
Abstract
The formation of membrane pores by cleaved N-terminal gasdermin D (GSDMD-NT) results in the release of cytokines and inflammatory cell death, known as pyroptosis. Blocking GSDMD-NT pores is an attractive and promising strategy for mitigating inflammation. Here we demonstrate that SK56, an artificial intelligence-screened peptide, effectively obstructs GSDMD-NT pores and inhibits pyroptosis and cytokine release in macrophages and human peripheral blood leukocyte-induced pyroptosis. SK56 prevents septic death induced by lipopolysaccharide or cecal ligation and puncture surgery in mice. SK56 does not influence cleavage of interleukin-1β or GSDMD. Instead, SK56 inhibits the release of cytokines from pyroptotic macrophages, mitigates the activation of primary mouse dendritic cells triggered by incubation with pyroptotic cytomembranes and prevents widespread cell death of human alveolar organoids in an organoid–macrophage coculture model. SK56 blocks GSDMD-NT pores on lipid-bilayer nanoparticles and enters pyroptotic macrophages to inhibit mitochondrial damage. SK56 presents new therapeutic possibilities for counteracting inflammation, which is implicated in numerous diseases.
Similar content being viewed by others

Main
The gasdermin (GSDM) family, which encompasses members GSDMA to GSDME in humans, forms membrane pores, which drive pyroptotic cell death1, and members of the GSDM family are involved in genetic diseases characterized by inflammation and autoimmunity. GSDMD serves as the ultimate effector in the lipopolysaccharide (LPS)-induced canonical NLRP3 inflammasome, which triggers caspase-1 activation, and noncanonical inflammasome pathway (for example, caspase-4/caspase-5 in humans or caspase-11 in mice) and is cleaved by inflammatory caspases to release its N-terminal domain (GSDMD-NT) from the autoinhibitory C-terminal domain2. GSDMD-NT binds phospholipids of the mitochondrion and plasma membrane, forming oligomeric pores that release interleukin-1β (IL-1β) and IL-18 (refs. 3,4) and disrupt membrane integrity via ninjurin-1 filaments5,6. Calcium influx signals mediated by GSDMD-NT pores can activate the cell-intrinsic endosomal sorting complexes required for transport (ESCRT; a membrane repair system) to repair the damaged cytomembrane7,8,9,10.
Inflammasome-induced pyroptosis is essential to physiological immune responses, but excessive pyroptosis can cause sepsis, cytokine release syndrome, severe inflammation and tissue damage11,12,13,14. Gsdmd-deficient mice are resistant to mortality due to LPS-induced sepsis15. Lipids from dying cells (such as pyroptotic cells) are captured and internalized by the pattern recognition receptor CD14 on dendritic cells (DCs), triggering inflammasome-dependent hyperactivation and inducing sustained release of IL-1, a phenomenon that contributes to the maintenance of a chronic inflammatory response16. Chronic dysregulated inflammasomes contribute to various human diseases, such as gout17, inflammatory bowel disease18, diabetes19, atherosclerosis20 and Alzheimer’s disease21.
Although targeting mature GSDMD-NT pores represents a therapeutic approach in all these conditions, it presents substantial challenges. Cryoelectron microscopy structural analysis of human GSDMD-NT pores22 has revealed that the pores possess a smooth surface rich in negative charges, enabling the selective release of mature IL-1β into the extracellular space through electrostatic filtering. The major structural changes during GSDMD-NT pore formation make it difficult to obtain stable GSDMD-NT pore proteins, hindering traditional high-throughput screening methods (for example, phage display) from identifying lead molecules for pore blockade. Additionally, the diameter of the GSDMD-NT pore, which is 21 nm, limits the effects of small-molecule inhibitors, even if they can bind to the pore region.
Artificial intelligence (AI) has revolutionized protein design, leading to the development of numerous methods, such as RFDiffusion23, MaSIF24, RifDock25 and SCUBA26, which enable the creation of high-affinity proteins. Here we used a deep learning-based interactive interface atomic generative model to conduct virtual peptide screening and refine and characterize SK56, a potent blocker of the GSDMD-NT pore. Our experiments demonstrated that SK56 effectively delays pyroptosis by selectively targeting the GSDMD-NT pore, inhibiting cytokine release, preventing mitochondrial damage, averting activation of DCs, reducing widespread cell death and exhibiting notable therapeutic effects in a mouse model of sepsis. Our findings could provide new therapeutic options for treating human diseases caused or exacerbated by excessive uncontrolled inflammation.
Results
Pyroptosis occurs in individuals with sepsis and mouse models
In mice, Gsdmd deletion inhibits pyroptosis and reduces cytokine release15. Gsdmd−/− mice showed a significantly better 7-day survival rate (90–100%) than wild-type mice (40–60%) in models of sepsis induced by LPS (15 mg per kg (body weight) intraperitoneal (i.p.); Extended Data Fig. 1a) or cecal ligation and puncture (CLP; Extended Data Fig. 1b). To examine the extent of tissue damage, we analyzed hematoxylin and eosin (H&E) staining of cryosections of lung, kidney, liver, intestine and spleen tissue and performed histopathological scoring on lung and kidney tissues. H&E staining and pathological scores showed that wild-type mice exhibited severe lung inflammatory lesions, whereas Gsdmd−/− mice showed reduced thickening of the alveolar septum and less leukocyte infiltration (Extended Data Fig. 1c) in both LPS and CLP models. Pathological scores in the affected kidneys and H&E staining in kidney, liver, intestine and spleen tissues revealed less injury in Gsdmd−/− mice than in wild-type mice (Extended Data Fig. 1d,e) for both LPS and CLP conditions. IL-1β expression in blood peaked at around 36 h after CLP and 24 h after LPS treatment in wild-type mice and at 36 h in Gsdmd−/− mice in both models (Extended Data Fig. 1f), and levels of IL-1β in Gsdmd−/− mice were lower than that observed in wild-type mice (Extended Data Fig. 1f). A multiplex bead-based immunoassay indicated that serum levels of CSF2, interferon-γ (IFNγ), TNF, IL-1β, IL-2 and IL-4 were significantly reduced in Gsdmd−/− mice at day 2 after LPS treatment, whereas levels of IL-10, IL-4, CSF2, IL-1β and IL-5 were decreased in Gsdmd−/− mice at day 2 after CLP compared to in wild-type mice (Extended Data Fig. 2a). Wild-type mice lost more weight than Gsdmd−/− mice in both LPS and CLP conditions (Extended Data Fig. 2b). Levels of blood biochemical indicators (aspartate aminotransferase (AST), blood urea nitrogen (BUN), alanine aminotransferase (ALT) and creatine kinase (CK)), reflecting heart, liver, muscle and kidney function, were significantly lower in Gsdmd−/− mice than in wild-type mice in both sepsis models (Extended Data Fig. 2c).
To analyze the link between cell pyroptosis and sepsis in a clinical setting, we collected blood samples from 88 healthy volunteers (n = 58 females, n = 30 males, median age 42.5 years (range 19–89 years)) and individuals diagnosed with mild (sequential organ failure assessment (SOFA) score of ≥2, n = 47 females, n = 90 males, median age 59 years (range 1–96 years)) or severe sepsis (SOFA score of >11, n = 10 females, n = 16 males, median age 61.5 years (range 22–84 years)) from January 2018 to May 2023 at the Daping Hospital of Army Medical University (Supplementary Table 1). Serum IL-1β levels were fivefold higher in individuals with mild sepsis than in healthy volunteers at 24 h after diagnosis, with severe cases showing a further twofold increase compared to healthy volunteers (Extended Data Fig. 2d). GSDMD-NT was detected in peripheral blood leukocytes from individuals with sepsis, but not in healthy volunteers (Extended Data Fig. 2e). These results indicate that GSDMD activation is associated with inflammation and IL-1β release during sepsis in humans and in mouse models.
AI-screened SK56 delays pyroptosis and IL-1β secretion
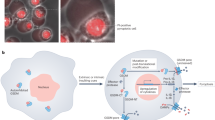
To identify molecules that interact with the smooth surface area of the GSDMD-NT pore, we adapted the deep learning model Transformer27 to process protein structures based on atomic coordinates, atom type and charge information and generate surface interactions as output (Fig. 1a). We trained the Transformer model by collecting structural data from real protein complexes (obtained by X-ray diffraction or cryoelectron microscopy) from the Protein Data Bank (PDB) database. This yielded approximately 40,000 experimentally determined natural interaction interfaces (paired sets of atomic points, atomic distance of ≤4 Å; Fig. 1b), which were used to train the modified Transformer model (Extended Data Fig. 3a). After 100,000 iterations, the loss converged (Extended Data Fig. 3b), indicating that the model parameters stabilized, and further optimization yielded minimal improvement. The target region atomic points from GSDMD-NT were then used as input for our model to obtain a matching set of atomic points (defined as an ideal interaction interface; Fig. 1c), followed by a search in a precomputed charges miniprotein scaffold library25 for matches to the output set of atomic points. The top 30% of candidate backbones were clustered by sequence diversity, and the directly targeted interacting residues were refined by using the Rosetta FastDesign module28. The 12 candidate peptide coding sequences (Supplementary Table 2 and Extended Data Fig. 3c) were cloned into pSmart-I vector with an N-terminal His6 small ubiquitin-like modifier (SUMO) tag, and all plasmids were transfected into Escherichia coli BL21 (DE3) for expression and purification.

a, Workflow showing the modifications to the Transformer model, which involve the splitting and processing of input and output data and the use of atomic-level information of the interface, including coordinates, charge and atom type as input. b, Flow chart of data extraction and processing for interaction atomic training data showing how ~20,000 pairs of protein complex data from the PDB database were used to extract atoms at interaction interfaces (distance of ≤4 Å), calculate charge using a force field (ff14SB) and perform model training. c, Schematic showing how a slightly modified Transformer model as in a was used for interference peptide design in the target surface region of GSDMD-NT (PDB ID: 6VFE; this model was selected to obtain the atomic coordinates on the surface). Charges were calculated using a force field (ff14SB), combined with the atomic coordinates, typed into a matrix and used as the input of the model after removing hydrogen atom information. The model outputs the atomic coordinates, charges and types of the theoretical interaction interface corresponding to the input surface and uses the interface atomic information to perform a point registration search (coherent point drift) in the precomputed charge scaffold database. Scaffolds with higher scores obtained from the search were optimized using the Rosetta FastDesign module for residue optimization to obtain peptides that interact with the target protein.
Candidate peptides (15 µM) were administered to phorbol 12-myristate 13-acetate (PMA)-differentiated human THP-1 cells (hereafter, THP-1 cells) that were primed with LPS and treated with nigericin to activate the canonical NLRP3 inflammasome. Peptide 11 (hereafter, SK56) exhibited the strongest inhibition of both IL-1β and IL-18 release in THP-1 cells compared to other tested peptides (Fig. 2a,b). Live-cell imaging showed that SK56 delayed LPS + nigericin-induced pyroptosis by about 40 min in THP-1 cells (Fig. 2c) and mouse bone marrow-derived macrophages (BMDMs) compared to PBS control treatment (Extended Data Fig. 3d and Supplementary Video 1). ATP assays showed that SK56 increased the amount of cellular ATP levels by 22–73% in a concentration-dependent manner in BMDMs (Extended Data Fig. 3e). Three-dimensional (3D) live-cell imaging showed delayed cell rupture by about 30 min in THP-1 cells compared to PBS (Extended Data Fig. 3f and Supplementary Video 2). For THP-1 or BMDM cells treated with LPS + nigericin for 180 min, the half-maximal inhibitory concentrations of SK56 were 1.38 ± 0.48 μM and 1.12 ± 0.37 μM, respectively (Extended Data Fig. 3g).

a, Immunoblots (left) and quantification (right) of IL-1β release in THP-1 cells differentiated with 120 nM PMA and treated with PBS, 1 μg ml−1 LPS + 5 mM ATP (L + A), 1 μg ml−1 LPS + 10 μM nigericin (L + N), 30 μM DSF or 15 μM of 12 candidate peptides (1–12; SK56 is peptide 11) at 2 h after the addition of LPS + nigericin; n = 3 repeats. b, ELISA of IL-1β and IL-18 release from THP-1 cells treated with DSF or peptides 1–12 in the presence of LPS + nigericin as in a. The red arrow indicates SK56 (peptide 11); n = 4 repeats; purple, **P = 8 × 10−7; black, **P = 1 × 10−8. c, Representative images from live-cell imaging experiments (see Supplementary Video 1; left) and the percentage of PI+ cells (right) among THP-1 cells incubated with LPS + nigericin + PBS or LPS + nigericin + SK56 (L + N + SK56; 15 µM) at 0, 20, 40, 60 and 80 min after the addition of LPS + nigericin; scale bar, 50 µm; n = 3 repeats. d,e, Representative immunoblotting of IL-1β and GSDMD cleavage (d; n = 2 repeats) and quantification of IL-1β release (e; n = 4 repeats) in THP-1 cells incubated with PBS or 1 μg ml−1 LPS + 10 μM nigericin together with 100 µM Z-VAD-fmk, PBS or 15 µM SK56 (d) or PBS, LPS or LPS + nigericin together with 100 µM Z-VAD-fmk or 0.6, 1.6, 5 and 15 µM SK56 (e) for 2 h. Sup, supernatant. f–h, Representative images from live-cell imaging (see Supplementary Video 3; f) and relative fluorescence intensity of MitoTracker (MT; g) or SYTOX (h) in THP-1 cells incubated with 15 µM SK56–FITC at 0, 20, 30, 40, 50, 60 and 80 min after the addition of LPS + nigericin as in a; scale bar, 50 µm; n = 3 repeats. i, Representative TEM image showing mitochondrial morphology (arrows) in PMA-differentiated THP-1 cells treated with PBS, 15 µM SK56, LPS + nigericin or LPS + nigericin + 15 µM SK56 for 1.5 h; scale bar, 500 nm; n = 3 repeats. j, Real-time single-cell biochemistry showing ROS generation in THP-1 cells treated with 1 μg ml−1 LPS at 0 min, 10 μM nigericin at 30 min, and PBS, SK56 or DSF at 0 min. DCFH-DA is a ROS indicator. Data in a–c, e and g were analyzed by two-tailed Student’s t-test and are shown as mean ± s.d.
SK56 (15 μM) inhibited pyroptosis and decreased IL-1β secretion in BMDMs transfected with LPS compared to PBS control (Extended Data Fig. 3h,i), suggesting that SK56 inhibited both canonical and noncanonical inflammasome-triggered pyroptosis. SK56 (15 µM) did not inhibit necroptosis in human enterocyte HT-29 cells treated with TNF, second mitochondria-derived activator of caspases (SMAC) mimetic and a pan-caspase inhibitor benzyloxycarbonyl-Val-Ala-Asp (OMe) fluoromethylketone (z-VAD-fmk) cocktail, although high concentrations (45 μM) of SK56 inhibited HT-29 cell necrosis for reasons yet unclear (Extended Data Fig. 3j). SK56 did not affect other non-GSDMD-regulated forms of cell death, such as apoptosis, cuproptosis and ferroptosis, in mouse macrophage RAW264.7 cells, AC16 human myocytes and MAD-MB-231 human epithelial cells treated with H2O2, elesclomol-CuCl2 and erastin, respectively (Extended Data Fig. 3k–m). SK56 (15 μM) inhibited SYTOX green influx (90% reduction, 70 min) in LPS + nigericin-treated THP-1 cells (Extended Data Fig. 4a). Immunofluorescence imaging and immunoblotting showed that SK56 did not affect cleavage of IL-1β and GSDMD (Fig. 2d,e) or GSDMD-NT translocation to the membrane (Extended Data Fig. 4b) but reduced the release of IL-1β (Fig. 2d,e) in THP-1 cells treated with LPS + nigericin compared to PBS. SK56 did not directly inhibit caspase-1 activation (Extended Data Fig. 4c).
To investigate whether SK56 interacted with GSDMD-NT and gained entry into pyroptotic cells, we conjugated fluorescein isothiocyanate (FITC) to the SK56 C terminus. Live-cell imaging in LPS + nigericin-treated THP-1 cells in which mitochondrial membrane potential was assessed with MitoTracker red indicated that SK56–FITC did not enter cells within 0–30 min but colocalized with the cell membrane and the mitochondria at 30 min after the addition of LPS + nigericin (Fig. 2f and Supplementary Video 3). Incubation with SK56–FITC inhibited the decline in MitoTracker red fluorescence by 40% in LPS + nigericin-treated THP-1 cells compared to PBS at 30 min after the addition of LPS + nigericin (Fig. 2f,g and Extended Data Fig. 4d). The timing for SK56–FITC and SYTOX entry into THP-1 cells was similar (Fig. 2h and Extended Data Fig. 4d), suggesting that SK56 might enter through GSDMD-NT pores and subsequently bind to mitochondria. GSDMD-NT forms pores on mitochondria, leading to reactive oxygen species (ROS) release and mitochondrial damage29. Transmission electron microscopy (TEM) indicated that SK56 reduced mitochondrial damage in LPS + nigericin-treated THP-1 cells compared to PBS (Fig. 2i). Kinetic analysis of ROS and ATP in single THP-1 cells showed that SK56 markedly inhibited 30–60% ROS accumulation (Fig. 2j and Extended Data Fig. 4e) and mitigated 10–40% of ATP decline in LPS + nigericin-treated THP-1 cells compared to PBS (Extended Data Fig. 4f). SK56 (30 μM) delayed lactate dehydrogenase release by 1 h in LPS + nigericin-treated THP-1 cells compared to PBS (Extended Data Fig. 4g), similar to the GSDMD-NT pore formation inhibitor disulfiram (DSF)30. Thus, SK56 did not interfere with GSDMD-NT membrane translocation or IL-1β cleavage but reduced IL-1β release, mitochondrial damage and membrane disintegration.
SK56 inhibits pyroptosis by blocking GSDMD-NT pore function
Next, we used biolayer interferometry to analyze the effect of SK56 on pyroptotic cytomembranes. Cell membrane fragments were collected from LPS + nigericin-treated THP-1 cells (hereafter, pyroptotic cytomembranes, which contain GSDMD-NT pores) and from THP-1 cells treated with or without LPS (hereafter, cytomembranes, which do not contain GSDMD-NT pores) by using ultracentrifugation. Biotinylated SK56 (N terminus) bound to pyroptotic cytomembranes with an estimated Kd of 10.9 nM, but did not bind to LPS-treated or untreated cytomembranes (Fig. 3a). No interactions were detected between biotinylated SK56 and PBS or free GSDMD-NT protein (Fig. 3a). N-terminal SUMO-tagged SK56 (SUMO–SK56) coimmunoprecipitated with assembled GSDMD-NT pores in THP-1 cells treated with LPS + nigericin for 4 h compared to the no-SUMO–SK56 control (Fig. 3b). Live-cell imaging in mouse BMDMs transfected with GSDMD tagged with blue fluorescent protein (BFP) at the N-terminal (BFP–GSDMD-NT) or C-terminal (GSDMD-CT–BFP) side and treated with LPS + nigericin to induce pyroptosis showed that SK56–FITC colocalized exclusively with BFP–GSDMD-NT, but not GSDMD-CT–BFP (Fig. 3c). To examine the selectivity of SK56, human HEK293 cells were transfected with plasmids expressing GSDMA-NT–green fluorescent protein (GSDMA-NT–GFP), GSDMB-NT–GFP, GSDMC-NT–GFP, GSDMD-NT–GFP and GSDME-NT–GFP, and the nondenatured lysates containing the fusion proteins were collected for interaction assays. Microscale thermophoresis (MST) indicated that SK56 had strong affinity for GSDMC-NT–GFP (Kd of ~0.22 µM) and GSDMD-NT–GFP (Kd of ~0.25 µM) and weaker affinity for other GSDM-NT–GFP subtypes (Fig. 3d). Incubation of polydiacetylene (PDA) nanoparticle hydrogel, which emits red fluorescence when pores form on their phospholipid layer31,32 (Extended Data Fig. 5a), with 1 μM GSDMD-NT for 30 min resulted in a red fluorescent signal, whereas incubation with the membrane protein ACE2 (which does not form pores) did not induce a fluorescent signal (Fig. 3e). Incubation of PDA nanoparticles containing GSDMD-NT with 15 μM SK56 for 15 min diminished the red fluorescence compared to those incubated without SK56 (Fig. 3e,f), suggesting that SK56 blocks the pores formed by GSDMD-NT in the PDA nanoparticle hydrogel.

a, Representative TEM images of cytomembranes from THP-1 cells (top) and representative biolayer interferometry traces from biotin–SK56 (bottom) incubated with PBS, 1 μg ml−1 LPS or 1 μg ml−1 LPS + 10 μM nigericin (top) or PBS, GSDMD-NT protein or cytomembranes from THP-1 cells incubated with PBS (CPBS), 1 μg ml−1 LPS (CLPS) or 312 nM–5 μM LPS + nigericin (CL + N) for 2 h (n = 2 repeats); scale bar, 200 nm. b, GSDMD immunoblotting of immunoprecipitated SUMO–SK56 in THP-1 cells treated with LPS + nigericin as in a; n = 2 repeats; IB, immunoblot; IP, immunoprecipitate. c, Representative live-cell images of mouse wild-type BMDMs transfected with BFP–GSDMD-NT or GSDMD-CT–BFP plasmids and incubated with 15 µM FITC–SK56 at 0, 40 and 80 min after LPS + nigericin treatment; scale bar, 2 µm. Arrowheads indicate colocalization; n = 2 cells. d, MST showing the binding affinity of SK56 to mature GSDMA–GSDME (n = 1 repeat). e,f, Representative images (e) and quantification of fluorescence (f) in PDA nanoparticle hydrogel incubated with PBS, 15 µM SK56, 5 μg ACE2 or 1 μM GSDMD-NT for 45 min or 1 μM GSDMD-NT for 30 min and then incubated with 15 μM SK56 for an additional 15 min; scale bar, 70 µm; n = 3 repeats. g, Cell viability in THP-1 cells incubated with PBS, 1 μg ml−1 LPS or LPS + nigericin in addition to PBS, 1.5 µM SK56 or 1.5 µM SK56 mutant peptides for 120 min; n = 4 repeats. h, Docking assay showing SK56–GSDMD-NT interaction. Critical residues (Arg 22–Glu 174 electrostatic interaction, Met 29–Pro 103 hydrophobic interaction and Tyr 26–Thr 63 hydrogen bond) are indicated. i, Proteomic analysis showing differentially expressed proteins (DEPs) in THP-1 cells treated with PBS, LPS + nigericin or LPS + nigericin + 15 µM SK56 for 90 min; n = 3 samples. Data in f and g were analyzed by two-tailed Student’s t-test and are shown as mean ± s.d.
To determine the binding site between SK65 and GSDMD-NT, we conducted point mutation experiments and molecular dynamics (MD) simulations to analyze binding energy contributions. The MD simulation revealed that eight residues on SK56 contribute the majority of the binding energy in its interaction with GSDMD-NT (Extended Data Fig. 5b). When mutant SK56, in which key residues in SK56 (Ser 1, Glu 11, Arg 22, Tyr 26, Met 29, Leu 32, Met 37 and Arg 42) were individually mutated to glycine, was incubated with THP-1 cells (at 2 h after the addition of LPS + nigericin), mutations at positions 22, 29 and 37 in SK56 completely abolished the ability to inhibit pyroptosis (Fig. 3g,h). Structural insights into the GSDMD-NT pore suggest that the pore is an acidic conduit, which electrostatically filters the release of IL-1β, with four critical acidic patches (AP1–AP4) predominantly determining its surface charge properties22. Based on docking assays, SK56 binding to the GSDMD-NT pore altered the surface charge characteristics between AP1 and AP3 (Extended Data Fig. 5c), suggesting that SK56 might deactivate the GSDMD-NT pore by modifying its surface charge properties. We also examined proteome changes in LPS + nigericin-treated THP-1 cells with or without SK56 treatment for 90 min by liquid chromatography–tandem mass spectrometry. One hundred and five proteins were differentially expressed (64 upregulated and 41 downregulated; Supplementary Table 3) in LPS + nigericin-treated compared to PBS-treated THP-1 cells (Fig. 3i), with a partial restoration (65% of upregulated and 60% of downregulated proteins) of expression observed in LPS + nigericin-treated THP-1 cells incubated with SK56 (Fig. 3i). These findings indicate that SK56 primarily targets the GSDMD-NT pore, rather than upstream cleavage events in the inflammasome pathway.
SK56 reduces pyroptosis by recruiting ESCRT and inhibits DC activity
GSDMD-NT-mediated cell membrane damage results in calcium ion influx and activates the ESCRT system to repair the membrane and prevent pyroptosis. During this process, the ESCRT protein CHMP4 targets the membranes and forms punctate patterns in dying cells8,33. To investigate whether SK56 delayed pyroptosis and, as such, allowed more time for the ESCRT system to repair the cell membrane, we assessed the effect of SK56 on the formation of CHMP4–GFP puncta in BMDMs treated with LPS + nigericin for 80 min to induce pyroptosis. Confocal imaging showed that 15 µM SK56 had no effect on the proportion of CHMP4–GFP+ puncta in LPS + nigericin-treated BMDMs, but reduced the number of Annexin V+ BMDMs by 68% compared to PBS (Fig. 4a). The addition of EDTA, an inhibitor of ESCRT, restored the proportion of Annexin V+ BMDMs to the level seen in BMDMs without SK56 treatment (Fig. 4a), suggesting that SK56-mediated inhibition of pyroptosis may require the ESCRT system.

a, Representative images of CHMP4–GFP puncta (arrowheads; left) and percentage of CHMP4 speckle+ (top right) or Annexin V+ (bottom right) cells in BMDMs incubated with PBS, 15 µM SK56, 2 mM EDTA or EDTA + SK56 at 2 h after the addition of 1 μg ml−1 LPS + 10 μM nigericin; scale bar, 50 µm; n = 5 repeats. b, Immunoblots (top) and quantification (bottom) showing GSDMD-NT in the supernatant from THP-1 cells treated with PBS, 30 µM DSF, 15 µM SK56 or 15 µM SK56 at 120 min after LPS + nigericin treatment; n = 3 repeats. c, Representative images (left) and percentage of GSDMD-NT–BFP/cytomembrane-CellMask Orange+ (right) cells in calcein-AM-labeled (green) mouse wild-type BMDCs incubated with 2 μg ml−1 pyroptotic cytomembrane fragments from mouse wild-type BMDMs transfected with a GSDMD-casp–BFP construct and incubated with LPS + nigericin (PCFBFP), 20 µM SK56 (PCFBFP + SK56) or 20 µM SK56scrambled (synthetic SK56 scrambled peptide; PCFBFP + SK56scrambled), pyroptotic cytomembrane fragments from mouse Gsdmd−/− BMDMs incubated with LPS + nigericin and PBS (PCFGsdmd−/−), cytomembranes from wild-type BMDMs incubated with PBS (NCF) or pyroptotic cytomembrane fragments from mouse BMDMs incubated with LPS + nigericin and 10 µg ml−1 BFP (PCF + BFP), 10 µg ml−1 GSDMD-NT–BFP or 10 µg ml−1 BFP for 2 h; green, calcein-AM+ BMDCs; red, CellMask Orange+ NCF, PCF or PCFGsdmd−/−; blue, GSDMD-NT–BFP, BFP or PCFBFP. The white arrow indicates cytomembrane outside BMDCs, and the red arrow indicates phagocytosed cytomembrane; scale bar, 25 μm; n = 3 repeats. d, ELISA of secreted (left) and cell-associated (right) IL-1β from wild-type BMDCs treated with PBS, 10 μg ml−1 BFP, 1 μg ml−1 GSDMD-NT, 20 µM SK56, SK56 + GSDMD-NT, 120 µM oxPAPC, SK56 + oxPAPC or 2 μg ml−1 pyroptotic cytomembrane fragments from mouse wild-type or Gsdmd−/− BMDMs as in c (NCF, PCF, PCF + SK56, PCFGsdmd−/−, PCFGsdmd−/− + SK56), treated or not treated with Pam3 for 12 h; n = 4 repeats. All data are shown as mean ± s.d., and P values were determined by two-tailed Student’s t-test; NS, not significant (P > 0.05).
Pyroptosis leads to cytomembrane rupture, releasing fragments containing oxidized phospholipids (oxPAPC) and GSDMD-NT pores16,34. To assess whether SK56 inhibits the release of pyroptotic cytomembrane fragments, we treated THP-1 cells with LPS + nigericin with or without SK56 for 2 h to induce pyroptosis, followed by supernatant collection and immunoblotting to detect the levels of GSDMD-NT in the medium. In this assay, SK56 (45 μM) inhibited the release of GSDMD-NT in the supernatant by 80% compared to PBS (Fig. 4b).
DCs can phagocytose cytomembrane fragments released by pyroptotic cells, a process that induces DC activation and sustained release of IL-1 (refs. 16,35), which perpetuates the inflammatory response. To investigate whether SK56 had an effect on the phagocytosis of pyroptotic cytomembranes by DCs, we transfected wild-type mouse BMDMs with GSDMD in which BFP was inserted before the caspase cleavage site (GSDMD-casp–BFP), a construct that allows the release of GSDMD-NT–BFP following activation of pyroptosis and labeling of the pyroptotic cytomembrane released in the supernatant. GSDMD-casp–BFP-transfected BMDMs were treated with LPS + nigericin for 4 h to induce pyroptosis, and pyroptotic cytomembrane fragments that contained GSDMD-NT–BFP pores (hereafter, PCFBFP) were collected from the supernatant via ultracentrifugation. As controls, we collected cytomembrane fragments from LPS + nigericin untreated BMDMs (referred to as native cell membrane fragments (NCFs)), GSDMD-casp–BFP nontransfected BMDMs treated with LPS + nigericin for 4 h (PCF) and GSDMD-casp–BFP nontransfected Gsdmd−/− BMDMs treated with LPS + nigericin for 4 h (PCFGsdmd−/−) by sonication and ultracentrifugation, followed by labeling with CellMask Orange (red fluorescence). Wild-type mouse bone marrow-derived dendritic cells (BMDCs) labeled with calcein-acetoxymethyl (green fluorescence; BMDCGF) incubated with NCF, PCF, PCFBFP, PCFGsdmd−/−, BFP or GSDMD-NT–BFP for 2 h were analyzed by confocal microscopy to assess phagocytosis. BMDCGF exhibited a 15% higher phagocytosis rate for GSDMD-NT–BFP than for BFP (Fig. 4c). Phagocytosis of PCFBFP or PCF by BMDCGF was 5-fold higher than that of NCF, GSDMD-NT–BFP protein or PCFGsdmd−/− and was 20-fold higher than that for BFP (Fig. 4c). SK56 (20 µM) inhibited BMDCGF phagocytosis of PCFBFP by 60%, whereas scrambled SK56 showed no inhibitory effect (Fig. 4c). These results suggest that the presence of GSDMD-NT pores on pyroptotic cytomembrane fragments promotes their phagocytosis by BMDCs, whereas SK56 inhibits this process.
To assess the impact of SK56 on IL-1β secretion in BMDCs, we treated Pam3CSK4-primed BMDCs for 12 h with PCF, NCF, PCFGsdmd−/−, BFP, GSDMD-NT protein or oxPAPC, followed by supernatant and cell collection and assessment of IL-1β levels by enzyme-linked immunosorbent assay (ELISA). GSDMD-NT increased IL-1β release by 10-fold compared to BFP, while PCF increased IL-1β release by 5-fold compared to NCF, 2-fold compared to GSDMD-NT and 20-fold compared to BFP (Fig. 4d). SK56 (20 µM) inhibited IL-1β release by ~50% compared to PCF, GSDMD-NT protein and oxPAPC but had no effect on PCFGsdmd−/− (Fig. 4d). None of these treatments affected the levels of intracellular IL-1β (Fig. 4d), indicating that SK56 blocks IL-1β release from BMDCs without impacting its production. These findings indicate that SK56 inhibits pyroptosis through the ESCRT-mediated membrane repair system, suppresses BMDC phagocytosis of pyroptotic cytomembrane fragments and reduces IL-1β secretion in activated BMDCs.
SK56 suppresses pyroptosis in organoids and blood
Following macrophage pyroptosis, GSDMD-NT pores propagate through extracellular vesicles and trigger cell death in nearby cells34. This process has been observed in individuals with sepsis and causes damage particularly in the lungs36,37. To evaluate whether SK56 inhibits widespread cell death and protects lung tissue, we established a coculture system using human alveolar organoids and THP-1 cells. Human lung CD45−CD31−LysoTracker+EPCAM+ alveolar epithelial type 2 cells sorted from tumor-adjacent tissues of individuals undergoing lung cancer resection were cultured in Matrigel with alveolar maintenance and differentiation medium (containing FGF10 and EGF) for 15 days to form alveolar organoids and then cocultured with THP-1 cells (Extended Data Fig. 6a–c) at a 1:10 ratio for 4 h before LPS + nigericin treatment for 4 h to induce macrophage pyroptosis. SYTOX was added to the medium to indicate pyroptosis, cathodoluminescent dil labeling was used to detect THP-1 cells, calcein-acetoxymethyl green labeling was used to indicate viable cells, and propidium iodide (PI) labeling was used to detect dead cells. Live-cell imaging showed that LPS + nigericin-treated alveolar organoids alone did not undergo pyroptosis but that 87% of SYTOX+ THP-1 cells and organoid cells underwent pyroptosis compared to PBS treatment when alveolar organoids cocultured with cathodoluminescent dil-labeled THP-1 cells were treated with LPS + nigericin (Extended Data Fig. 6d). When the LPS + nigericin-treated alveolar organoid–THP-1 cell coculture system was treated with SK56 (15 μM) or the GSDMD-NT pore formation inhibitors DSF, 25 μM dimethyl fumarate (DMF) or 20 μM necrosulfonamide (NSA), followed by 18 h of continuous live-cell imaging, SK56 caused a delayed decrease in calcein-acetoxymethyl fluorescence in calcein-acetoxymethyl+ organoid and THP-1 cells by 50% and a delayed increase in PI fluorescence in PI+ organoid and THP-1 cells by approximately 8 h compared to PBS-treated controls, whereas DSF, DMF and NSA showed no effect (Fig. 5a and Supplementary Video 4). H&E staining indicated that SK56 reduced macrophage infiltration in alveolar organoids by 60% compared to PBS at 8 h (Fig. 5b and Extended Data Fig. 6e). Additionally, immunofluorescence imaging showed that SK56 decreased the percentage of GSDMD-NT+ cells based on labeling with anti-GSDMD-NT by 50% compared to PBS at 12 h (Extended Data Fig. 6f), whereas ELISA of supernatants indicated that SK56 inhibits IL-1β release by approximately 60% compared to PBS at 12 h (Fig. 5c).

a, Representative images from live-cell imaging (see Supplementary Video 4; left) and relative PI stain intensity (top right) and calcein-AM staining (bottom right) in human alveolar organoids (calcein-AM+, green) cocultured with GSDMD-casp–BFP-transfected THP-1 cells treated with LPS + nigericin and incubated with PBS, 30 µM DSF, 25 µM DMF, 20 µM NSA or 15 μM SK56 at 0.5, 4, 8, 12 and 16 h after treatment with LPS + nigericin; scale bar, 40 μm; n = 3 repeats. b, Representative H&E staining (top) and percentage of the infiltration area (bottom) in fixed alveolar organoids + THP-1 cell cocultures as in a treated with PBS or LPS + nigericin together with PBS (L + N + PBS) or 15 μM SK56 at 8 h after the addition of LPS + nigericin; scale bar, 75 μm; n = 5 repeats. c, ELISA showing IL-1β release in the supernatant of alveolar organoids + THP-1 cell cocultures as in a treated with PBS, 15 μM SK56 or LPS + nigericin together with PBS or 15 μM SK56 or at 12 h after LPS + nigericin treatment; n = 4 repeats. d, Representative images (left) and percentage relative to DAPI+ cells (right) of GSDMD-NT+ (green) cells in human blood leukocytes incubated with PBS or LPS + nigericin together with PBS or 15 μM SK56 at 1 h after nigericin + LPS treatment; n = 20 repeats. e, Representative immunoblots of IL-1β and GSDMD-NT in whole human blood leukocytes treated as in d; n = 3 repeats. f, Heat map illustrating inflammatory cytokine profiles in whole human blood treated with normal saline (NSal) and LPS + nigericin together with normal saline (L + N + NS) or 15 μM SK56; *P < 0.05 L + N + SK56 versus L + N + NS, n = 4 samples. Data are shown as mean ± s.d., and P values were calculated by two-tailed Student’s t-test; *P < 0.05.
To test whether SK56 could block pyroptosis in human blood cells, we stimulated whole blood from healthy volunteers with LPS + nigericin for 1 h and measured the percentage of GSDMD-NT+ leukocytes compared to PBS treatment (Fig. 5d,e). Immunofluorescence staining of cell smears with anti-GSDMD-NT indicated that 20% of leukocytes were GSDMD-NT+ in LPS + nigericin-treated blood samples compared to 2% in PBS-treated samples and 10% in LPS + nigericin-treated blood samples treated with 15 μM SK56 (Fig. 5d). Immunoblotting indicated that SK56 did not affect the cleavage of GSDMD or IL-1β (Fig. 5e) but reduced the secretion of IL-1β and IL-18 by 80% (Extended Data Fig. 6g), whereas DSF completely blocked the secretion of IL-1β and IL-18 (Extended Data Fig. 6g) compared to PBS in LPS + nigericin-treated blood. Multiplex bead-based immunoassays used to quantify 48 cytokines in LPS + nigericin-treated human whole blood with or without 15 μM SK56 treatment indicated that SK56 significantly reduced the amount of 37 cytokines, including IL-1β and IL-18, compared to the no-SK56 control (Fig. 5f).
We also assessed the effect of SK56 on peripheral blood collected within 72 h of diagnosis from six individuals with sepsis (n = 2 females, n = 4 males, median age 56 years (range 51–69 years)) and seven healthy volunteers (n = 3 females, n = 4 males, median age 32 years (range 26–38 years)). At baseline, we found a significant increase in 7-aminoactinomycin D+ dead cells (1% compared to 28%) and a notable decrease in CD45+ immune cell counts (80% compared to 92%), particularly in CD45+CD14−CD16++ nonclassical monocytes (45% compared to 57%) and CD45+CD11b+CD15+CD16+ neutrophils (19% compared to 51%) in individuals with sepsis compared to healthy volunteers (Extended Data Fig. 7). Treatment of whole blood from healthy volunteers with LPS + nigericin for 4 h did not replicate the B cell, T cell, monocyte and neutrophil population changes observed in individuals with sepsis but increased the percentage of 7-aminoactinomycin D+ dead cells to 40% (Extended Data Fig. 7), whereas the addition of 15 μM SK56 or 30 μM DSF reduced the percentage of 7-aminoactinomycin D+ cells to 7% (Extended Data Fig. 7). These results indicate that SK56 inhibits LPS + nigericin-induced cell death in tissue organoids and whole-blood cell cultures.
SK56 protects against LPS- and CLP-induced sepsis
To investigate whether SK56 delays pyroptosis in vivo, we induced low-dose LPS (15 mg per kg (body weight)) or CLP-mediated sepsis in mice as described above. In vivo, in mice, SK56 had a half-life of approximately 2.6 h (Extended Data Fig. 8a and Supplementary Table 4), whereas no significant degradation occurred in human blood at room temperature over 12 h (Extended Data Fig. 8b). Based on the fact that IL-1β levels in blood peaked at around 24 h after LPS treatment in mice (Extended Data Figs. 1f and 8c), we intravenously (i.v.) injected wild-type or Gsdmd−/− mice with 1 mg per kg (body weight) SK56, 50 mg per kg (body weight) DSF, 50 mg per kg (body weight) DMF or 20 mg per kg (body weight) NSA at 16 h after initiation of LPS treatment. Wild-type mice treated with SK56, Gsdmd−/− mice treated with or without SK56, but not mice treated with DSF, DMF or NSA, exhibited a significant reduction in mortality (from 55% to 0%) at 7 day compared to saline-treated wild-type control mice (Fig. 6a). Pathologic analysis indicated that SK56 reduced kidney, liver, intestine, spleen and lung injury (Extended Data Fig. 8d,e), decreased blood biochemical markers (AST, BUN, ALT and CK) of heart, liver, muscle and kidney damage (Fig. 6b) and reduced the amount of CSF2, IFNγ, IL-1β, IL-2, IL-10 and TNF in the peripheral blood (Fig. 6c) at day 2 after treatment initiation in LPS-treated mice compared to in saline-treated wild-type mice. NSA treatment inhibited AST, BUN, ALT and CK, whereas DSF and NSA suppressed AST, ALT and CK but not BUN (kidney injury marker) at day 2 after LPS treatment (Fig. 6b). In the CLP model, SK56-treated wild-type mice and both treated and nontreated Gsdmd−/− mice showed higher survival rates (Extended Data Fig. 9a), less organ damage (Extended Data Figs. 8e and 9b,c) and reduced levels of cytokines (CSF2, IL-1β, IL-4, IL-10 and TNF; Extended Data Fig. 9d) and organ damage markers in the blood (Extended Data Fig. 9e) compared to saline-treated control mice. Wild-type mice treated with SK56 and Gsdmd−/− mice showed less weight loss in both sepsis models (Extended Data Fig. 9f). SK56 showed no efficacy in Gsdmd−/− mice compared to saline in both sepsis models (Fig. 6a–c and Extended Data Figs. 8d,e and 9a–d,f), indicating that the anti-inflammatory effect of SK56 was GSDMD dependent.

a, Kaplan–Meier analysis of survival in wild-type (WT) and Gsdmd−/− mice challenged i.p. with 15 mg per kg (body weight) LPS and treated with normal saline, SK56 (1 mg per kg (body weight) i.v.) or 50 mg per kg (body weight) DSF, 50 mg per kg (body weight) DMF or 20 mg per kg (body weight) NSA i.p. at 16 h after LPS injection; n = 10 mice per group. b, Blood AST, BUN, ALT and CK levels in wild-type and Gsdmd−/− mice treated as in a at day 2 after LPS injection; n = 6 mice. c, Heat map displaying the expression of cytokines in the plasma of wild-type and Gsdmd−/− mice treated with normal saline or SK56 as in a at day 2 after LPS injection; n = 10 mice. d, Kaplan–Meier analysis of survival in wild-type mice challenged with 25 mg per kg (body weight) LPS i.p. and treated with SK56 (2 mg per kg (body weight) i.v.) or 50 mg per kg (body weight) DSF, 50 mg per kg (body weight) DMF or 20 mg per kg (body weight) NSA i.p. at 5 h after LPS injection; n = 20 mice per group. e, ELISA showing plasma expression of IL-1β every 3 h up to 48 h after LPS i.p. injection in wild-type mice treated with normal saline or SK56 (2 mg per kg (body weight) i.v.) as in d. f, Kaplan–Meier analysis of survival in wild-type mice challenged with 50 mg per kg (body weight) LPS i.p. and treated with normal saline, SK56 (4 mg per kg (body weight) i.v.) or 50 mg per kg (body weight) DSF, 50 mg per kg (body weight) DMF or 20 mg per kg (body weight) NSA i.p. at 4 h after LPS injection; n = 20 mice per group. g, ELISA showing plasma expression of IL-1β every 3 h up to 48 h after LPS i.p injection in mice treated with normal saline or SK56 as in f. Data in b, e and g were analyzed by two-tailed Student’s t-test and are shown as mean ± s.d. Data in a, d and f were analyzed by log-rank (Mantel–Cox) test. Arrows indicate treatment time.
To investigate the effect of SK56 in severe sepsis, we injected mice i.p. with a single dose of 25 mg per kg (body weight) or 50 mg per kg (body weight) LPS to induce moderate or severe sepsis, respectively, followed by i.v. injection with 2 mg per kg (body weight) SK56 for moderate sepsis and 4 mg per kg (body weight) SK56 for severe sepsis or i.p. injection with 50 mg per kg (body weight) DSF, 50 mg per kg (body weight) DMF or 20 mg per kg (body weight) NSA at 5 h (moderate sepsis) or 4 h (severe sepsis) after LPS injection. SK56 increased mouse survival from 5% to 45% in the moderate sepsis LPS model (Fig. 6d), whereas DSF, DMF and NSA improved mouse survival, but the results were not statistically significant (Fig. 6d). Continuous monitoring of IL-1β in the blood at 0, 1, 3, 6, 9, 12, 24, 36 and 48 h after injection of 25 mg per kg (body weight) LPS showed that SK56 delayed IL-1β release from peak levels at 6 h in saline-treated mice to 9 h in SK56-treated mice (Fig. 6e). SK56 did not notably improve survival (from 0% to 10%) in the severe sepsis LPS model (Fig. 6f) but delayed the first peak levels of IL-1β from 6 h in saline-treated control mice to 9 h after LPS injection, while having no effect on the second IL-1β peak (Fig. 6g). In mice with mild, moderate and severe sepsis, SK56, DSF and DMF treatment reduced plasma IL-1β levels at 24, 6 and 5 h, respectively, compared to saline-treated control mice (Extended Data Fig. 9g). NSA treatment had no effect on IL-1β levels in severe sepsis (Extended Data Fig. 9g). SK56, DMF and NSA treatment lowered levels of organ damage markers in the plasma at 6 and 5 h in mice with moderate and severe sepsis, whereas DSF failed to suppress BUN but inhibited the other three organ damage markers in mice with severe sepsis (Extended Data Fig. 9h). Wild-type mice treated i.p. with a single dose of 15 mg per kg (body weight) LPS or PBS for 24 h or followed by i.v. injection with 1 mg per kg (body weight) SK56 at 16 h after LPS injection. Compared to PBS-treated control mice, mice treated with 15 mg per kg (body weight) LPS for 24 h showed alterations in spleen CD45+CD11b+Ly6G+Ly6C++ monocyte (+114%), pulmonary CD45+CD3+CD19− T cell (+97%), total blood CD45+ immune cell (–21%) and CD45+CD3−CD19+ B cell (–89%) populations (Extended Data Fig. 10), whereas SK56 treatment in these mice reduced the LPS-induced increase in spleen monocytes by 30%, inhibited the increase in T cells in the lung by 60% and restored total immune cell counts by 50% and B cell counts by 30% in peripheral blood at 24 h after LPS treatment initiation (Extended Data Fig. 10). Thus, SK56 reduces excessive inflammation and organ damage and improves survival in models of sepsis in mice.
Discussion
Here we showed that an AI-screened GSDMD-NT pore blocker SK56 inhibits pyroptosis and ROS release from mitochondria in THP-1 cells, improves survival and reduces organ damage in models of sepsis in mice. SK56 selectively blocks GSDMD-NT pores to suppress IL-1β release, but does not interfere with upstream inflammasome assembly or caspase activation.
The conservation of GSDMD pore formation in all vertebrates38 suggests that GSDMD pores may have nonredundant roles beyond cytokine release. GSDMD-NT pore-mediated 11,12-epoxyeicosatrienoic acid secretion may contribute to tissue repair39. However, whether SK56 may negatively impact tissue repair remains to be investigated. We found that mouse BMDCs exhibited fivefold increased phagocytosis of pyroptotic cytomembrane fragments that contained mature GSDMD-NT pores compared to NCF or pyroptotic cytomembrane fragments from Gsdmd−/− mice. SK56, which is expected to bind to GSDMD-NT pores on these pyroptotic cytomembrane fragments, inhibited BMDC phagocytosis by 60%, suggesting that GSDMD-NT pores are likely recognized directly by DCs. Furthermore, phagocytosis of pyroptotic cytomembrane fragments induced IL-1β release in BMDCs40. Macrophages activated by oxidized lipids also release IL-1β through GSDMD-NT pores3. Although it remains unclear whether the pyroptotic cytomembrane fragment-activated BMDCs released IL-1β through GSDMD-NT pores, the process was inhibited by SK56.
SK56 also protected human alveolar organoids from inflammation caused by pyroptotic macrophage-derived cytomembrane fragments and extracellular vesicles34 containing GSDMD-NT pores. DMF, DSF and NSA have been reported to inhibit the formation of GSDMD-NT pores and have promising therapeutic effects in sepsis models in mice30,41,42. These compounds are all cysteine-reactive drugs that covalently modify Cys 191 in GSDMD to block the formation of mature pores. However, bystander pyroptosis, which is due to mature GSDMD-NT pores released from pyroptotic cells, was found to be insensitive to DSF34. Our tests indicate that NSA and DMF also have no notable inhibitory effects on widespread cell death in alveolar organoids. By contrast, SK56 directly targeted mature GSDMD-NT pores and may be a valuable tool for comprehensive analysis of the role of bystander pyroptosis in inflammatory conditions.
SK56 protected mice against lethal LPS- and CLP-induced sepsis. Although the protective effect of GSDMD deficiency in LPS-challenged mice is well known, its role in CLP-induced sepsis remains controversial. Although systemic GSDMD deficiency43,44,45, platelet GSDMD deficiency46 and neutrophil-specific GSDMD deficiency47 reduced CLP-induced sepsis mortality in some studies due to attenuated inflammatory responses, impaired neutrophil extracellular trap formation and diminished platelet pyroptosis43,44,45,46,47, other studies reported that neutrophil-specific GSDMD deficiency compromised bacterial clearance, worsened infection severity and diminished survival in CLP48. We found that systemic GSDMD deficiency reduced mortality in LPS and CLP models of sepsis, in agreement with the former observations43,44,45. Reports48 of worsened outcomes in GSDMD-deficient mice during CLP sepsis may stem from variable CLP severity or microbiome variability. Resolving this controversy would require conditional deletion of Gsdmd in specific immune cell populations to dissect its cell-type-specific roles in CLP versus LPS sepsis.
The observation that SK56 inhibited GSDMD-NT pores and mitigated organelle damage without abolishing pyroptosis aligns with emerging paradigms of ‘damage containment’ as a therapeutic goal, which makes SK56 a promising therapeutic candidate for diverse inflammatory conditions, including cytokine release syndromes, autoimmune disorders and chronic inflammatory diseases.
Methods
Ethics statement
Human paracancerous lung tissue was collected at the Daping Hospital of Army Medical University. Blood from 163 individuals with sepsis (n = 57 females, n = 106 males, median age 59 years (range 1–96 years)) or 88 healthy volunteers (n = 58 females, n = 30 males, median age 42.5 years (range 19–89 years)) was collected for analysis of IL-1β levels. Blood from six individuals with sepsis (n = 2 females, n = 4 males, median age 56 years (range 51–69 years)) and seven healthy volunteers (n = 3 females, n = 4 males, median age 32 years (range 26–38 years)) was collected to detect changes in immune cell populations. The SOFA score49 was used for the assessment of individuals with sepsis. Samples were obtained after receiving written informed consent from all participants, and all procedures were ethically approved by the Ethics Committee of Hospital (2023178). All animal studies were conducted at a specific pathogen-free facility under sterile conditions and were performed in strict accordance with the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health. All study protocols were approved by the Laboratory Animal Welfare and Ethics Committee of the Army Medical University (AMUWEC20237397).
Cell treatment
The human monocyte THP-1 (TIB-202, ATCC) and MDA-MB-231 (CRM-HTB-26, ATCC) cell lines were cultured in RPMI-1640 medium (10-040-CV, Corning). HT-29 (HTB-38, ATCC) cells were maintained in McCoy’s 5a modified medium (16600082, Gibco). AC16 (CRL-3568, ATCC) and RAW264.7 (TIB-71, ATCC) cells were cultured in DMEM (11965092, Gibco). Male C57BL/6 mice (8 weeks of age) were anesthetized via i.p. injection of sodium pentobarbital at a dose of 70 mg per kg (body weight). The abdominal area was disinfected with an iodine tincture for 5 min, followed by disinfection with 75% ethanol for another 5 min. A midline incision was made to expose the abdominal cavity, and the skin was carefully retracted. The femurs were removed, as well as the surrounding muscle tissue. The bones were then disinfected with 75% ethanol for 30 s and washed three times with PBS. Using fine scissors, the joints at both ends of the femur were clipped, and bone marrow was extracted by flushing the cavity with precooled PBS using a 2-ml syringe. The collected cell suspension was filtered through a 200-μm mesh and centrifuged at 3,300g for 5 min to collect the cell pellet. The pellet was resuspended in 1 ml of red blood cell lysis buffer (00-4333-57, Thermo Fisher Scientific) and incubated at room temperature for 5 min to lyse the erythrocytes. The suspension was then centrifuged again at 300g for 5 min, and the resulting cell pellet was resuspended in DMEM/F12 containing 10 ng ml−1 recombinant mouse macrophage colony-stimulating factor (416-ML-010/CF, R&D Systems). The medium was refreshed every other day, and the cells were cultured until day 6 to obtain mature BMDMs. All culture media were supplemented with 10% fetal bovine serum (FBS; 10099141C, Gibco) and 10 U ml−1 penicillin–streptomycin and were maintained in a humidified atmosphere containing 5% CO2 in an incubator at 37 °C. Cells were verified to be free of mycoplasma contamination.
For canonical inflammasome activation, THP-1 cells were first differentiated by incubation with 150 nM PMA (HY-18739, MCE) for 24 h and then primed with LPS (1 μg ml−1; O55:B5; L2880, Sigma-Aldrich) for 3 h before treatment with nigericin (10 μM; HY-100381, MCE) or 2 mM ATP. To induce noncanonical and inflammasome-mediated pyroptosis, primed BMDMs (1 μg ml−1 Pam3CSK4, 12 h) were transfected with 2 μg of LPS (0111:B4; L3024, Sigma-Aldrich) using DOTAP (HY-112754A, MCE) for 16 h. Necroptosis was induced in HT-29 cells by adding TNF, SM-164 and z-VAD-fmk cocktail (1:1,000; C1058, Beyotime) for 4 h. Concentrations of IL-1β (DLB50 or MLB00C) and IL-18 (DBP180 or DY122-05) in culture supernatants and mouse sera were measured using ELISA kits (R&D Systems) according to the manufacturer’s instructions. Apoptosis in RAW264.7 cells was assessed using Annexin V-APC/PI double staining. Following treatment with H2O2 (200 μM) and SK56 (15 μM and 45 μM) for 18 h, the cells were resuspended in 500 μl of 1× binding buffer and mixed with Annexin V-APC/PI (2361973, Invitrogen). After a 10-min incubation, the cells were analyzed using flow cytometry. For cuproptosis assays, AC16 cells were cultured in 96-well plates at a density of 1 × 105 cells per well. The cells were treated with elesclomol (40 nM; HY-12040, MCE), CuCl2 (10 μM; 751944, Sigma-Aldrich) and EDTA (1.2 mM; HY-Y0682, MCE), followed by the addition of SK56 (15 μM and 45 μM) under complete medium conditions for 24 h. Control cells were maintained under identical culture conditions without treatment. Subsequently, 10 μl of CCK-8 reagent (HY-K0301, MCE) was added to each well and incubated for 1 h. The absorbance was measured at 450 nm. For ferroptosis assays, MAD-MB-231 cells were cultured in 96-well plates at a density of 1 × 105 cells per well. The cells were treated with erastin (10 μM; HY-15763, MCE), ferrostatin-1 (1 μM; HY-100579, MCE) and subsequently with SK56 (15 μM and 45 μM) under complete medium conditions for 24 h. Control cells were maintained under identical culture conditions without treatment. Afterward, 10 μl of CCK-8 reagent (HY-K0301, MCE) was added to each well and incubated for 1 h. The absorbance was measured at 450 nm.
Design and training of interactive interface generative models
To obtain the training data, we collected real interactions from the PDB database and used these interaction data to train a slightly modified Transformer model (Fig. 1a). The self-attention operator, which serves as the basis for transformer networks, makes the transformer model highly suitable for processing 3D points. This is because self-attention operates on sets, meaning that it is not influenced by the order or number of input elements. Given that 3D point sets (for example, protein atoms) can be viewed as sets within the framework of 3D space, it is natural to use self-attention to process these point sets27. Therefore, we directly used the x–y–z coordinates as input.
Charge is an important influencing factor in protein interactions, and we quickly obtained the charge of all atoms in ~40,000 interaction interfaces using the ff14SB force field50. Atom type is also important information, and common atom types in proteins are limited (C, O, N and S). We tallied all possible combinations of atomic charges and types in all interfaces and discovered a total of 630 combinations (as vocabulary). We embedded the charge and atom-type combinations ([e, M]) into a 29-dimensional vector and combined it with coordinates as input for the model training. Because there are two types of data involved, only the [e, M] needs to be processed through a Softmax layer, whereas the coordinates do not. For the loss calculation, we used two different loss functions: cross-entropy KLdiv for the [e, M] loss and SmoothL1Loss for the coordinate losses.
The trained model can be used to output the ideal atomic coordinates, charges and atom types of an interaction interface. We then used the obtained ideal interaction interface to search the charge precomputed scaffold database (from the laboratory of D. Baker at the University of Washington or a custom-built peptide/cyclic-peptide database can also be used) using the coherent point drift algorithm51 for point set registration. After registration, the atomic distances, charge differences and atom-type matches were scored according to the following scoring function:
The atomic types that correspond to the ideal interface are represented by m, nmatch represents the number of atoms in the peptide that have the same charge polarity to the corresponding atoms in the ideal interface, ne represents the number of atoms in the peptide that have opposite charge polarity to the atoms in the ideal interface, n represents the total number of atoms in the ideal interface, es represents the difference in charges between the atoms in the peptide and the corresponding atoms in the ideal interface that have the same polarity, and d represents the difference in distances between the atoms in the peptide and the corresponding atoms in the ideal interface. Afterward, the top 30% of sequences with the highest scores were clustered based on sequence diversity. Subsequently, the surface charge distributions of the peptide sequences were analyzed, and 12 peptides with distinct features were ultimately selected. The peptides interacting directly with the target protein surface residues were optimized using the Rosetta FastDesign module, followed by expression, purification and further functional validation. All source code and database information are publicly available at Zenodo (https://doi.org/10.5281/zenodo.12527708)52 or GitHub (https://github.com/snhoau/TransForPep).
Recombinant protein expression
Plasmids (pSmart-I) encoding peptides, GSDMD-NT–BFP and SK56 mutants (S1G, E11G, R22G, Y26G, M29G, L32G, M37G and R42G) were prepared, and BL21 (DE3) E. coli cells were transformed with these vectors. An overnight preculture was added to Luria–Bertani medium with an original optical density of 0.05–0.1. Cells were grown at 37 °C and 220 rpm until the optical density reached 0.8–1, and the culture was then induced with 0.2 mM isopropyl β-d-thiogalactoside (final concentration; I6758, Sigma-Aldrich). Cells were further grown overnight at 37 °C and 220 rpm. The next day, the cells were collected at 6,000g for 5 min at 4 °C and resuspended in 40 ml of PBS–urea buffer (PBS with 0.5 M urea, pH 7.4). Cells were lysed by ultrasonication, and the soluble proteins were isolated from the cell debris by centrifugation at 12,000g for 30 min at 4 °C. Proteins were purified by affinity chromatography using an Ni-NTA resin column (30210, Qiagen) with wash buffer (PBS–urea buffer with 0, 20 and 50 mM imidazole) and eluting buffer (PBS–urea buffer with 500 mM imidazole). The obtained His6–SUMO-tagged protein was treated with SUMO protease (ULP1; 12588018, Thermo Fisher Scientific) to remove the tag (Extended Data Fig. 3c).
LPS- and CLP-induced sepsis in mice
All mice were kept under a 12-h light/12-h dark cycle in a specific pathogen-free facility at the Animal Resource Center. Age- and sex-matched mice were assigned randomly to the experimental and control groups. The investigators were not blinded to the experiments and outcome assessments. Sepsis was induced in C57BL/6J and Gsdmd−/– mice (8–10 weeks old, 50:50 female:male ratio) by i.p. injection of LPS (O55:B5; L2880, Sigma-Aldrich) at 15, 25 or 50 mg per kg (body weight), respectively. For CLP-induced sepsis, after anesthetizing male mice weighing 20–25 g with 3% isoflurane, a 1- to 2-cm longitudinal incision was made along the abdomen to expose the cecum. The cecum was ligated with a sterile 2–0 silk suture at a distance of 1.0 cm from the tip. Two punctures were made with a number 19 needle at the middle and top of the cecum to induce septic injury. After puncture, the cecum was gently compressed and returned to the abdominal cavity, and the abdominal cavity was sutured. All mice were subcutaneously injected with 1 ml of prewarmed 0.9% saline and 0.05 mg per kg (body weight) buprenorphine for fluid supplementation and postoperative pain relief. Mice were treated with SK56 or GSDMD-NT pore formation inhibitors (50 mg per kg (body weight) DSF; HY-B0240, MCE; 50 mg per kg (body weight) DMF; HY-17363, MCE; 20 mg per kg (body weight) NSA; HY-100573, MCE) or vehicle by i.v. injection at 4, 5 and 16 h after sepsis induction, respectively. To measure circulating cytokines, blood samples were collected by tail vein or posterior orbital vein bleed after LPS or CLP challenge and allowed to clot at room temperature. Sera obtained after centrifugation at 2,000g for 10 min were analyzed for inflammatory cytokines by ELISA, bead-based immunoassay or biochemical (IDEXX Catalyst One) analysis. Primary organs (heart, liver, kidney, gut and lung) were collected to make sections for pathologic analysis.
Construction of coculture of organoids and macrophages
Cancer-adjacent tissues were collected from clinical lung cancer resection samples. After being washed three times with PBS, the tissues were minced using ophthalmic scissors and digested with 10 U ml−1 dispase (354235, Corning) and 100 µg ml−1 DNase I (D5025, Sigma-Aldrich) to obtain a single-cell suspension of lung tissue. The suspension was subjected to sorting using a MoFlo Astrios EQ flow cytometer (Beckman Coulter) based on CD45−CD31−LysoTracker+EPCAM+ markers. After centrifugation, primary alveolar epithelial type 2 cells were collected and diluted to a cell density of 1 × 106 cells per ml and mixed with Matrigel (354234, Corning) at a 1:1 (vol:vol) ratio. The mixture was seeded in a 96-well plate (100 μl) and placed in a cell culture incubator for 30 min. Subsequently, 100 μl of alveolar maintenance medium (AMM), which was preheated to 37 °C, was slowly added along the well wall. The AMM was composed of 10 μM SB431542 (HY-10431, MCE), 3 μM CHIR99021 (HY-10182, MCE), 1 μM BIRB796 (HY-10320, MCE), 1 μM DMH-1 (HY-12273, MCE), 10 μM Y-27632 (HY-10071, MCE), 50 ng ml−1 human EGF (236-EG-200, R&D Systems), 10 ng ml−1 human FGF10 (345-FG-025, R&D Systems), 10 ng ml−1 mouse lL-1β (211-11B-10UG, PeproTech), 10 ng ml−1 human noggin protein (6057-NG-025, R&D Systems), 5 μg ml−1 heparin (HY-17567, MCE), 1× B-27 supplement (12587010, Thermo Fisher Scientific), 15 mM HEPES (15630106, Gibco), 1.25 mM N-acetylcysteine amide (5619, Tocris), 1× GlutaMAX (35050061, Gibco), 10% FBS (10099141C, Gibco) and DMEM/F12 (1:1; 11320033, Gibco). The organoids were cultured for 4 days before the medium was replaced with AMM without Y-27632 and IL-1β. The organoids were further cultured until day 10, after which they were switched to alveolar differentiation medium (ADM) for differentiation for an additional 5 days. ADM was composed of 5 ng ml−1 human EGF, 1 ng ml−1 human FGF10, 5 μg ml−1 heparin, 1× B-27 supplement, 15 mM HEPES, 1.25 mM N-acetylcysteine amide, 1× GlutaMAX, 10% FBS and DMEM/F12 (1:1). Mature alveolar organoids were collected using Cultrex Organoid Harvesting Solution (3700-100-01, R&D Systems) to remove the matrix gel. After centrifugation, the organoids were collected, and for every 500 organoids, 5 × 104 macrophages (THP-1 cells induced by 150 nM PMA) were added and cocultured in ultralow attachment culture plates for 4 h. Subsequently, 1 μg ml−1 LPS and 10 μM nigericin (with or without SK56) were added and incubated for 8–12 h. Finally, the organoids were fixed with 4% paraformaldehyde and subjected to immunofluorescence staining. For long-term experiments, organoids were first labeled with calcein-acetoxymethyl and then co-incubated with macrophages (THP-1 cells induced by 150 nM PMA) transfected with GSDMD-insert–BFP (insertion at Asp 275), and 0.1 μM PI was added at the start of recording to indicate dead cells. GSDMD-NT pore formation inhibitors (30 µM DSF, 25 µM DMF and 20 µM NSA) were used as controls.
Detection of cell membrane repair
BMDMs were cultured to 80% confluence and transfected with CHMP4–GFP plasmid using Advanced DNA/RNA Transfection Reagent (AD600150, Zeta Life) for 12 h. After transfection, the reagent was removed, and the cells were further cultured in fresh medium for 48 h. Successfully transfected BMDMs were used for experimental treatments. The cells were incubated at 37 °C for 80 min, followed by fixation with 4% paraformaldehyde for 15 min. Images were captured using a Nikon (Ti2-A) microscope.
Phagocytosis of pyroptotic membranes and activation of DCs
To differentiate DCs, bone marrow cells were cultured in RPMI-1640 medium supplemented with 25 ng ml−1 recombinant mouse granulocyte–macrophage colony-stimulating factor (GM-CSF; 315-03-50UG, PeproTech) and 10 ng ml−1 mouse IL-4 (214-14-50UG, PeproTech). On days 2 and 4, three-quarters of the supplemented medium was replaced and supplemented with sufficient GM-CSF and IL-4. The cells were cultured for 6 days, and DCs were collected and replated to promote DC maturation. After 48 h, LPS (1 μg ml−1) was added for 48 h to obtain mature BMDCs. The DC-enriched preparations routinely contained 75–85% CD11c+ cells. To obtain pyroptotic membrane fragments marked with BFP (PCFBFP), we constructed a plasmid with BFP inserted before the GSDMD cleavage site (Asp 275; GSDMD-casp–BFP). This plasmid was transfected (AD600150, Zeta Life) into BMDMs, and 36 h after transfection, pyroptosis was induced using 1 µg ml−1 LPS and 10 µM nigericin for 4 h. The supernatant was collected and centrifuged at 1,000g for 15 min, and the supernatant was further centrifuged at 20,000g for 30 min to collect the precipitated cell membrane fragments for subsequent experiments. BMDMs from Gsdmd−/− (PCFGsdmd−/−) or wild-type (PCF) mice were treated similarly with 1 µg ml−1 LPS and 10 µM nigericin for 4 h, followed by sonication to disrupt the cells. The supernatant was collected and centrifuged at 1,000g for 15 min and then at 150,000g for 1 h to collect cell membranes. The collection of NFCs was performed in the same manner as for PCFs, except that no LPS + nigericin treatment was used to activate pyroptosis. The collected cell membranes were subsequently fluorescently labeled using CellMask Orange (C10045, Thermo Fisher). Pretreated membrane fragments (2 µg ml−1) or proteins (10 µg ml−1) were added to the DCs and cocultured for 2 h. Phagocytosis efficiency was assessed using a Stellaris 5 confocal microscope (Leica Microsystems) for imaging and statistical analysis. To analyze the effect of SK56 on the activation of DCs, DCs were seeded at a density of 1 × 106 cells in 6-cm dishes and treated with 1 µg ml−1 Pam3CSK4 (HY-P1180A, MCE) for 3 h in a cell culture incubator. Following treatment, the cells were washed three times with PBS and subsequently stimulated according to their respective experimental groups with oxPAPC (120 µM; tlrl-oxp1, InvivoGen), GSDMD-NT (1 µg ml−1; P9442, FineTest), NFC (2 µg ml−1), PCF (2 µg ml−1), BFP (10 µg ml−1; P08114, Solarbio), SK56 (20 µM) and scrambled SK56 (SK56scrambled). After 12 h of stimulation, cell supernatants and lysates were collected. An ELISA for IL-1β was performed according to the manufacturer’s instructions provided with the IL-1β ELISA kit (BSK12015, Bioss). Data were collected using a microplate reader at a wavelength of 450 nm.
Quantification and statistical analysis
Statistical analyses were performed using GraphPad Prism 9.5 (GraphPad Software). No statistical methods were used to predetermine sample sizes, but our sample sizes are similar to those reported in previous publications30,41,42. Data normality was assessed using the d’Agostino-Pearson normality test. Student’s t-tests or nonparametric Mann–Whitney tests were used for comparing groups depending on the distribution of the data. Statistical comparisons between groups were conducted using ordinary one-way analysis of variance with multiple comparisons tests.
Additional methods and references are available in the Supplementary Information.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The mass spectrometry proteomics data have been deposited at the ProteomeXchange Consortium (PXD053323). All supporting data are available at Figshare (https://doi.org/10.6084/m9.figshare.26105632)53. Source data are provided with this paper.
Code availability
All source code and database information are publicly available at Zenodo (https://doi.org/10.5281/zenodo.12527708)52 and GitHub (https://github.com/snhoau/TransForPep).
Change history
17 September 2025
In the version of the article initially published, Jianhui Sun was incorrectly listed with two affiliations and is now listed with only one affiliation (Department of Trauma Medical Center, Daping Hospital, State Key Laboratory of Trauma and Chemical Poisoning, Army Medical University, Chongqing, China) in the HTML and PDF versions of the article.
References
Ding, J. J. et al. Pore-forming activity and structural autoinhibition of the gasdermin family. Nature 535, 111–116 (2016).
Shi, J. J. et al. Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature 526, 660–665 (2015).
Evavold, C. L. et al. The pore-forming protein gasdermin D regulates interleukin-1 secretion from living macrophages. Immunity 48, 35–44 (2018).
Heilig, R. et al. The gasdermin-D pore acts as a conduit for IL-1β secretion in mice. Eur. J. Immunol. 48, 584–592 (2018).
Degen, M. et al. Structural basis of NINJ1-mediated plasma membrane rupture in cell death. Nature 618, 1065–1071 (2023).
Kayagaki, N. et al. NINJ1 mediates plasma membrane rupture during lytic cell death. Nature 591, 131–136 (2021).
Andrews, N. W., Almeida, P. E. & Corrotte, M. Damage control: cellular mechanisms of plasma membrane repair. Trends Cell Biol. 24, 734–742 (2014).
Jimenez, A. J. et al. ESCRT machinery is required for plasma membrane repair. Science 343, 1247136 (2014).
Scheffer, L. L. et al. Mechanism of Ca2+-triggered ESCRT assembly and regulation of cell membrane repair. Nat. Commun. 5, 5646 (2014).
Tam, C. et al. Exocytosis of acid sphingomyelinase by wounded cells promotes endocytosis and plasma membrane repair. J. Cell Biol. 189, 1027–1038 (2010).
Karki, R. et al. Synergism of TNF-α and IFN-γ triggers inflammatory cell death, tissue damage, and mortality in SARS-CoV-2 infection and cytokine shock syndromes. Cell 184, 149–168 (2021).
Rathinam, V. A. K., Zhao, Y. & Shao, F. Innate immunity to intracellular LPS. Nat. Immunol. 20, 527–533 (2019).
Wu, C. Q. et al. Inflammasome activation triggers blood clotting and host death through pyroptosis. Immunity 50, 1401–1411 (2019).
Cheng, K. T. et al. Caspase-11-mediated endothelial pyroptosis underlies endotoxemia-induced lung injury. J. Clin. Invest. 127, 4124–4135 (2017).
Kayagaki, N. et al. Caspase-11 cleaves gasdermin D for non-canonical inflammasome signalling. Nature 526, 666–671 (2015).
Zanoni, I., Tan, Y., Di Gioia, M., Springstead, J. R. & Kagan, J. C. By capturing inflammatory lipids released from dying cells, the receptor CD14 induces inflammasome-dependent phagocyte hyperactivation. Immunity 47, 697–709 (2017).
Szekanecz, Z., Szamosi, S., Kovacs, G. E., Kocsis, E. & Benko, S. The NLRP3 inflammasome–interleukin 1 pathway as a therapeutic target in gout. Arch. Biochem. Biophys. 670, 82–93 (2019).
Chao, K. L., Kulakova, L. & Herzberg, O. Gene polymorphism linked to increased asthma and IBD risk alters gasdermin-B structure, a sulfatide and phosphoinositide binding protein. Proc. Natl Acad. Sci. USA 114, E1128–E1137 (2017).
Cao, Z. H. et al. Pyroptosis in diabetes and diabetic nephropathy. Clin. Chim. Acta 531, 188–196 (2022).
Pan, J. Y. et al. AIM2 accelerates the atherosclerotic plaque progressions in Apoe−/– mice. Biochem. Biophys. Res. Commun. 498, 487–494 (2018).
Flores, J. et al. Caspase-1 inhibition alleviates cognitive impairment and neuropathology in an Alzheimer’s disease mouse model. Nat. Commun. 9, 3916 (2018).
Xia, S. Y. et al. Gasdermin D pore structure reveals preferential release of mature interleukin-1. Nature 593, 607–611 (2021).
Watson, J. L. et al. De novo design of protein structure and function with RFdiffusion. Nature 620, 1089–1100 (2023).
Gainza, P. et al. De novo design of protein interactions with learned surface fingerprints. Nature 617, 176–184 (2023).
Cao, L. X. et al. Design of protein-binding proteins from the target structure alone. Nature 605, 551–560 (2022).
Huang, B. et al. A backbone-centred energy function of neural networks for protein design. Nature 602, 523–528 (2022).
Vaswani, A. et al. Attention is all you need. In Proc. 31st International Conference on Neural Information Processing Systems 6000–6010 (Curran Associates, 2017).
Fleishman, S. J. et al. Computational design of proteins targeting the conserved stem region of influenza hemagglutinin. Science 332, 816–821 (2011).
Miao, R. et al. Gasdermin D permeabilization of mitochondrial inner and outer membranes accelerates and enhances pyroptosis. Immunity 56, 2523–2541 (2023).
Hu, J. J. et al. FDA-approved disulfiram inhibits pyroptosis by blocking gasdermin D pore formation. Nat. Immunol. 21, 736–745 (2020).
Tao, J. et al. Polydiacetylene-nanoparticle-functionalized microgels for topical bacterial infection treatment. ACS Macro. Lett. 8, 563–568 (2019).
Gou, M. L. et al. Bio-inspired detoxification using 3D-printed hydrogel nanocomposites. Nat. Commun. 5, 3774 (2014).
Ruhl, S. et al. ESCRT-dependent membrane repair negatively regulates pyroptosis downstream of GSDMD activation. Science 362, 956–960 (2018).
Wright, S. S. et al. Transplantation of gasdermin pores by extracellular vesicles propagates pyroptosis to bystander cells. Cell 188, 280–291 (2024).
Holley, C. L. et al. Pyroptotic cell corpses are crowned with F-actin-rich filopodia that engage CLEC9A signaling in incoming dendritic cells. Nat. Immunol. 26, 42–52 (2025).
Weng, L. et al. National incidence and mortality of hospitalized sepsis in China. Crit. Care 27, 84 (2023).
Rudd, K. E. et al. Global, regional, and national sepsis incidence and mortality, 1990–2017: analysis for the Global Burden of Disease Study. Lancet 395, 200–211 (2020).
Angosto-Bazarra, D. et al. Evolutionary analyses of the gasdermin family suggest conserved roles in infection response despite loss of pore-forming functionality. BMC Biol. 20, 9 (2022).
Chi, Z. et al. Gasdermin D-mediated metabolic crosstalk promotes tissue repair. Nature 634, 1168–1177 (2024).
Zanoni, I. et al. An endogenous caspase-11 ligand elicits interleukin-1 release from living dendritic cells. Science 352, 1232–1236 (2016).
Rathkey, J. K. et al. Chemical disruption of the pyroptotic pore-forming protein gasdermin D inhibits inflammatory cell death and sepsis. Sci. Immunol. 3, eaat2738 (2018).
Humphries, F. et al. Succination inactivates gasdermin D and blocks pyroptosis. Science 369, 1633–1637 (2020).
Chen, H. et al. RIPK3 collaborates with GSDMD to drive tissue injury in lethal polymicrobial sepsis. Cell Death Differ. 27, 2568–2585 (2020).
Kang, R. et al. Lipid peroxidation drives gasdermin D-mediated pyroptosis in lethal polymicrobial sepsis. Cell Host Microbe 24, 97–108 (2018).
Silva, C. M. S. et al. Gasdermin D inhibition prevents multiple organ dysfunction during sepsis by blocking NET formation. Blood 138, 2702–2713 (2021).
Su, M. et al. Gasdermin D-dependent platelet pyroptosis exacerbates NET formation and inflammation in severe sepsis. Nat. Cardiovasc. Res 1, 732–747 (2022).
Zhu, C. L. et al. PD-L1 promotes GSDMD-mediated NET release by maintaining the transcriptional activity of STAT3 in sepsis-associated encephalopathy. Int. J. Biol. Sci. 19, 1413–1429 (2023).
Liu, F. et al. Neutrophil-specific depletion of gasdermin D does not protect against murine sepsis. Blood 141, 550–554 (2023).
Vincent, J. L. et al. The SOFA (sepsis-related organ failure assessment) score to describe organ dysfunction/failure. Intensive Care Med. 22, 707–710 (1996).
Maier, J. A. et al. ff14SB: improving the accuracy of protein side chain and backbone parameters from ff99SB. J. Chem. Theory Comput. 11, 3696–3713 (2015).
Myronenko, A. & Song, X. B. Point set registration: coherent point drift. IEEE Trans. Pattern Anal. Mach. Intell. 32, 2262–2275 (2010).
Wang, G. & Meng, P. TransForPrep. Zenodo https://doi.org/10.5281/zenodo.12527708 (2023).
Wang, G. et al. Data for ‘Delaying Pyroptosis With AI-Screened Gasdermin D Pore Blocker Mitigates Inflammatory Response’. figshare https://doi.org/10.6084/m9.figshare.26105632 (2025).
Acknowledgements
We greatly appreciate receiving the Gsdmd−/− mice from F. Shao at the National Institute of Biological Sciences. This work was supported by National Natural Science Foundation of China (82222038), Outstanding Young Talents of National Defense Biotechnology (01-SWKJYCJJ06) and Chongqing Outstanding Youth Fund (CSTB2022NSCQ-JQX0017) to L.Z. and the National Natural Science Foundation of China (82360075), Science and Technology Department of Yunnan Province (202405AJ310005, 2018DG008 and 202201AS070070) to P.M. and the National Natural Science Foundation of China (32471178 and 81903666) and Science and Technology Department of Yunnan Province (202101AT070301) to G.W. The funders had no role in study design, data collection and analysis, decision to publish or preparation of the manuscript.
Author information
Authors and Affiliations
Contributions
G.W., P.M. and L.Z. designed the study and wrote the paper. J.S., G.W. and J.Y. conducted most of the experiments and analyzed the results. J.T. conducted the nanoparticle experiments. R.W., S.Z., B.D. and S.T. contributed clinical samples and analyzed the clinical data. Y.Y. conducted the peptide purification experiments. H.Z. and W.L. conducted the MST experiments. D.L. and C.G. conducted peptide mutation experiments. Y.L., H.Q., J.J. and P.L. provided technical assistance and contributed to the preparation of the figures. R.S., Y.H. and J.L. provided feedback on the paper revisions. All authors analyzed the results and approved the final version of the paper.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Immunology thanks the anonymous reviewers for their contribution to the peer review of this work. Peer reviewer reports are available. Primary Handling Editor: Ioana Staicu, in collaboration with the Nature Immunology team.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data
Extended Data Fig. 1 GSDMD deficiency protects mice from sepsis-induced organ damage.
a-b, Kaplan–Meier analysis of survival rates in wild-type (WT) C57BL/6 and Gsdmd−/− mice (n = 10 mice per group) challenged with LPS (15 mg/kg i.p., a) and cecal ligation and puncture (CLP) surgery (b). c, Pathology assay showing representative lung H&E staining (upper left) and lung injury scores (upper right, n = 15 samples) from WT and Gsdmd−/− mice 2 days post-LPS (15 mg/kg i.p.) treatment. representative lung H&E staining (lower left) and pathology scores (lower right, n = 16 samples) in WT and Gsdmd−/− mice 2 days post-CLP. Scale bars 1 mm (overview), 50 µm (zoomed-in). d, Pathology assay showing kidney injury scores in WT and Gsdmd−/− mice 2 day after LPS (15 mg/kg i.p. n = 15 samples) or CLP (n = 16 samples). e, Representative H&E staining showing kidney, liver, intestine and spleen in WT and Gsdmd−/− mice at 2 day post-LPS or CLP. n = 10 mice. f, ELISA assay showing IL-1β levels in blood from WT and Gsdmd−/− mice after LPS (left) or CLP (right). n = 3 samples. Data in c, d and f were analyzed using two-tailed Student’s t-test; NS (P > 0.05, not significant); means ± s.d. Data in a, b were analyzed by log-rank (Mantel-Cox) test.
Extended Data Fig. 2 GSDMD deficiency reduces systemic inflammation and is activated in sepsis patients.
a, Heat map showing inflammatory cytokine profiles in peripheral blood of WT and Gsdmd−/− mice (n = 10 samples) at 2 days post LPS (15 mg/kg i.p.) or CLP. b, Changes in body weight of WT and Gsdmd−/− mice after LPS (15 mg/kg i.p.) or CLP (n = 10 mice per group). c, Biochemistry assay showing serum enzyme activity in WT and Gsdmd−/− mice 2 days post-LPS or CLP (n = 6 samples). d, Clinical assay showing serum IL-1β levels in healthy volunteers (n = 88), mild sepsis (SOFA ≥ 2, n = 137), and severe sepsis (SOFA > 11, n = 26) patients. e, Immunofluorescence image (left) and percentage of GSDMD-NT stained cells (right) in whole blood leukocytes from healthy volunteers (n = 5) and sepsis patients (n = 16). Scale bar, 25 µm. All graphs in this figure present mean ± s.d., and P values were determined using two-tailed Student’s t-test, NS (P > 0.05, not significant).
Extended Data Fig. 3 SK56 inhibits pyroptosis in vitro.
a, The atom frequency distribution histogram of the number at the interface (with H) involved in nearly 40,000 pairs of interaction interfaces. About 90% of the interfaces involved in the PDB database contain fewer than 350 atoms. b, The variation in loss during model training (total of 153k steps). c, SDS-PAGE image showing the prokaryotic expression and purification of SK56 (including tag cleavage). d, Live-cell imaging showing pyroptosis-induced cell rupture delay in BMDM cells treated with or without SK56 (15 µM). n = 3 repeats. Scale bar, 50 µm. e, ATP assay showing concentration-dependent suppression of pyroptosis by SK56 in BMDM cells. n = 3 repeats. f, 3D imaging showing THP-1 cell pyroptosis treated with or without SK56. Scale bar, 20 µm. g-h, ATP assay showing IC50 of SK56 in classical pyroptosis pathway (g) in THP-1 cells or nonclassical pyroptosis pathway in BMDM cells (h) at 3 h post-induction. n = 3 repeats. i, ELISA results showing the release of IL-1β during nonclassical pyroptosis treated with or without SK56 in BMDM cells. n = 3 repeats. j-m, Cell death assays showing HT29 (j, necrosis, TSZ: TNF-α + SM-164 + Z-VAD-FMK, n = 3 repeats), RAW264.7 (k, apoptosis, n = 4 repeats), AC16 (l, cuproptosis, n = 3 repeats), and MDA-MB-231 (m, ferroptosis, n = 3 repeats) cell responses to SK56. All graphs present mean ± s.d., and P values were determined using two-tailed Student’s t-test, NS (P > 0.05, not significant).
Extended Data Fig. 4 SK56 targets GSDMD-NT pore during pyroptosis.
a, Live-cell imaging showing SYTOX uptake in pyroptotic cells with or without SK56 (15 µM), n = 3 views. Scale bars, 10 µm. b, Immunofluorescence image showing GSDMD-NT (red arrow) localization in THP-1 treated with SK56 (15 µM). n = 5 views. c, Caspase-1 activity results showing SK56 effect on caspase-1. n = 4 repeats. d, Live-cell imaging showing mitochondrial membrane potential in pyroptotic cells. n = 3 cells. Scale bars, 4 µm. e-f, Single-cell biochemical assays showing ROS (e) and ATP (f) changes in THP-1 cells during pyroptosis with SK56 (15 µM) or DSF (30 µM) (n = 4 cells). g, lactate dehydrogenase (LDH) assay showing LDH release in pyroptosis THP-1 cells with SK56 or disulfiram (30 µM) (n = 3 repeats). All graphs present mean ± s.d., and P values were determined using two-tailed Student’s t-test, *P < 0.05, **P < 0.01, NS (P > 0.05, not significant).
Extended Data Fig. 5 Structural analysis of SK56 interaction with the GSDMD-NT pore.
a, Polydiacetylene (PDA) nanoparticles solidified by hydrogel were used to determine if SK56 has a repairing effect on preformed holes. Schematic presentation of installing PDA nanoparticles in the network of Poly (ethylene glycol) diacrylate (PEGDA) hydrogel. PDA nanoparticles can be chemically linked to the network of PEGDA hydrogels by photocrosslinking PEGDA monomers and acrylamide-modified PDA nanoparticles via addition polymerization. NPs, nanoparticles. b, MD simulation depicting residue-wise MM-GBSA binding free energy decomposition for SK56-GSDMD-NT interaction. n = 1 run. c, Structural analysis showing SK56-induced surface charge alterations in GSDMD-NT acidic patches (AP1/AP3).
Extended Data Fig. 6 SK56 protects human alveolar organoid-macrophage co-cultures from pyroptosis.
a, Image showing LysoTracker+ alveolar type 2 cells in 15-day human alveolar organoids. b, RNA-scope/IF images showing alveolar type 1 (AQP5 or Aqp5) and alveolar type 2 (SPC or Sftpc) in 15-day organoids. n = 2 repeats, Scale bar 25 µm. c, Morphology assay showing growth progression of human alveolar organoids over 20 days. n = 1 repeat. d, Co-culture assay showing CM-DiI-labeled THP-1 derived macrophages (red) with organoids treated with 1 μg/ml LPS + 10 μM nigericin (L + N) or PBS for 4 hours. SYTOX+ pyroptotic macrophages or organoid cells compromise organoid integrity. n = 3 repeats. Hoechst 33342 (blue) is used to indicate nucleus. Scale bar, 25 µm. e, Histology assay showing H&E-stained organoids with or without PMA-differentiated THP-1 at 8 hours post-L + N added. n = 5 repeats. Scale bar, 75 µm. f, Representative immunofluorescence image showing GSDMD-NT membrane translocation (red arrow) in organoid-macrophage cocultures (12 hours post-inducing pyroptosis, n = 7 repeats). Scale bar, 25 µm. Graphs present mean ± s.d., and P values were determined using two-tailed Student’s t-test.g, Immunoblots of whole blood from healthy volunteers treated with PBS, SK56 or DSF 1 hour post L + N added. n = 3 repeats.
Extended Data Fig. 7 SK56 inhibits cell death in human blood under pyroptosis condition.
Flow cytometry assay show changes in cell populations in peripheral blood samples from healthy volunteers (n = 7) and sepsis patients (n = 6; within 72 hours of sepsis confirmation). Blood from healthy volunteers was treated at 37 °C for 4 hours with 1 μg/ml LPS, 10 μM nigericin to induce pyroptosis, and DSF (disulfiram, 30 μM added 30 min before inducing pyroptosis). All graphs present mean ± s.d. Statistical significance was assessed using two-way ANOVA (NS, P > 0.05, no significance; *P < 0.05, **P < 0.01).
Extended Data Fig. 8 Pharmacokinetics and organ protection of SK56 in septic mice.
a, Serum concentrations of SK56 were measured following intravenous administration (i.v.) of 1 mg/kg SK56 in mice. n = 2 mice. b, Stability of SK56 in human whole blood was evaluated at 37 °C for 12 hours (n = 4 repeats). c, ELISA results showing plasma IL-1β dynamics in low-dose LPS (15 mg/kg, i.p.) sepsis mice with or without SK56 (n = 10 mice). The purple arrow indicates the time of SK56 administration. d, H&E-stained histopathological sections showing damage to lung tissues (2-day post-treatment) LPS-treated WT and Gsdmd−/− mice with or without SK56 (1 mg/kg i.v. n = 15 samples), along with the corresponding pathological score (right). Scale bars, 1 mm (whole section) and 50 µm (zoomed-in). e, H&E-stained histopathological sections illustrating damage to the kidneys, livers, intestines and spleens in WT or Gsdmd−/− mice treated with or without SK56 (1 mg/kg, i.v.), along with the pathological score (n = 30 in LPS; n = 27 samples in CLP) of the kidneys. All graphs show mean ± s.d., and P values were calculated using two-tailed Student’s t-test, NS (P > 0.05, not significant).
Extended Data Fig. 9 SK56 protects against sepsis induced by CLP.
a, Kaplan–Meier analysis of survival rates in WT and Gsdmd−/− mice challenged with CLP surgery and treated with SK56 (i.v. 1 mg/kg, 16 hours after CLP surgery) (n = 10 mice per group). b-c, Histopathology assay showing H&E-stained lung tissues (b) and pathological scores (c, n = 15 samples) in SK56-treated WT and Gsdmd−/− mice 2 days after CLP. Scale bars, 1 mm (whole section) and 50 µm (zoomed-in). d, Cytokine assay showing plasma cytokines level in SK56 treated or untreated CLP wild-type or Gsdmd−/− mice (n = 10 mice). e, Biochemistry assay indicating blood levels of organ damage markers in SK56 treated or untreated CLP mice (n = 6 mice). f, Body weight changes were monitored in mice subjected to LPS or CLP surgery with or without SK56 (n = 10 mice per group). g-h, SK56 and three GSDMD-NT pore formation inhibitors (DSF, disulfiram; DMF, dimethyl fumarate; NSA, necrosulfonamide) were evaluated for their impact on IL-1β levels (g) and biochemical markers (h) in LPS-treated mice blood (n = 10 mice). Data in c-e and g-h were analyzed using two-tailed paired Student’s t-test; NS (P > 0.05, not significant); means ± s.d. Data in a was analyzed by log-rank (Mantel-Cox) test.
Extended Data Fig. 10 SK56 modulates immune cell populations in septic mice.
Flow cytometry assay showing immune cell populations changes in the spleen, lung, liver, and peripheral blood of LPS-treated mice with or without SK56 treatment (n = 5 mice). All graphs present mean ± s.d. Statistical significance was evaluated using two-way ANOVA (NS, P > 0.05, no significance; *P < 0.05).
Supplementary information
Supplementary Information
Supplementary Methods.
Supplementary Table 1
Supplementary Tables 1–4.
Supplementary Video 2
Extended Data Fig. 3f. Live-cell imaging of the THP-1 cell line using a 3D Cell Explorer microscope (Nanolive). Cells were seeded onto a 35-mm glass-bottom dish 1 day before imaging. The 3D reconstruction images are displayed below.
Supplementary Video 3
Fig. 2f. Live-cell imaging showing the process of SK56 entering pyroptotic cells. FITC–SK56 was not bound to the cell in the early stages of pyroptosis. As pyroptosis progressed, the mitochondrial membrane potential decreased. Around 30 min later, fluorescently labeled SK56 entered the cell and colocalized with mitochondria and the cell membrane. As the peptide SK56 bound to the mitochondria, the mitochondrial membrane potential did not continue to decrease.
Supplementary Video 4
Fig. 5a. Long-term live-cell imaging revealing that in a coculture system of alveolar organoids and macrophages, the structure of the organoid is disrupted after inducing pyroptosis using LPS and nigericin. However, adding SK56 can effectively protect the organoid and delay its damage, whereas GSDMD-NT pore formation inhibitors (30 µM DSF, 25 µM DMF and 20 µM NSA) have no obvious protective effect on the organoids.
Source data
Source Data Figs. 2–5 and Extended Data Fig. 6
Unprocessed western blots.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Sun, J., Yang, J., Tao, J. et al. Delaying pyroptosis with an AI-screened gasdermin D pore blocker mitigates inflammatory response. Nat Immunol 26, 1660–1672 (2025). https://doi.org/10.1038/s41590-025-02280-x
Received:
Accepted:
Published:
Issue date:
DOI: https://doi.org/10.1038/s41590-025-02280-x
