluted phage prep was incubated at 95 °C for 20 min, used as a template in quantitative PCR using primers oYZ4215 and oYZ4216 to amplify the circular junction of MDAΦ and compared with the reference plasmid pYZ2852 encoding this junction sequence. All quantitative polymerase chain reactions (qPCRs) in this study were done on a CFX Opus 384 Real-Time PCR instrument using SsoAdvanced Universal SYBR Green Supermix (Bio-Rad; 1725271).
Phage transduction assay
Phage transduction was performed, as described earlier27 with minor modifications. Recipient strains grown overnight on GCB plates were resuspended to OD600 of 1.0 in GCB liquid medium containing 2.5 mM MgCl2, 2.5 mM MgSO4, 25 μg ml−1 of DNase I (Roche; 10104159001) and Kellogg’s supplements I and II. In one well of a 24-well plate (Falcon; 353047), 200 μl of this suspension was mixed with MDAΦ prep at a ratio of five MDAΦ particles per host cell. Adsorption was performed for 30 min at 37 °C and 5% CO2 with gentle shakes every 5 min, before the addition of 800 μl GCB liquid medium (with 2.5 mM MgCl2, 2.5 mM MgSO4, 25 μg ml−1 of DNase I and Kellogg’s supplements I and II) to each well. The plates were further incubated for 2.5 h at 37 °C and 5% CO2 without shaking. Serial dilutions were spread on GCB–Kan plate. Transductants were reported as Kan-resistant CFU per millilitre, compared with total CFU per millilitre from three independent experiments (mean ± s.d.).
Phage DNA copy number determination for transductants
MDAΦ copy number was determined by quantifying the MDA circular junction using published qPCR methods53,54. Briefly, total DNA was isolated from the transductant pool using QIAamp DNA Mini Kit (QIAGEN; 51306) with the following minor modifications. After cell lysis at 56 °C for 40 min, RNase A (1 mg ml−1; QIAGEN; 19101) treatment was performed at room temperature for 2 min. This total DNA was used as a template for the first qPCR to determine the MDAΦ DNA copy number from the standard curve (see next section) on the basis of the respective cycle threshold (Ct) value. This calculated copy number X is considering both dsDNA (replicative form) and single-stranded DNA (ssDNA) genome of MDAΦ. For the second qPCR, the total DNA treated with S1 nuclease (Promega; M5761; to remove the ssDNA) was used as a template to quantify copy number Y of MDAΦ dsDNA. Note that the ssDNA template needs an extra round of amplification compared with the dsDNA template; hence, the total MDAΦ DNA copy number equals (X − Y) × 2 + Y, whereas the ssDNA copy number equals (X − Y) × 2. In addition, the copy number of the genomic prophage form of MDAΦ was determined using total DNA as a qPCR template to amplify the MDAΦ end-dRS3 junction with primers oYZ4627 and oYZ4271.
Real-time qPCR standard curve generation
To generate a standard curve for quantifying circular MDAΦ junction, pYZ2852 was used as the reference plasmid because it contains both the MDAΦ circular junction sequence and a segment of the N. meningitidis genome control locus NMV1851 (amplified using the primers oYZ4217 + 4218). The pYZ2852 concentration was measured using Qubit dsDNA HS Kits (Thermo Fisher Scientific; Q32854) to calculate its copy number on the basis of molecular weight. The Ct values obtained from the serial dilutions were plotted against the logarithm of the known DNA copy numbers, and a linear regression analysis was performed to generate the standard curve. Similarly, to generate a standard curve for quantifying the genomic MDAΦ prophage form, pYZ2936 was used as reference plasmid because it contains both the MDAΦ–dRS3 junction and the NMV1851 genomic control sequence.
Meningococcal mutant strain construction
N. meningitidis 8013 derivatives were created by natural transformation, as previously described26, and all manipulations were achieved on the chromosome through double-crossover homologous recombination. Donor DNA (0.1–3 μg of linearized shuttle plasmid, 50-ng chromosomal DNA or 1-μg PCR fragment) encoding the intended allele was transformed into the recipient strain, followed by selection on an appropriate GCB–antibiotic plate. Four to eight representative transformants from each reaction were verified by re-streaking on selective plates twice and then genotyping PCR with Taq DNA Polymerase (NEB; M0267L). Genomic DNA (10 ng; 25 cycles) or 0.5-μl cell lysis solution (see below) extract (35 cycles) was used as a template, with appropriate PCR primer sets to amplify two genomic junctions and the full integration product whenever possible. Key amplicons were validated using Sanger sequencing. Cell lysis solution extract was made by lysing half of a single colony in 1% Triton X-100 (Sigma; T8787), 20 mM Tris (Thermo Fisher Scientific; BP152; pH 8.3) and 2 mM EDTA (Sigma; E6758) at 94 °C for 15 min. For each strain, two independent final candidates were kept at −80 °C in 1-ml GCB liquid medium supplemented with 20% glycerol.
Detailed construction strategies for all strains used in this study are listed in Supplementary Table 3. For strains used in Figs. 1d–g, 2 and 5 and Extended Data Figs. 2–4, 6, 8 and 9, pLac–Nmecas1–2 overexpressing cassette was inserted into iga–trpB locus by transforming pYZ008 into appropriate recipient strains and selecting for CmR transformants. To complement the tracrRNA gene at the NICS locus, we transformed Δtracr derivatives with pGCC2-based plasmids encoding tracrRNA and then selected for ErmR transformants. These two types of genetic manipulations at independent loci were combined to create more strains.
For strains used in Figs. 3 and 4 and Extended Data Figs. 5 and 10, pLac–Nmecas1–2 overexpressing cassette was inserted into the NICS locus by transforming pYZ1075 into receipt strains, followed by CmR selection. Cas9 complements, with or without small RNA, were done at iga–trpB locus by transforming Δcas9 or ΔtracrΔcas9Δcripsr::R26 derivatives with pMR68-based plasmids and selecting for ErmR transformants. These pMR68-based plasmids encode cas9 variants driven from pTet, with or without an extra tracrRNA/sgRNA driven from a tracr promoter (pTR).
Unmarked clean deletion strains were created using a two-step counter-selection approach, as previously described26. For Δtracr and ΔtracrΔcas9, we first transformed a StrepR background strain NYZ-11 with pYZ1019 to replace the tracrRNA locus with a CAT–rpsL dual marker. A resulting StrepSCmR transformant was colony-purified, PCR-verified and used as the intermediate receipt strain for subsequent transformation with pYZ1020 or pYZ1021, respectively, followed by StrepRCmS selection and PCR/Sanger confirmation. To create the endogenous dcas9 strain, a PCR fragment was transformed into NYZ-11 to replace the 7-590 aa coding region of NmeCas9 with a CAT–rpsL dual marker. The resulting StrepSCmR intermediate strain was then transformed with pYZ1111 and selected for StrepRCmS transformants that carry endo dCas9.
To create single-repeat and array-truncated alleles at the endogenous crispr locus, appropriate background strains were first transformed with pYZEJS068 to replace the full CRISPR array with CAT–rpsL dual marker. The resulting StrepSCmR intermediate strain was transformed with pYZ1043, pYZ2756, pYZ2757, pYZ2758, pYZ2759, pYZ2760, pYZ2934, pYZ2935, pYZ2762, pYZ2763 or pYZ2764 and selected for StrepRCmS transformants for further validation.
CRISPR interference assay
CRISPR interference was assayed by natural transformation, as previously described26. Antibiotic-resistant CFU per millilitre and total CFU per millilitre were reported from three independent experiments (mean ± s.d.). The results for pYZ1107-derived transforming DNAs with or without CRISPR target were compared to evaluate interference phenotype.
Transduction–adaptation PCR assay
MDAΦ–Kan transductants established in the infection assay were diluted in GCB liquid medium (with 2.5 mM MgCl2, 2.5 mM MgSO4 and Kellogg’s supplements I and II) to OD600 of 0.1. This culture, with or without 0.1 mM IPTG (Fisher; BP1755) to induce the cas1–cas2 overexpressing cassette, was incubated at 37 °C in a 5% CO2 humidified atmosphere overnight. Genomic DNA was isolated using QIAamp DNA Mini Kit according to the manufacturer’s instructions, with further RNase A (1 mg ml−1; QIAGEN; 19101) treatment at room temperature for 2 min after the cell lysis step at 56 °C for 40 min.
To detect spacer acquisition, 10-ng genomic DNA was used as the template to amplify the leader end of CRISPR using Q5 DNA Polymerase with PCR primers described in Supplementary Table 1. The PCR conditions used were 98 °C for 30 s; 25 cycles of 98 °C for 10 s, 60 °C for 10 s and 72 °C for 20 s; and final extension at 72 °C for 2 min. The PCR products were resolved on 2% agarose gel, stained by SYBR Safe (Invitrogen; S33102) and visualized on Bio-Rad ChemiDoc MP Imager (see Supplementary Fig. 1 for uncropped gel images). The adapted +1 and +2 bands are 66 nt and 132 nt longer than the unadapted 0 product derived from WT CRISPR owing to the addition of repeat–spacer units (66 nt each). Quantification of PCR band intensity was performed using Image Lab 6.0 software (Bio-Rad), and adaptation efficiency was calculated as the percentage of total CRISPR amplicons with new spacers (normalized to amplicon sizes). Although this method is used for comparison purposes, we acknowledge its limitation relative to next-generation sequencing (NGS)-based quantification, which was used in native acquisition experiments in Extended Data Fig. 7 to quantify low-frequency acquisition events in unengineered strains.
High-throughput sequencing and Illumina NGS library construction
Unique molecular identifier incorporation
For strains used in Figs. 1g and 2 and Extended Data Figs. 3h, 4f, 6j and 8, a FatI site in spacer 25 or a BsmAI site downstream of repeat 1, both of which were close enough to the newly added spacers, was used to add eight-base unique molecular identifiers (UMIs) as part of the library building procedure. FatI or BsmAI digestion and Y-adaptor ligation allow for UMI incorporation before PCR to control for amplification bias. Y-shaped adaptors containing UMI were prepared by annealing oligo pair oYZ591/592 (for FatI) or oYZ592/4055 (for BsmAI) in 1x annealing buffer (100 mM NaCl and 10 mM Tris (pH 8.0)) at 100 μM and stored at −20 °C in 2.5-μl aliquots. Genomic DNA (500 ng) from a transduction–adaptation assay was FatI or BsmAI digested at 55 °C for 3 h and purified with QIAquick PCR Purification Kit (QIAGEN; 28104) with final elution in 20 μl. Half of the DNA eluate was ligated to 10 μM Y-shaped adaptor in a 25-μl reaction at room temperature for 1 h using T4 DNA Ligase (NEB; M0202L). The reaction was purified with 0.9x AMPure XP beads (Beckman Coulter; A63881) and eluted with 20 μl of 10 mM Tris (pH 8.0).
UMI library construction
All PCRs for NGS library construction were performed using Q5 High-Fidelity Polymerase (NEB; M0493L). Ligated products from above were used as a template for the first step of Q5 PCR to amplify the leader end of CRISPR with primers OYZ3624/593 containing partial Illumina adaptors. The cycling conditions were 98 °C for 30 s; 25 cycles of 98 °C for 10 s, 60 °C for 10 s, 72 °C for 25 s; and a final extension at 72 °C for 2 min. Following 2% agarose gel electrophoresis, +1 adapted band was gel extracted with QIAquick Gel Extraction Kit (QIAGEN; 28706). The eluate served as a template for the second step of Q5 PCR using i5 and i7 index primers to add full Illumina adapters and indexes. The second PCR conditions were 98 °C for 30 s; 15 cycles of 98 °C for 10 s, 65 °C for 10 s and 72 °C for 25 s; and final extension at 72 °C for 2 min.
Non-UMI library construction
For strains used in Figs. 1e, 3 and 4 and Extended Data Figs. 6b and 10e,g, a non-UMI method was used because of the lack of any unique restriction site (such as FatI/BsmAI) close enough to new spacers. Genomic DNA (10 ng) from an adaptation experiment was used as a template for the first PCR step, with the appropriate primers described in Supplementary Table 1. The following steps for +1 adapted band gel extraction and the second step of Q5 PCR were performed as described in the previous paragraph. The final libraries from the second step of PCR were pooled together, cleaned up using QIAquick PCR Purification kit and gel extraction purified as one sample. The final pooled library was sequenced on Illumina NovaSeq (2 × 150 bp) at the Advanced Genomics Core at the University of Michigan. For the native acquisition test in Extended Data Fig. 7, non-UMI libraries were constructed in the same way, except that 100-ng genomic DNA was used as a template and that 1 μl of the first step of the PCR reaction was used directly as a template for the second step of PCR.
Bioinformatics analysis for NGS datasets
Read processing
Sequencing reads were assessed for quality using FastQC (v.0.11.8)55 and MultiQC (v.0.92)56 to ensure the general quality of the datasets. For new spacers, the read 1 sequences of the paired-end sequencing reads were trimmed to extract new spacer instances using Cutadapt (v.2.6) by identifying the flanking repeat 26 sequence (attgtagcactgcgaaatgagaaagggagctacaac)57.
For samples with UMIs, the read 2 sequences of the paired-end reads were processed using Cutadapt to extract the 8-nt UMI sequences by trimming known construct sequences. The new spacer–UMI sequence combinations were paired using fastq-pair (v.0.4)58 and processed using UMI-tools (v.1.0.1)59 in preparation for deduplication after alignment. For samples with no UMIs, no UMI processing was performed (see the paragraph below for the alternative approach that we used to minimize PCR bias/duplication artefacts).
The preprocessed spacer sequences were aligned using Bowtie 2 (v.2.4.1)60 with the ‘very-sensitive’ preset to the combined alignment reference containing the N. meningitidis 8013 genome sequence (RefSeq accession no. NC_017501.1; assembly: GCF_000026965.1) and the MDAΦ genome sequence27. For samples with UMIs, aligned spacers were deduplicated using UMI-tools with the directional-adjacency method, as noted above. For samples without UMIs, to handle technical artefacts such as PCR jackpot sites that can over-amplify a subset of spacer sequences, a maximum cap of 5% of the total detected spacer count on the corresponding Neisseria or phage genome was applied to each unique spacer sequence. More specifically, for each unique spacer sequence, if its abundance among the total spacers exceeded 5%, it was set to 5% of the total detected spacer count of the given source genome. For native acquisition samples in Extended Data Fig. 7, the interquartile range method was used to handle technical artefacts such as PCR jackpots. The identified outlier spacer sequences were assigned the average spacer counts of the remaining spacer sequences.
Reads that mapped ambiguously were removed from further analysis using Samtools (v.1.9)61. For each genomic location aligned with the 5′-end of any new spacer, the genomic context was extracted as the reference genome sequences flanking the alignment location, including the sequence upstream 10 bp from the alignment sites. The numbers of spacers at each unique spacer-aligned location were counted using bedtools2 (v.2.26.0)62, and custom codes were used to format the genomic context sequences for sequence logo inputs. For each adaptation NGS sample, a position frequency matrix was created by counting the occurrence of each nucleotide at each position upstream of the spacer alignment location.
PAM logo generation
All the PAM logos were generated through WebLogo 3 with custom parameters (--number-interval 1 --errorbars NO --stack-widt 90 --aspect-ratio 3 --color #CC0000 T ‘Pyrimidine’ --color #000DCC C ‘Pyrimidine’ --color #FFB300 G ‘Purine’ --color #00CC13 A ‘Purine’)63.
Viral protospacer distribution plot
For the NGS dataset of Fig. 1d (lane 4), phage-derived protospacers and their frequencies were extracted after sequence alignment. To create the distribution plot in Extended Data Fig. 2e, protospacers were grouped into 200-bp bins along the 8-kb viral genome. Reads per million viral reads (RPMviral) was calculated as viral protospacer reads within a bin per million total viral reads, as described in a previous study64. Finally, RPMviral values were plotted on the y axis with cubic spline smoothing.
4-mer PAM variant usage plot
For the NGS sample in Fig. 1d (lane 4), we counted the abundance of all viral spacer acquisition events for each of 256 possible 4-mer permutations at the 3′ 5–8 nt protospacer-flanking position (listed in Supplementary Table 5). A distribution plot was generated, ranking their frequencies in descending order, as shown in Extended Data Fig. 2h.
Nucleotide frequency plot
For the NGS sample in Fig. 1d (lane 4), the 8-nt 3′ flanks of protospacers were extracted, and the frequency of occurrence for each nucleotide base was calculated at each position. For visualization, a frequency plot was generated, with a baseline of 25% frequency included in the plot to reference random distribution.
All custom codes were run using Python (v.3.7.3)65 or R (v.3.6.2)66. A complete list of packages and their versions for a typical R session is included in the GitHub repository for source codes.
Large-scale analysis of natural spacers from the PubMLST database
All Neisseria isolate sequences were retrieved from the PubMLST database67, resulting in a collection of 53,174 strains. The CRISPR–Cas systems were identified using CRISPRCasFinder68,69,70, leading to 15,179 II-C arrays as having repeats with more than 80% identity to the repeat sequence of N. meningitidis strain 8013 (GTTGTAGCTCCCTTTCTCATTTCGCAGTGCTACAAT). The spacer sequences were extracted to identify those with a 100% protospacer match to their own host genome. Flanking regions of the protospacers were then analysed to determine the presence or absence of a functional PAM for the respective Cas9.
Recombinant protein expression and purification
NmeCas9 protein was expressed and purified, as previously described71, except for the omission of the ion exchange chromatography step. To purify NmeCas1 and Cas2 individually, plasmid pYZ22 or pYZ1515 was transformed into E. coli BL21(DE3) competent cells. Protein expression was induced, as described for NmeCas9 (ref. 71), except that 50 μg ml−1 of carbenicillin was used for pYZ22. Bacterial pellet for Cas1 or Cas2 was resuspended in lysis buffer (1x phosphate-buffered saline (PBS) (pH 7.5), 500 mM NaCl, 20 mM imidazole (Sigma; I202), 0.5 mM Tris(2-carboxyethyl)phosphine (Thermo Fisher Scientific; 77720) and 1x FastBreak Cell Lysis Reagent (Promega; V8571)) and lysed by means of sonication. Clarified lysates were bound to nickel–nitrilotriacetic acid resin (QIAGEN; 30210) and eluted with 1x PBS (pH 7.5), 500 (for Cas1) or 350 (for Cas2) mM NaCl, 500 mM imidazole and 0.5 mM Tris(2-carboxyethyl)phosphine. For His–NmeCas1, the eluate was concentrated and loaded onto a Sephacryl S-200 column (Cytiva; 17116601) with buffer 1x PBS (pH 7.5), 350 mM NaCl and 0.5 mM dithiothreitol (DTT; Thermo Fisher Scientific; BP17225). Fractions containing His–NmeCas1 were pooled, concentrated to 5 mg ml−1, aliquoted and stored at −80 °C. A 1-ml aliquot was used for custom antibody production (GenScript; affinity-purified polyclonal antibody rabbit).
For His–SUMO–NmeCas2, the nickel–nitrilotriacetic acid resin eluate was mixed with SUMO protease and dialysed overnight at 4 °C in dialysis buffer (1x PBS (pH 7.5), 350 mM NaCl and 0.5 mM DTT). To remove the SUMO protease, the resulting mixture was loaded onto a HiTrap Heparin column (Cytiva; 17040703) and eluted with a step gradient of NaCl (1x PBS (pH 7.5) with 600 mM, 850 mM and 2 M NaCl). Fractions containing NmeCas2 (cleaved and uncleaved) but lacking SUMO protease were pooled, concentrated to 1 ml and further loaded onto a Sephacryl S-200 column for size exclusion chromatography. Cleaved NmeCas2 can be separated from uncleaved His–SUMO–NmeCas2 in this step. S-200 fractions containing only cleaved NmeCas2 were pooled, concentrated and stored at −80 °C. A 0.7-ml aliquot of the resulting 6.8 mg ml−1 protein was used for custom antibody production (GenScript; affinity-purified polyclonal antibody rabbit).
In vitro transcription
All crRNAs, tracrRNAs and sgRNAs used in the biochemical assays were generated by in vitro transcription using an AmpliScribe T7-Flash Transcription Kit (Lucigen; ASF3507) and gel-purified by means of 15% denaturing PAGE, as described in a previous study72. Gel slices were eluted with agitation in 0.3 M NaCl–TE (10 mM Tris and 1 mM EDTA (pH 7.5)) overnight and RNA recovered by means of isopropanol precipitation. The transcription templates used were gel-purified PCR amplicons or annealed DNA oligo pairs.
Biotin-16-UTP-labelled RNA probes for northern blot were in vitro transcribed using an AmpliScribe T7-Flash Transcription Kit, along with a Biotin RNA Labeling Mix (Roche; 11685597910) according to the manufacturer’s instructions. The RNA products were purified using RNA Clean & Concentrator-5 (ZYMO; R1015). Annealed oligo pairs OYZ1166/1167, OYZ1185/1186 and OYZ1183/1184 were used as templates to synthesize anti-tracr, anti-repeat and anti-5S probes, respectively. All DNA oligos used for in vitro transcription contained T7 promoter sequence (see Supplementary Table 1 for details).
RNA electrophoretic mobility shift assay
The in vitro binding reactions were assembled and resolved on a 6% native PAGE–0.5x Tris–borate–EDTA (TBE), as previously described71, except that the final concentration for apoCas9 was 500 nM, small RNAs were each at 50 nM and no DNA targets were included. The 1x binding buffer is 20 mM HEPES (Sigma; H4034; pH 7.5), 150 mM KCl (Sigma; P9541), 0.1 mM EDTA, 0.5 mM DTT and 30 μg ml−1 of heparin (Sigma; H4784, as nonspecific competitor). The gels were stained with SYBR Gold (Invitrogen; S11494) for nucleic acid visualization on a Bio-Rad ChemiDoc MP Imager (see Supplementary Fig. 1 for uncropped gel images).
Limited proteolysis of NmeCas9
Limited proteolysis was conducted in 10-μl reactions using 5 μM NmeCas9 with or without in vitro transcribed small RNA partners (1:1.5 molar ratio; 7.5 μM). NmeCas9 and RNAs were pre-incubated at room temperature for 30 min in 1x binding buffer (20 mM HEPES (pH 7.5), 150 mM KCl, 0.1 mM EDTA, 0.5 mM DTT and 15 μg ml−1 of heparin). Trypsin proteolysis was initiated by adding 13-ng trypsin (NEB; P8101S) to each 10-μl reaction for 5-min, 10-min and 30-min digestion and then quenched using 2x Laemmli Sample Buffer (Bio-Rad; 1610737). The final products were resolved on a 4–15% gradient SDS–PAGE, stained with Coomassie blue (Bio-Rad; 1610786) and imaged on Bio-Rad ChemiDoc MP Imager (see Supplementary Fig. 1 for uncropped gel images).
Total RNA extraction and northern blot
Total RNA, including small RNA, was extracted from bacterial strains using miRNeasy Mini Kit (QIAGEN; 217004), as described previously26. Northern blot was performed, as described previously26 with minor modifications. Total RNA (3 μg) for each sample was separated by means of electrophoresis using 15% polyacrylamide/7 M urea/1x TBE gel in an 18-well cassette (Bio-Rad; 3459902). After electroblotting at 4 °C overnight at 14 V to a Zeta-Probe Membrane (Bio-Rad; 1620165) in 1x TBE, RNAs were crosslinked to the membrane by ultraviolet irradiation without any chemical treatment. Blots were prehybridized with 8 ml ULTRAhyb Buffer (Invitrogen; AM8670) at 60 °C for 30 min and then probed at 42 °C overnight with 300 ng of in vitro transcribed biotin-16-UTP-labelled RNA probe. The blots were washed at room temperature twice with 2x saline–sodium citrate (SSC)/0.1% SDS for 5 min and then twice with 0.1x SSC/0.1% SDS for 15 min. A 20x SSC solution contains 3 M NaCl and 0.3 M sodium citrate (Sigma; S4641) (pH 7.0). The hybridized biotin probes were detected using the Chemiluminescent Nucleic Acid Detection Module Kit (Thermo Fisher Scientific; 89880) and Clarity Max ECL Substrate (Bio-Rad; 1705062) according to the manufacturers’ instructions, followed by visualization on Bio-Rad ChemiDoc MP Imager. The same blot was then stripped by means of incubation in 80 ml of 1% SDS at 70 °C for 1 h with agitation and rehybridized with a different probe for other RNAs or 5S loading control (see Supplementary Fig. 1 for uncropped images).
Transcriptome profiling library construction
Total RNAs (2 µg), resulted from the previous section, were subjected to ribosomal RNA depletion using the Ribo-Zero Magnetic Kit (Epicentre; MRZMB126), followed by transcriptome profiling library construction using the NEBNext Ultra II Directional RNA Library Prep Kit for Illumina (NEB; E7760S) following the manufacturer’s instructions. Libraries were quantified using Qubit dsDNA HS Kits (Thermo Fisher Scientific; Q32854) and sequenced on a NextSeq 1000 instrument using XLEAP P1 flow cell (Illumina) with 300-bp single-end reads.
Western blot
Neisseria cells were collected from GCB plates or in-liquid adaptation assay. Cell resuspension equivalent to 200 μl of OD600 of 1.0 culture was pelleted, washed once with 1x PBS and lysed and denatured in 200 μl of lysis buffer (1x PBS, 1x Laemmli Sample Buffer and 50 mM DTT) by heating at 95 °C for 5 min. Subsequently, 15 μl of each denatured sample, along with 5-μl molecular weight markers (Bio-Rad; 1610374), was separated on a 10% (for Cas9) or 15% (for Cas1 or Cas2) SDS–PAGE and transferred to 0.2-μm pore polyvinylidene fluoride membrane (Cytiva; 10600021) using Trans-Blot SD Semi-Dry Transfer Cell (Bio-Rad; 1703940) in blotting buffer (25 mM Tris, 192 mM glycine, 0.03% SDS and 20% methanol (pH 8.3)) at room temperature for 30 min at 15 V. After transfer, the membrane was soaked in blocking buffer (3% non-fat dry milk in 1x Tris-buffered saline with Tween 20 (TBST)) at room temperature for 30 min and then incubated with the primary antibody in blocking buffer at 4 °C overnight. The primary antibodies used were rabbit anti-NmeCas1 (custom; 1:370), anti-NmeCas9 (Abcam; ab202638; 1:5,000) and anti-FLAG M2 (Cell Signaling Technology; 2368S; 1:5,000). After washing three times with 1x TBST at room temperature for 10 min, the membrane was incubated with horseradish peroxidase-conjugated anti-rabbit immunoglobulin G secondary antibody (Promega; W401B; 1:5,000) in blocking buffer at room temperature for 1 h. The blot was washed three times with 1x TBST at room temperature for 10 min, treated with Clarity Western ECL Substrate (Bio-Rad; 1705060) and imaged on Bio-Rad ChemiDoc MP Imager (see Supplementary Fig. 1 for uncropped images). For loading control, the same blot was rinsed with 1x TBST and stripped with 2-ml OneMinute Plus Western Blot Stripping Buffer (GM Biosciences; GM6011). The stripped blot was re-probed with rabbit anti-GroEL (Abcam; ab90522; 1:10,000).
Nanopore library construction
All steps were performed using wide-bore pipette tips to minimize DNA shearing. Total DNA was purified from Neisseria cells equivalent to 10 ml of OD600 of 1.0 culture. The cell pellet was resuspended in lysis buffer (30 mM Tris (pH 8.0), 1 mM EDTA (pH 8.0) and 1.5 mg ml−1 of lysozyme (Sigma; L6876)), supplemented with 40 μl of 20 mg ml−1 of proteinase K (Promega; MC500B) and incubated at room temperature for 10 min. Thereafter, 20 μl of 100 mg ml−1 of RNase A (QIAGEN; 19101) was added, followed by 15-min incubation at 37 °C. DNA was then purified by phenol–chloroform extraction and ethanol precipitation and resuspended in 10 mM Tris (pH 8.5). Nanopore sequencing library was prepared from this DNA using Oxford Nanopore Ligation Sequencing Kit (SQK-LSK109) with native barcodes (EXP-NBD104) according to the manufacturer’s instructions. The library was sequenced using a MinION 9.4.1 flow cell, followed by high-quality base calling.
Nanopore informatic analysis
Reads were aligned to N. meningitidis 8013 reference genome and MDAΦ–Kan genome separately using minimap2 (ref. 73). MDAΦ integrations (that is, reads with alignments to both genomes) were classified as ‘complete’ if the full phage genome was observed flanked by the host genome on either side (junctions identified by minimap2 within 200 bp of the genome start/end), ‘canonical’ if a single such junction was observed or ‘other’ if no junction near the end of the phage genome could be detected. Potential dRS3 sites were defined by taking the set of sites showing the best scores observed in the genome for the motif ATTCCCGCCTGCGCGGGAAT, assessed using FIMO74 and cross-referenced with the end points of the phage integrations using bedtools62.
Adaptation-MDAΦ defence assay
For the experiments shown in Extended Data Fig. 9, we assayed strains lacking MDAΦ-targeting spacers by conducting 3–4 h MDAΦ infection in the absence or presence of IPTG-induced Cas1–2 overexpressing to trigger acquisition simultaneously. The reactions were then plated on KanR-selective versus non-selective GCB plates to quantify the MDAΦ infection levels. The ratios of transductant counts comparing conditions with or without IPTG were calculated to assess adaptation-triggered MDAΦ defence.
Phylogenetic analysis
For Cas9 phylogenetic analyses in Extended Data Fig. 10b, multiple alignments were performed using MSAProbs75 at https://toolkit.tuebingen.mpg.de. Phylogenetic trees were created using the maximum likelihood method with PhyML (substitution model, WAG; number of categories, four) available on the web server Phylogeny.fr (http://www.phylogeny.fr/)76.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
All demultiplexed raw adaptation NGS sequencing reads and processed new spacer reads, raw RNA sequencing reads and processed results, nanopore raw sequencing reads and processed data are available at the NCBI Gene Expression Omnibus (accession no. GSE302092). Uncropped gel images are shown in Supplementary Fig. 1. All other data supporting the findings of this study are available within the paper and Supplementary Information. All plasmids and custom antibodies are available upon request. Full plasmid sequences and maps are not included but are available upon request.
Code availability
Source codes for sequencing data analysis are available at GitHub (https://github.com/freddolino-lab/2023_CRISPR_adaptation).
References
Barrangou, R. et al. CRISPR provides acquired resistance against viruses in prokaryotes. Science 315, 1709–1712 (2007).
Heler, R. et al. Cas9 specifies functional viral targets during CRISPR–Cas adaptation. Nature 519, 199–202 (2015).
Wei, Y., Terns, R. M. & Terns, M. P. Cas9 function and host genome sampling in type II-A CRISPR–Cas adaptation. Genes Dev. 29, 356–361 (2015).
Brouns, S. J. et al. Small CRISPR RNAs guide antiviral defense in prokaryotes. Science 321, 960–964 (2008).
Garneau, J. E. et al. The CRISPR/Cas bacterial immune system cleaves bacteriophage and plasmid DNA. Nature 468, 67–71 (2010).
Deveau, H. et al. Phage response to CRISPR-encoded resistance in Streptococcus thermophilus. J. Bacteriol. 190, 1390–1400 (2008).
Mojica, F. J., Diez-Villasenor, C., Garcia-Martinez, J. & Almendros, C. Short motif sequences determine the targets of the prokaryotic CRISPR defence system. Microbiology 155, 733–740 (2009).
Marraffini, L. A. & Sontheimer, E. J. Self versus non-self discrimination during CRISPR RNA-directed immunity. Nature 463, 568–571 (2010).
Yosef, I., Goren, M. G. & Qimron, U. Proteins and DNA elements essential for the CRISPR adaptation process in Escherichia coli. Nucleic Acids Res. 40, 5569–5576 (2012).
Datsenko, K. A. et al. Molecular memory of prior infections activates the CRISPR/Cas adaptive bacterial immunity system. Nat. Commun. 3, 945 (2012).
Nunez, J. K. et al. Cas1–Cas2 complex formation mediates spacer acquisition during CRISPR–Cas adaptive immunity. Nat. Struct. Mol. Biol. 21, 528–534 (2014).
Kieper, S. N. et al. Cas4 facilitates PAM-compatible spacer selection during CRISPR adaptation. Cell Rep. 22, 3377–3384 (2018).
Lee, H., Zhou, Y., Taylor, D. W. & Sashital, D. G. Cas4-dependent prespacer processing ensures high-fidelity programming of CRISPR arrays. Mol. Cell 70, 48–59 (2018).
Shiimori, M., Garrett, S. C., Graveley, B. R. & Terns, M. P. Cas4 nucleases define the PAM, length, and orientation of DNA fragments integrated at CRISPR loci. Mol. Cell 70, 814–824 (2018).
Hu, C. et al. Mechanism for Cas4-assisted directional spacer acquisition in CRISPR–Cas. Nature 598, 515–520 (2021).
Wang, J. Y. et al. Genome expansion by a CRISPR trimmer-integrase. Nature 618, 855–861 (2023).
Kim, S. et al. Selective loading and processing of prespacers for precise CRISPR adaptation. Nature 579, 141–145 (2020).
Shiriaeva, A. A. et al. Host nucleases generate prespacers for primed adaptation in the E. coli type I-E CRISPR-Cas system. Sci. Adv. 8, eabn8650 (2022).
Jinek, M. et al. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 337, 816–821 (2012).
Deltcheva, E. et al. CRISPR RNA maturation by trans-encoded small RNA and host factor RNase III. Nature 471, 602–607 (2011).
Gasiunas, G., Barrangou, R., Horvath, P. & Siksnys, V. Cas9–crRNA ribonucleoprotein complex mediates specific DNA cleavage for adaptive immunity in bacteria. Proc. Natl Acad. Sci. USA 109, E2579–E2586 (2012).
Wilkinson, M. et al. Structure of the DNA-bound spacer capture complex of a type II CRISPR-Cas system. Mol. Cell 75, 90–101 (2019).
Shmakov, S. et al. Diversity and evolution of class 2 CRISPR–Cas systems. Nat. Rev. Microbiol. 15, 169–182 (2017).
Mir, A., Edraki, A., Lee, J. & Sontheimer, E. J. Type II-C CRISPR-Cas9 biology, mechanism, and application. ACS Chem. Biol. 13, 357–365 (2018).
Garrett, S. C. Pruning and tending immune memories: spacer dynamics in the CRISPR array. Front. Microbiol. 12, 664299 (2021).
Zhang, Y. et al. Processing-independent CRISPR RNAs limit natural transformation in Neisseria meningitidis. Mol. Cell 50, 488–503 (2013).
Meyer, J. et al. Characterization of MDAPhi, a temperate filamentous bacteriophage of Neisseria meningitidis. Microbiology 162, 268–282 (2016).
Bille, E. et al. A virulence-associated filamentous bacteriophage of Neisseria meningitidis increases host-cell colonisation. PLoS Pathog. 13, e1006495 (2017).
Hay, I. D. & Lithgow, T. Filamentous phages: masters of a microbial sharing economy. EMBO Rep. 20, e47427 (2019).
Sternberg, S. H., Richter, H., Charpentier, E. & Qimron, U. Adaptation in CRISPR-Cas systems. Mol. Cell 61, 797–808 (2016).
Sun, W. et al. Structures of Neisseria meningitidis Cas9 complexes in catalytically poised and anti-CRISPR-inhibited states. Mol. Cell 76, 938–952 (2019).
Edraki, A. et al. A compact, high-accuracy Cas9 with a dinucleotide PAM for in vivo genome editing. Mol. Cell 73, 714–726 (2019).
Wei, Y., Chesne, M. T., Terns, R. M. & Terns, M. P. Sequences spanning the leader-repeat junction mediate CRISPR adaptation to phage in Streptococcus thermophilus. Nucleic Acids Res. 43, 1749–1758 (2015).
Liao, C. et al. Spacer prioritization in CRISPR-Cas9 immunity is enabled by the leader RNA. Nat. Microbiol. 7, 530–541 (2022).
Jiang, F. & Doudna, J. A. CRISPR–Cas9 structures and mechanisms. Annu. Rev. Biophys. 46, 505–529 (2017).
Jiang, F., Zhou, K., Ma, L., Gressel, S. & Doudna, J. A. A Cas9-guide RNA complex preorganized for target DNA recognition. Science 348, 1477–1481 (2015).
Jiang, W. et al. Dealing with the evolutionary downside of CRISPR immunity: bacteria and beneficial plasmids. PLoS Genet. 9, e1003844 (2013).
Gudbergsdottir, S. et al. Dynamic properties of the Sulfolobus CRISPR/Cas and CRISPR/Cmr systems when challenged with vector-borne viral and plasmid genes and protospacers. Mol. Microbiol. 79, 35–49 (2011).
Hamilton, H. L. & Dillard, J. P. Natural transformation of Neisseria gonorrhoeae: from DNA donation to homologous recombination. Mol. Microbiol. 59, 376–385 (2006).
Fonfara, I. et al. Phylogeny of Cas9 determines functional exchangeability of dual-RNA and Cas9 among orthologous type II CRISPR-Cas systems. Nucleic Acids Res. 42, 2577–2590 (2014).
Fedorova, I. et al. PpCas9 from Pasteurella pneumotropica — a compact type II-C Cas9 ortholog active in human cells. Nucleic Acids Res. 48, 12297–12309 (2020).
Wei, J. et al. Closely related type II-C Cas9 orthologs recognize diverse PAMs. eLife 11, e77825 (2022).
Westra, E. R., Buckling, A. & Fineran, P. C. CRISPR–Cas systems: beyond adaptive immunity. Nat. Rev. Microbiol. 12, 317–326 (2014).
Louwen, R., Staals, R. H., Endtz, H. P., van Baarlen, P. & van der Oost, J. The role of CRISPR-Cas systems in virulence of pathogenic bacteria. Microbiol. Mol. Biol. Rev. 78, 74–88 (2014).
Workman, R. E. et al. A natural single-guide RNA repurposes Cas9 to autoregulate CRISPR-Cas expression. Cell 184, 675–688 (2021).
Ratner, H. K. et al. Catalytically active Cas9 mediates transcriptional interference to facilitate bacterial virulence. Mol. Cell 75, 498–510(2019).
Pourcel, C. et al. CRISPRCasdb a successor of CRISPRdb containing CRISPR arrays and cas genes from complete genome sequences, and tools to download and query lists of repeats and spacers. Nucleic Acids Res. 48, D535–D544 (2020).
Heidrich, N. et al. The primary transcriptome of Neisseria meningitidis and its interaction with the RNA chaperone Hfq. Nucleic Acids Res. 45, 6147–6167 (2017).
Camara-Wilpert, S. et al. Bacteriophages suppress CRISPR–Cas immunity using RNA-based anti-CRISPRs. Nature 623, 601–607 (2023).
Bhattarai-Kline, S. et al. Recording gene expression order in DNA by CRISPR addition of retron barcodes. Nature 608, 217–225 (2022).
Sheth, R. U., Yim, S. S., Wu, F. L. & Wang, H. H. Multiplex recording of cellular events over time on CRISPR biological tape. Science 358, 1457–1461 (2017).
Shipman, S. L., Nivala, J., Macklis, J. D. & Church, G. M. CRISPR–Cas encoding of a digital movie into the genomes of a population of living bacteria. Nature 547, 345–349 (2017).
Jian, H. et al. The origin and impeded dissemination of the DNA phosphorothioation system in prokaryotes. Nat. Commun. 12, 6382 (2021).
Jian, H., Xu, J., Xiao, X. & Wang, F. Dynamic modulation of DNA replication and gene transcription in deep-sea filamentous phage SW1 in response to changes of host growth and temperature. PLoS ONE 7, e41578 (2012).
Andrews, S. et al. FastQC v.0.11.8 (Babraham Institute, 2010).
Ewels, P., Magnusson, M., Lundin, S. & Kaller, M. MultiQC: summarize analysis results for multiple tools and samples in a single report. Bioinformatics 32, 3047–3048 (2016).
Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J. 17, 10–12 (2011).
Edwards, J. A. & Edwards, R. A. Fastq-pair: efficient synchronization of paired-end fastq files. Preprint at bioRxiv https://doi.org/10.1101/552885 (2019).
Smith, T., Heger, A. & Sudbery, I. UMI-tools: modeling sequencing errors in Unique Molecular Identifiers to improve quantification accuracy. Genome Res. 27, 491–499 (2017).
Langmead, B. & Salzberg, S. L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359 (2012).
Li, H. et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079 (2009).
Quinlan, A. R. & Hall, I. M. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842 (2010).
Crooks, G. E., Hon, G., Chandonia, J. M. & Brenner, S. E. WebLogo: a sequence logo generator. Genome Res. 14, 1188–1190 (2004).
Modell, J. W., Jiang, W. & Marraffini, L. A. CRISPR–Cas systems exploit viral DNA injection to establish and maintain adaptive immunity. Nature 544, 101–104 (2017).
van Rossum, G. & Drake, F. Python 3 Reference Manual (CreateSpace, 2009).
R Core Team. R: A Language and Environment for Statistical Computing (R Foundation for Statistical Computing, 2019).
Jolley, K. A., Bray, J. E. & Maiden, M. C. J. Open-access bacterial population genomics: BIGSdb software, the PubMLST.org website and their applications. Wellcome Open Res. 3, 124 (2018).
Couvin, D. et al. CRISPRCasFinder, an update of CRISRFinder, includes a portable version, enhanced performance and integrates search for Cas proteins. Nucleic Acids Res. 46, W246–W251 (2018).
Grissa, I., Vergnaud, G. & Pourcel, C. CRISPRFinder: a web tool to identify clustered regularly interspaced short palindromic repeats. Nucleic Acids Res. 35, W52–W57 (2007).
Abby, S. S., Neron, B., Menager, H., Touchon, M. & Rocha, E. P. MacSyFinder: a program to mine genomes for molecular systems with an application to CRISPR-Cas systems. PLoS ONE 9, e110726 (2014).
Rousseau, B. A., Hou, Z., Gramelspacher, M. J. & Zhang, Y. Programmable RNA cleavage and recognition by a natural CRISPR-Cas9 system from Neisseria meningitidis. Mol. Cell 69, 906–914 (2018).
Gramelspacher, M. J., Hou, Z. & Zhang, Y. Biochemical characterization of RNA-guided ribonuclease activities for CRISPR-Cas9 systems. Methods 172, 32–41 (2020).
Li, H. Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics 34, 3094–3100 (2018).
Grant, C. E., Bailey, T. L. & Noble, W. S. FIMO: scanning for occurrences of a given motif. Bioinformatics 27, 1017–1018 (2011).
Gonzalez-Dominguez, J., Liu, Y., Tourino, J. & Schmidt, B. MSAProbs-MPI: parallel multiple sequence aligner for distributed-memory systems. Bioinformatics 32, 3826–3828 (2016).
Ipoutcha, T. et al. Multiple origins and specific evolution of CRISPR/Cas9 systems in minimal bacteria (Mollicutes). Front. Microbiol. 10, 2701 (2019).
Acknowledgements
We thank E. Bille and X. Nassif for sharing MDAΦ stocks and protocols; A. Pawluk for helpful feedback on the paper; H. S. Seifert, E. J. Sontheimer and A. Ke for critical reading of the paper; J. P. Dillard for providing Neisseria genomic complementation constructs; Y. Liu, J. Heo and T. Tripathi for technical support; University of Michigan Advanced Genomics Core Facility for NGS support; and all Zhang laboratory members for thoughtful discussions. X.L. was supported by a Rackham International Student Fellowship; M.J.G. and X.L. were supported by a Rackham Graduate Student Research Grant; and C.A.Z. was supported by the National Science Foundation Fellowship (DGE1256260) and NIH Michigan Predoctoral Training in Genetics (T32GM007544). This study was supported by the National Institutes of Health grants GM117268 and GM137883 to Y.Z., GM128637 and AI134678 to L.F. and the University of Michigan Institutional Fund and Biological Scholar Award to Y.Z.
Author information
Authors and Affiliations
Contributions
X.Z., X.L., Z.H. and Y.Z. conceived the project. X.L. performed phage production, characterization and transduction. X.Z., X.L., C.A.Z., Z.H. and Y.Z. created strains, performed adaptation assays and constructed NGS libraries. R.D., X.L., Z.H. and L.F. conducted informatic analyses. X.Z. and X.L. performed interference assays, western blot, northern blot and biochemical assays. M.J.G. purified all proteins. L.F., Z.H. and Y.Z. supervised the project. X.Z., R.D., X.L., L.F., Z.H. and Y.Z. analysed the data and wrote the paper. All authors read, edited and approved the paper.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature thanks Chase Beisel and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data figures and tables
Extended Data Fig. 1 MDAΦ lysogenization of Nme strain 8013.
a, MDAΦ lysogenization requires major pilin pilE, but not the minor pilin pilX or other host factors for natural transformation. To validate host factor requirements for MDAΦ infection, we tested a panel of host gene deletion strains. Only ΔpilE abolished MDAΦ transduction, confirming type IV pili’s role as phage receptor and that KanR colonies are indeed transductants, not transformants caused by genomic DNA contamination in phage prep. MDAΦ transduction frequencies % (transductants per CFU of total host cells) in mutants are normalized to that of WT strain. Data are mean ± s.d., n = 3. b, Natural transformation test in the same set of strains as in a. Transformants were abolished by ΔRecA, ΔcomA, ΔdprA, ΔcomP, and ΔpilE, consistent with these host factors’ known roles in competency. c, Illustration of MDAΦ life cycle in Nme. MDAΦ infects mainly as episomes, including circular ssDNA viral genome and dsDNA replication form (RF). It can also integrate into host chromosome as a prophage (green) via conserved dRS3 sites (orange). d, Nanopore sequencing of total DNA from MDAΦ transductant pool to confirm the existence of MDAΦ prophage. e, Diagnostic PCRs detecting the circular junction of MDAΦ episome and prophage-host integration junction from total DNA of MDAΦ transductant pool. WT strain without MDAΦ infection is the negative control. Green and orange arrows indicate PCR primers used with annealing sites depicted in d. f, h, Real-time PCR determination of the relative copy numbers of circular MDAΦ (f) and dRS3-MDAΦ junction (h), normalized to Neisseria host gene NMV_1851 copy set as 1. On average, 10 copies of MDAΦ episome exist per host genome as shown by panel f and about 25% of the transductants’ genome have a MDAΦ prophage as shown in panel h. g, i, PCR standard curves created on serial 10-fold dilutions of reference plasmids. CT values plotted against the log scale of known initial copy numbers. Data are mean values from 3 independent experiments.
Extended Data Fig. 2 New spacers are preferentially acquired from MDAΦ and NmeCas9’s PID dictates PAM enrichment for acquisition.
a, SDS-PAGE of purified NmeCas1 and Cas2 proteins used for custom antibody production. b, Cas1 protein induced from a cas1-2 OE cassette integrated into the host genome. 0.1 mM IPTG is used in subsequent experiments. Top, anti-Cas1 western blot using custom antibody; bottom, GroEL probed as loading control. c, Overview of the informatic analysis for new spacers. d, MDAΦ vs. host genome breakdown for DNA source of new spacers from “+1” band in lanes 1 and 4 of Fig. 1d and lane 4 of Fig. 1f. Red dashed line, 3.5 % should be MDAΦ-derived if there is no viral versus self- discrimination by acquisition. Data are mean ± s.d., n = 3. e, New spacers from “+1” band in lane 4 of Fig. 1d are analyzed in e, g, h. Distribution of spacer abundance (reads per million of phage-matching across the MDAΦ genome. f, Transcriptome profiling of MDA transuctants shows that protospacer sampling is not driven by viral transcript abundance. g, Nucleotide frequency plot for viral protospacers. Black line, 25% G/A/T/C equal frequency. h, All 256 possible 4-mers for positions 5-8 nts 3′ of protospacers ranked in descending order of abundance. N4GATT PAM, red dot; all other 4-mers, blue dots. i, Domain architecture of WT, dcas9, cas9H1024A and cas9PID-swap alleles, with D16A and H588A mutations denoted by black stars and H1024A PAM-binding-disrupting mutation by red star. j, Genomically-complemented cas9 mutants are expressed to similar level as WT control in strains in Fig. 1f, g. Top, anti-NmeCas9 western blot; bottom, GroEL as loading control. k, Interference assay showing that dcas9 and cas9H1024A are interference defective. cas9PID-swap altered PAM to 3′-N4CC in interference. Data are log-scale of CFU/mL (mean ± s.d., n = 3) for transformants (red bars) and total cells (blue bars). NS, not significant (P ≥ 0.05), * 0.005 ≤ P < 0.05, ** P < 0.005; P values calculated by two-tailed Welch’s t-tests.
Extended Data Fig. 3 Super-adaptation in Δtracr is not due to lack of interference or changes to cas expressions.
a, Schematic of Nme strains used in Fig. 2b, c and Extended Data Fig. 3b–h. b, Northern blot confirming tracrRNA’s absence and complementation. Total RNAs were probed for tracrRNA (top), crRNA (bottom), and 5S rRNA (loading control, middle). Mature crRNAs disappeared in Δtracr as expected, as they must co-load into Cas9 along with tracrRNA to stably exist. c, MDAΦ versus host breakdown for DNA source of new spacers from “+1” bands of lanes 2 and 4 in Fig. 2b. In super-adaptation, viral DNA remained preferred over the host genome. Red dashed line, theoretical random breakdown without any viral vs. self- discrimination. Data are mean ± s.d., n = 3. d-e, Super-adaptation is not due to changes in Cas9 or Cas1 protein levels. Anti-Cas9 (d) and anti-Cas1 (e, w/ or w/o IPTG induction) western blots showed comparable protein levels between Δtracr and WT strains. GroEL, loading control. f, Interference assay for strains used in Fig. 2b. Interference is abolished by Δtracr and restored by tracr complementation. Data are shown as in Extended Data Fig. 2k. g, Strains isogenic to those in Fig. 2b but encode dcas9 instead of WT cas9 showed similar super-adaptation phenotypes. Top, a representative adaptation PCR gel; bottom, quantification of adaptation efficiencies. Data are mean ± s.d., n = 3. NS, not significant (P ≥ 0.05), * 0.005 ≤ P < 0.05, ** P < 0.005; P values calculated by two-tailed Welch’s t-tests. h, 3′ motif analysis for new viral spacers from panel g.
Extended Data Fig. 4 R26 allele is crRNA null.
a, Different crRNA biogenesis pathways in types II-A and II-C systems. Left, II-A crRNA is transcribed from an external promoter within the leader, producing crRNAs and a leader-derived extraneous crRNA. Right, II-C crRNA is transcribed from repeat-embedded internal promoters and initiates from the 6th nt of each spacer. b, II-C R26 allele is predicted to not produce any mature crRNA or crRNA mimic. c, northern blot validating b. Total RNAs of the R26 strain were probed for crRNA, tracrRNA, and 5S loading control. ΔCRISPR, Δtracr, and Δcas9 strains are controls. d, Schematic of dcas9-encoding trains used in panels e-f. e, R26 triggered super-adaptation is independent of interference or Cas9’s nuclease activity. Strains isogenic to those in Fig. 2e but encode WT cas9 were assayed for MDAΦ infection-acquisition, with or without IPTG induction of cas1-2. Data are shown as in Fig. 2e. Data are mean ± s.d., n = 3. NS, not significant (P ≥ 0.05), * 0.005 ≤ P < 0.05, ** P < 0.005; P values calculated by two-tailed Welch’s t-tests. f, 3′ flanking motif for new spacers from panel e. g-h, Super-adaptation in Fig. 2e is not due to changes in Cas9 or Cas1 protein levels. Anti-Cas1 (g, +/- IPTG induction) and anti-Cas9 (h) western blots showed comparable protein levels in FL CRISPR vs. R26 strains. GroEL is loading control.
Extended Data Fig. 5 ApoNmeCas9 exhibits acquisition-stimulatory activity and has distinct domain requirements as interference.
a, Interference assay showing that a crRNA-tracrRNA pair or sgRNA is required for Cas9 to block transformation. Data are shown as in Extended Data Fig. 2k. b, Cas9 is expressed to comparable levels in strains used in Fig. 3. Top, anti-Cas9 western blot, leaky expression from uninduced Tet promoter; bottom, GroEL, loading control. c, NmeCas9 has two states: an apo- state that stimulates acquisition and an RNA-loaded state that ensures low acquisition. d, RNA EMSA showed that NmeCas9 engage cognate crRNA and tracrRNA as a pair in vitro. Binding was performed at 37 °C for 30 min, in the absence (“-”) or presence (“+”) of NmeCas9. e, NmeCas9 is protected from trypsin proteolysis by its crRNA-tracr pair. Purified apoCas9 was pre-incubated with various RNAs, trypsin digested for 5-, 15-, or 30- min, then analyzed by SDS-PAGE. Input, without trypsin. f, Schematic of strains used in Fig. 4. Blue box, cas9 deletion mutants complemented to genomic iga-trpB locus, with or without an sgRNA. g, All but one Cas9 mutant (ΔRuvCIΔBH) is expressed in Neisseria. Top, anti-Flag western blot to track Cas9; bottom, GroEL probed as a loading control. h, All domain deletion mutants tested exhibited major defects in CRISPR interference. Data are shown as in Extended Data Fig. 2k.
Extended Data Fig. 6 Characterization of the evolution mimic, co-existing arrays, and interference escaper strains.
a, The evolution mimic series of strains with CRISPR arrays of varying lengths expressed Cas9 to comparable levels. Top, anti-Cas9 western blot; bottom, GroEL loading control. b, flanking motif analysis for new viral spacers from Fig. 5c. c, For each array length, crRNA levels quantified from Fig. 5b and acquisition efficiencies from Fig. 5c are plotted. d, CrRNA abundance and acquisition efficiency are inversely correlated, fitted with a one-phase decay non-linear regression model in Prism. e, Schematics of array-coexistence strains used for MDAΦ infection-acquisition test under cas1-2 induction in f. Genotypes at endogenous CRISPR locus and the 2nd CRISPR array inserted at the NICS genomic site are depicted. f, Total acquisition efficiency was evenly distributed between co-existing arrays, with longer 2nd arrays progressively reducing acquisition at both CRISPR loci within the same cell. Data shown are mean ± s.d., n = 3. NS, not significant (P ≥ 0.05), * 0.005 ≤ P < 0.05, ** P < 0.005; P values calculated by two-tailed Welch’s t-tests. g, Donut chart of interference escaping mechanisms. Purple, cas9 mutations (38/62); blue, CRISPR array collapse (13/62); red, target mutation or deletion (8/62); green, unclear (5/62). h, Interference escapers with mutated cas9 (e2, e3, e4) are permanently defective for interference, as revealed by interference re-test using transforming DNA bearing protospacers 6 or 25. i, Array-contracted escapers expressed Cas9 protein to comparable levels as the WT array. j, 3′ flanking motif for new viral spacers of Fig. 5i.
Extended Data Fig. 7 Native acquisition efficiency, albeit low, is elevated in shorter-array-strains, including natural escapers, evolutionary mimics, and naturally short-array isolates.
a, Un-engineered natural escapers from Fig. 5e,f assayed for native MDAΦ acquisition by NGS and exhibited elevated acquisition efficiencies. Notably, no Cas1-2 OE cassette was introduced and endogenous crispr-cas locus remained unchanged apart from natural array compactions. Acquisition efficiency was determined by NGS of all CRISPRs without gel extraction to enrich for the adapted arrays. Data are mean ± s.d., n = 3. NS, not significant (P ≥ 0.05), * 0.005 ≤ P < 0.05, ** P < 0.005; P values calculated by two-tailed Welch’s t-tests. b, 3′ flanking motifs for new viral spacers from panel a. c, Native acquisition efficiencies assayed and plotted as in panel a, for evolutionary mimics from Fig. 5a,b and strains encoding natural short arrays of Neisseria isolates CA40755 and M10 240569 (w/2-, 3-spacers). d, 3′ flanking motifs for new viral spacers from panel c.
Extended Data Fig. 8 ApoCas9 stimulates acquisition for collapsed arrays that arise via homologous recombination between CRISPR repeats.
a, Diagram of the repeat recombination experiment. WT 8013 strain is transformed with DNA that contains a repeat, a kanamycin-resistant marker, and a genomic sequence downstream of CRISPR locus. KanR selects for recombination events between the incoming repeat and recipient cells’ CRISPR array. b, Diagnostic PCR of representative, randomly selected KanR transformants showing that most have undergone array compaction. c, Representative shortened arrays (marked in red in panel b) have lost blocks of spacer-repeat units, revealed by sanger sequencing. d, Array-collapsed colonies elicit elevated MDAΦ-acquisition, with the efficiency increasing as array length decreases. Top, a representative adaptation PCR gel. Bottom, quantification of adaptation efficiencies. Data are mean ± s.d., n = 3. NS, not significant (P ≥ 0.05), * 0.005 ≤ P < 0.05, ** P < 0.005; P values calculated by two-tailed Welch’s t-tests. e, 3′ flanking motif analysis for new viral spacers of panel d.
Extended Data Fig. 9 Strains with shorter CRISPR arrays exhibited stronger resistance to MDAΦ lysogenization upon acquisition induction.
a, Nme 8013 strain derivatives, including those with varying-sized arrays and natural escaper e5, were infected with MDAΦ in the presence or absence of IPTG to induce Cas1-Cas2 OE. Plotted are ratios of CFU/mL comparing conditions with versus without IPTG-induced acquisition, with white bars representing transductants and grey bars representing total viable cells. Data are mean ± s.d., n = 3. NS, not significant (P ≥ 0.05), * 0.005 ≤ P < 0.05, ** P < 0.005; P values calculated by two-tailed Welch’s t-tests. b, Fold changes in transduction frequencies comparing IPTG-induced acquisition versus uninduced conditions from the same experiment in panel a. Transduction frequency is the percentage of recipient cells successfully transduced. Data are mean ± s.d., n = 3. NS, not significant (P ≥ 0.05), * 0.005 ≤ P < 0.05, ** P < 0.005; P values calculated by two-tailed Welch’s t-tests.
Extended Data Fig. 10 ApoCas9 and tracrRNA-crRNA play conserved roles in acquisition across divergent II-C systems.
a, Schematic of strains used. b, Phylogeny of II-C cas9 orthologs tested. Percentages of protein sequence identity relative to NmeCas9 are in parentheses. II-A SpyCas9 is included as an outgroup. c, Top, anti-Flag western blots confirmed the expression of II-C cas9 orthologs in Neisseria, and that their levels were not affected by the respective sgRNA. Asterisk, expression of CjeCas9, unlike other orthologs, required aTc induction of pTet to become detectable by western blot. GroEL, loading control. d, ApoCjeCas9 failed to induce super-adaptation. Left, a representative adaptation PCR. Right, quantification of adaptation efficiencies. Data are mean ± s.d., n = 3. NS, not significant (P ≥ 0.05), * 0.005 ≤ P < 0.05, ** P < 0.005; P values calculated by two-tailed Welch’s t-tests. e, 3′ flanking motifs analysis of new viral spacers from panel d. f, Multiple II-C Cas9s stimulated super-adaptation, which was repressed to baseline level by their respective sgRNAs. Top, representative adaptation PCR. Bottom, quantification of adaptation efficiencies, mean ± s.d., n = 3. NS, not significant (P ≥ 0.05), * 0.005 ≤ P < 0.05, ** P < 0.005; P values calculated by two-tailed Welch’s t-tests. g, 3′ flanking motifs for new viral spacers from panel f. h, CRISPR interference assay showed that WT Cas9 and sgRNA- expressing strains are competent for interference and requires the corresponding PAMs. Data are shown as in Extended Data Fig. 2k.
Supplementary information
Supplementary Information
This file contains Supplementary Fig. 1, Discussion and references. Supplementary Fig. 1 shows the uncropped images of DNA gels, western blots and northern blots.
Supplementary Tables
Supplementary Tables 1–5 contain lists of oligos, plasmids and their cloning strategies, N. meningitidis strains and their construction strategies, gBlocks used in the study and raw PAM counts for data shown in Extended Data Fig. 2h.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Zhou, X., Diao, R., Li, X. et al. Cas9 senses CRISPR RNA abundance to regulate CRISPR spacer acquisition. Nature (2025). https://doi.org/10.1038/s41586-025-09577-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41586-025-09577-9
