
Abstract
Duchenne muscular dystrophy (DMD) is a rare, X-linked neuromuscular disease caused by pathogenic variants in the DMD gene that result in the absence of functional dystrophin, beginning at birth and leading to progressive impaired motor function, loss of ambulation and life-threatening cardiorespiratory complications. Delandistrogene moxeparvovec, an adeno-associated rh74-viral vector-based gene therapy, addresses absent functional dystrophin in DMD. Here the phase 3 EMBARK study aimed to assess the efficacy and safety of delandistrogene moxeparvovec in patients with DMD. Ambulatory males with DMD, ≥4 years to <8 years of age, were randomized and stratified by age group and North Star Ambulatory Assessment (NSAA) score to single-administration intravenous delandistrogene moxeparvovec (1.33 × 1014 vector genomes per kilogram; n = 63) or placebo (n = 62). At week 52, the primary endpoint, change from baseline in NSAA score, was not met (least squares mean 2.57 (delandistrogene moxeparvovec) versus 1.92 (placebo) points; between-group difference, 0.65; 95% confidence interval (CI), −0.45, 1.74; P = 0.2441). Secondary efficacy endpoints included mean micro-dystrophin expression at week 12: 34.29% (treated) versus 0.00% (placebo). Other secondary efficacy endpoints at week 52 (between-group differences (95% CI)) included: Time to Rise (−0.64 (−1.06, −0.23)), 10-meter Walk/Run (−0.42 (−0.71, −0.13)), stride velocity 95th centile (0.10 (0.00, 0.19)), 100-meter Walk/Run (−3.29 (−8.28, 1.70)), time to ascend 4 steps (–0.36 (−0.71, −0.01)), PROMIS Mobility and Upper Extremity (0.05 (−0.08, 0.19); −0.04 (−0.24, 0.17)) and number of NSAA skills gained/improved (0.19 (−0.67, 1.06)). In total, 674 adverse events were recorded with delandistrogene moxeparvovec and 514 with placebo. There were no deaths, discontinuations or clinically significant complement-mediated adverse events; 7 patients (11.1%) experienced 10 treatment-related serious adverse events. Delandistrogene moxeparvovec did not lead to a significant improvement in NSAA score at week 52. Some of the secondary endpoints numerically favored treatment, although no statistical significance can be claimed. Safety was manageable and consistent with previous delandistrogene moxeparvovec trials. ClinicalTrials.gov: NCT05096221
Similar content being viewed by others


Main
Duchenne muscular dystrophy (DMD) is caused by pathogenic variants in the X-linked DMD gene, leading to an absence of functional dystrophin and continuous muscle damage, beginning from birth1. Impaired motor function can be observed by the age of 3 years and typically progresses to loss of ambulation during adolescence with standard-of-care corticosteroid treatment1,2,3. Current approved treatments, including therapies designed to produce low-level dystrophin expression, may provide benefit for a minority of patients with specific pathogenic variants, but there is an unmet need for therapies that can more effectively stabilize or slow disease progression and that could be applicable to most of the DMD patient population, which sparked the research of new innovative therapies, including gene therapy4,5,6,7,8,9,10.
Delandistrogene moxeparvovec is a single-administration recombinant adeno-associated virus rhesus isolate serotype 74 (rAAVrh74) vector-based gene transfer therapy, approved in the United States for the treatment of patients with DMD at least 4 years of age with a confirmed mutation in the DMD gene, regardless of ambulatory status11,12. It is also approved in other select countries13,14,15,16,17,18. Delandistrogene moxeparvovec is designed to address the absence of functional dystrophin in DMD by delivering a transgene encoding delandistrogene moxeparvovec micro-dystrophin, an engineered protein retaining key functional domains of dystrophin19.
Early-phase clinical studies demonstrated a manageable safety profile for delandistrogene moxeparvovec. In these studies, delandistrogene moxeparvovec micro-dystrophin expression was robust with sarcolemmal localization up to 60 weeks after treatment and demonstrated a sustained functional stabilization through 4 years in four males with DMD (mean age at treatment, 5.1 years; mean age at 4-year follow-up, 9.2 years)20,21,22. Here we report results from Part 1 (52 weeks) of EMBARK (ClinicalTrials.gov: NCT05096221), a large, phase 3, two-part, multinational, randomized, double-blind, placebo-controlled trial assessing delandistrogene moxeparvovec safety and efficacy in patients with DMD aged ≥4 years to <8 years23.
Results
Patient disposition
Between October 2021 and September 2022, 173 patients were screened, 131 were randomized and 125 patients were treated (delandistrogene moxeparvovec, n = 63; placebo, n = 62; Fig. 1). Of the 173 patients screened, 13.3% were excluded due to elevated antibody titers to rAAVrh74. Analysis was by original assigned group (modified intent-to-treat population). The mean (s.d.) age at randomization was 6.03 (1.05) years, and the mean (s.d.) baseline North Star Ambulatory Assessment (NSAA) total score was 22.96 (3.75). Baseline clinical characteristics were balanced between groups (Table 1, Extended Data Table 1 and Supplementary Table 1). The week 52 cutoff date was 13 September 2023.

aOne patient was enrolled in Japan as part of a regional extension and was too late for inclusion in the primary analysis.
Primary outcome
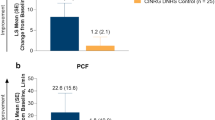
The primary outcome was change in NSAA total score from baseline to week 52 (Part 1). The NSAA is a categorical assessment of motor function in ambulatory patients with DMD, consisting of 17 items scored with a 0, 1 or 2 based on the patient’s ability to complete the task. At week 52 in the overall population, the least squares mean (LSM) change (95% confidence interval (CI)) from baseline in NSAA total score was 2.57 (1.80, 3.34) versus 1.92 (1.14, 2.70) points with delandistrogene moxeparvovec and placebo, respectively. The between-group difference was not statistically significant (0.65 (s.e. = 0.55) points; 95% CI, −0.45, 1.74; P = 0.2441; Fig. 2a,b). Results were consistent across pre-specified age subgroups and baseline NSAA total score subgroups (Supplementary Table 2).

a, Forest plot showing the primary endpoint (change from baseline to week 52 in NSAA total score, points) and key functional secondary endpoints (change from baseline to week 52 in TTR, seconds, and change from baseline to week 52 in 10MWR, seconds) for delandistrogene moxeparvovec and placebo groups in the modified intent-to-treat population. LSMs (of change from baseline) and CIs were standardized by dividing by the s.e. LSM differences are on original scale (without s.e. adjustment). TTR and 10MWR signs were reversed in the forest plot to align favorable directions among endpoints. Numerical results of LSM difference kept the original signs. One patient in the placebo group had missing data at week 52; functional tests were marked as invalid by the clinical evaluator due to back pain from compression fractures. b, Line graph showing LSM change from baseline to week 52 in NSAA total score, points, for delandistrogene moxeparvovec (n = 63) and placebo (n = 61) groups in the modified intent-to-treat population. Data are presented as LSM values ± 95% CI. c, Line graph showing LSM change from baseline to week 52 in TTR, seconds, for delandistrogene moxeparvovec (n = 63) and placebo (n = 61) groups in the modified intent-to-treat population. Data are presented as LSM values ± 95% CI. d, Line graph showing LSM change from baseline to week 52 in 10MWR, seconds, for delandistrogene moxeparvovec (n = 63) and placebo (n = 61) groups in the modified intent-to-treat population. Data are presented as LSM values ± 95% CI. a–d, The widths of the CIs have not been adjusted for multiplicity and cannot be used to infer definitive treatment effects. Negative values for TFTs (TTR and 10MWR) show an improvement in the time taken to achieve these endpoints.
Secondary outcomes
As defined per protocol, key secondary functional endpoints were Time to Rise (TTR) from the floor and 10-meter Walk/Run (10MWR) at week 52. The LSM change (95% CI) from baseline to week 52 on the TTR was −0.27 s (−0.56, 0.02) for delandistrogene moxeparvovec versus 0.37 s (0.08, 0.67) for placebo, with a between-group difference of −0.64 s (95% CI, −1.06, −0.23). Similarly, the LSM change (95% CI) from baseline to week 52 on the 10MWR was −0.34 s (−0.55, −0.14) for delandistrogene moxeparvovec versus 0.08 s (−0.13, 0.29) for placebo, with a between-group difference of –0.42 s (95% CI, −0.71, −0.13) (Fig. 2a,c,d). Subgroup analysis data are provided in Supplementary Table 2.
Other secondary functional endpoints assessed were stride velocity 95th centile (SV95C), 100-meter Walk/Run (100MWR) and time to ascend 4 steps. The LSM change (95% CI) from baseline to week 52 on SV95C was 0.06 meters per second (0.00, 0.13) for delandistrogene moxeparvovec versus –0.03 meters per second (−0.09, 0.03) for placebo, with a between-group difference of 0.10 meters per second (95% CI, 0.00, 0.19). The LSM change (95% CI) from baseline to week 52 on the 100MWR was −6.57 s (−10.05, −3.09) for delandistrogene moxeparvovec versus −3.28 s (−6.86, 0.29) for placebo, with a between-group difference of −3.29 s (95% CI, −8.28, 1.70). Analysis of time to ascend 4 steps showed LSM change (95% CI) from baseline to week 52 of −0.44 s (−0.69, −0.20) for delandistrogene moxeparvovec versus −0.08 s (−0.33, 0.17) for placebo, with a between-group difference of −0.36 s (95% CI, −0.71, −0.01) (Fig. 3). Subgroup analyses by age and baseline NSAA total scores are presented in Supplementary Table 2.

a, Forest plot showing other functional endpoints (change from baseline to week 52 in SV95C, meters per second; 100MWR, seconds; and time to ascend 4 steps, seconds) for delandistrogene moxeparvovec and placebo groups in the modified intent-to-treat population. LSMs (of change from baseline) and CIs were standardized by dividing by the s.e. Numerical results of the LSMs are on original scale (without s.e. adjustment). Signs of TFTs (100MWR and time to ascend 4 steps) were reversed in the forest plot to align favorable directions among endpoints. Numerical results of LSM difference kept the original signs. SV95C: a small number of patients did not have sufficient recorded hours at week 52 for analysis; 100MWR and time to ascend 4 steps: a small number of tests at either baseline or week 52 were marked as invalid by the clinical investigator; the most common reason was due to behavior. b, Line graph showing LSM change from baseline to week 52 in SV95C, meters per second, for delandistrogene moxeparvovec (n = 57) and placebo (n = 61) groups in the modified intent-to-treat population. Data are presented as LSM values ± 95% CI. c, Line graph showing LSM change from baseline to week 52 in 100MWR, seconds, for delandistrogene moxeparvovec (n = 59) and placebo (n = 57) groups in the modified intent-to-treat population. Data are presented as LSM values ± 95% CI. d, Line graph showing LSM change from baseline to week 52 in time to ascend 4 steps, seconds, for delandistrogene moxeparvovec (n = 62) and placebo (n = 60) groups in the modified intent-to-treat population. Data are presented as LSM values ± 95% CI. a–d, The widths of the CIs have not been adjusted for multiplicity and cannot be used to infer definitive treatment effects. Negative values for TFTs (100MWR and time to ascend 4 steps) show an improvement in the time taken to achieve these endpoints.
The LSM change (95% CI) from baseline to week 52 in the number of skills gained or improved as measured by the NSAA was 4.18 (3.58, 4.79) in the delandistrogene moxeparvovec group and 3.99 (3.37, 4.60) in the placebo group, with a between-group difference of 0.19 (−0.67, 1.06) (Supplementary Table 2 and Extended Data Table 2).
Analysis of Patient-Reported Outcomes Measurement Information System (PROMIS) Mobility showed an LSM (95% CI) change from baseline to week 52 of 0.05 (−0.05, 0.14) for delandistrogene moxeparvovec versus −0.01 (−0.10, 0.09) for placebo, with a between-group difference of 0.05 (−0.08, 0.19) (Supplementary Table 2). The LSM change (95% CI) from baseline for PROMIS Upper Extremity was 0.19 (0.05, 0.34) for delandistrogene moxeparvovec versus 0.23 (0.08, 0.37) for placebo, with a between-group difference of −0.04 (−0.24, 0.17).
Western blot analysis of week 12 biopsies in a subset of patients (n = 31) treated in trial sites where biopsies could be performed showed delandistrogene moxeparvovec micro-dystrophin expression in the treated group (mean (s.d.), 34.29% (41.04)) versus placebo (0.00% (0.00)) (Fig. 4 and Supplementary Fig. 1).

a, Delandistrogene moxeparvovec micro-dystrophin expression at week 12 as measured by western blot, percent normal (n = 17) and placebo (n = 14) groups in patients who had a muscle biopsy. Baseline data were not available as muscle biopsies were performed only at week 12. Each patient had two samples of biopsies taken, and all samples were analyzed. b, Representative western blots for delandistrogene moxeparvovec micro-dystrophin (left) and loading controls (right) from week 12 biopsies. Lane 1: DMD pool (negative control); Lanes 2–3: samples from placebo-treated patients; Lanes 4–5: samples from delandistrogene moxeparvovec–treated patients; Lanes 6–10: recombinant micro-dystrophin protein standard curve (21.85, 43.70, 87.39, 174.79 and 349.58 fmol mg−1). The faint upper lower molecular weight bands are non-specific. The 137-kDa band denotes the presence of delandistrogene moxeparvovec micro-dystrophin and was quantified. Each patient had two samples of biopsies taken, and all samples were analyzed.
Safety
Overall, 1,188 adverse events (AEs) were reported: 674 with delandistrogene moxeparvovec and 514 with placebo (Fig. 5). AEs are described as reported by the principal investigator at each study site. In the delandistrogene moxeparvovec group, 48 patients (76.2%) experienced 235 treatment-related treatment-emergent AEs (TR-TEAEs), with most occurring within the first 90 d of infusion; 83.3% were mild to moderate in severity, 98.3% of which resolved; and the events that were assessed as unresolved by the investigator are irritability (n = 2), decreased appetite (n = 1) and an erroneous laboratory value that was normal upon repeat (n = 1). Fourteen patients (22.2%) experienced 21 serious AEs (SAEs), and seven patients (11.1%) experienced 10 treatment-related SAEs (TR-SAEs) (Fig. 5 and Extended Data Table 3). There were no clinically significant complement-mediated AEs that triggered medical intervention as measured by C3, C4 and 50% hemolytic complement levels, and there were no cases of thrombotic microangiopathy. There were no AEs leading to study discontinuation or death. AEs of special interest are reported in Extended Data Table 4. A full list of TEAEs is reported in Supplementary Table 3. Post-baseline changes on electrocardiogram parameters and selected echocardiogram parameters were either normal or not clinically significant, and there were no remarkable findings in vital signs.

a, The timeline of events for the 10 TR-SAEs experienced by seven patients treated with delandistrogene moxeparvovec, detailing SAE symptom onset, hospital admission, hospital discharge and SAE resolution. See Extended Data Table 3 for a complete TR-SAE safety narrative. b, aGLDH increases were based on investigator assessment and their institution’s normal range. Shown are summaries of AEs, SAEs, TEAEs, TR-TEAEs, TR-SAEs, AEs leading to study discontinuation, deaths and TR-TEAEs occurring in more than 10% of patients. The safety population included all patients who received study treatment (excluding one patient enrolled under a regional addendum). Events are listed in descending order of frequency in the delandistrogene moxeparvovec group. AEs were classified according to the Medical Dictionary for Regulatory Activities.
In the placebo group, 17 patients (27.4%) experienced 43 TR-TEAEs; five patients (8.1%) experienced nine SAEs (coronavirus disease 2019, anal abscess, influenza, toxic shock syndrome, vomiting, arterial injury, upper limb fracture, left ventricular dysfunction and pyrexia); and there were no TR-SAEs.
The most common TR-TEAEs with delandistrogene moxeparvovec were vomiting (54.0%), nausea (31.7%) and decreased appetite (27.0%). TR-TEAEs of transient liver enzyme elevations (glutamate dehydrogenase (GLDH), gamma-glutamyl transferase, alanine transaminase and/or aspartate transaminase increases; total, 41.3%) occurred within the first 90 d after infusion (median, 42 d), resolved spontaneously or after an increase in peri-infusion corticosteroid treatment (10/26 (38.5%) patients with a liver event), and none progressed to liver failure. Post-infusion-added corticosteroid treatment for immunosuppression was increased if gamma-glutamyl transferase levels were confirmed to be ≥150 U L−1 or if there were any other clinically significant liver function abnormalities after infusion. The investigator may have made subsequent adjustments to immunosuppressive therapy in reaction to the subsequent course of acute liver injury or other AEs.
In the delandistrogene moxeparvovec group, seven patients (11.1%) experienced 10 TR-SAEs as reported by the principal investigator: acute liver injury (terms selected by investigators that refer to similar clinical patterns of liver biochemical markers included transient liver enzyme elevations (three events), hepatotoxicity and liver injury (one event of each)), myocarditis, nausea, vomiting, pyrexia and rhabdomyolysis (one event of each) (Fig. 5). The onset of the TR-SAEs were days 30–51 for acute liver injury, day 1 for myocarditis, nausea, vomiting and pyrexia and day 2 for rhabdomyolysis. All have resolved. For detailed narratives on TR-SAEs, see Extended Data Table 3.
There was a single event (1.6%) of myositis (TR-TEAE, separate from the event of rhabdomyolysis) reported in the delandistrogene moxeparvovec group that occurred on day 92 after infusion. The event occurred in a patient with a deletion of exons 46–50. There were no concurrent or recent illnesses or increased activity reported. Upon presentation, the patient was asymptomatic with a creatine kinase (CK) of 40,360 U L−1 (1.2× baseline) and an abnormal urinalysis (protein 1+, ketones 1+, hemoglobin 2+). The patient received intravenous (IV) fluids. By day 94, CK was down to 22,872 U L−1; however, CK increased to more than 40,000 U L−1 on day 96. The patient received a single IV dose of 18.125 mg of methylprednisolone and oral corticosteroid increase for 3 d. CK decreased to 19,315 U L−1 on day 99. The patient remained asymptomatic without any muscle tenderness, weakness or pain, and the mild event of myositis was assessed as recovered on day 108 after infusion. Myositis was reported by the investigator due to increased CK levels that were measured per protocol. The timing, severity and clinical course of this event differentiates it from previously observed events of immune-mediated myositis24.
Exploratory outcomes
Mean CK levels decreased with delandistrogene moxeparvovec versus placebo, with an LSM between-group difference in change from baseline to week 52 of −4,343.59 U L−1 (95% CI, −6,616.04, −2,071.15) (Extended Data Table 5 and Supplementary Fig. 2).
Sensitivity analyses
The pre-specified global statistical test on a composite of six functional endpoints (NSAA total score, TTR, 10MWR, SV95C, 100MWR and time to ascend 4 steps) conducted to analyze the totality of evidence for the treatment effect showed a difference (P = 0.0044) between delandistrogene moxeparvovec and placebo.
Post hoc analyses
A post hoc analysis showed that 3.2% of patients in the delandistrogene moxeparvovec group versus 16.4% of patients in the placebo group (odds ratio = 0.091; 95% CI, 0.01, 0.61) progressed to a TTR of over 5 s at week 52, a threshold of prognostic significance for loss of ambulation3,25.
Discussion
Results from EMBARK Part 1 confirmed that, at week 52, the safety profile of delandistrogene moxeparvovec is consistent with prior experience, and AEs were medically manageable with appropriate monitoring and treatment20,21,22. Immune reactions stimulated by the AAV vector are thought to be the primary cause of AEs in systemic AAV gene therapy, and each vector serotype may have a distinctive safety profile26,27. Delandistrogene moxeparvovec uses the rAAVrh74 vector, a clade E AAV28, distinct from the AAV9 clade F vector used in some DMD clinical trials and an approved gene therapy for spinal muscular atrophy29,30 (a clade being a phylogenetic group whose members share similarities in both function and serology)28,31,32. One of the challenges posed by the presence of pre-existing anti-AAV antibodies is the potential for activation of the complement system, which may lead to inflammation and safety concerns33,34. The particular characteristics of rAAVrh74 as well as the trial design may contribute to the absence of clinically significant complement-mediated AEs observed in the delandistrogene moxeparvovec clinical trials31,35. The rationale behind selection of the rAAVrh74 vector was that the non-human primate origin would decrease the likelihood of pre-existing immunity19,34. Seroprevalence analyses of patients with DMD in a previous study suggested that the presence of pre-existing antibodies against AAVrh74 was lower compared to AAV2, AAV8 and AAV9 seroprevalence36. Patients with elevated rAAVrh74 antibody titers (>1:400) were not eligible for the delandistrogene moxeparvovec clinical trials (excluding ongoing and upcoming trials designed to assess ways to overcome pre-existing immunity)34,37. Additionally, the use of MHCK7 as the promoter, associated with high levels of expression in skeletal muscles, and the inclusion of an enhancer to drive expression in the heart results in minimal off-target expression10,19. The safety profile observed thus far for delandistrogene moxeparvovec has supported the use of pre-infusion and post-infusion corticosteroid rather than a more intense prophylactic regimen of immunosuppressive drugs35.
Delandistrogene moxeparvovec did not show a statistically significant difference in the primary endpoint at week 52 versus placebo. Some of the key secondary and other functional endpoints that consisted of well-validated measures of ambulatory function in DMD numerically favored treatment, although no statistical significance can be drawn. Furthermore, the separation on TTR and 10MWR were consistent and similar in magnitude across the age groups.
The heterogeneity of disease progression is a challenge when designing DMD clinical trials, specifically trials of short duration38. Particularly, during the ages of 4–7 years, motor function and coordination, including ambulation, may be still improving, maintaining or starting to decline from peak function as patients may be in the maturational or the plateau/early-decline phase39. During the maturational phase, ambulatory function is still improving due to developmental changes in coordination and muscle growth and regeneration. Throughout the plateau and early-decline phase, peak motor function has generally been achieved with an onset of functional decline39. Furthermore, treatment with standard-of-care daily steroids may improve muscle strength and function over the short term in patients with DMD aged 4–7 years40, making demonstration of incremental short-term treatment benefit or further functional improvement in this patient population particularly challenging, especially in patients who had initiated steroids shortly before screening. In EMBARK, all patients were treated with daily corticosteroids, and baseline characteristics were well balanced across important prognostic variables, such as age, duration of steroid use, NSAA total score and timed function tests (TFTs), predicting similar disease progression between cohorts41.
This study highlights the value of objective and quantitative functional measures, such as TFTs and SV95C, for short-duration trials in younger patients with DMD treated with corticosteroids. The responsiveness of NSAA and TFTs, particularly in the younger population, is an area of recent investigation that will inform future trials in DMD3,42,43. In the present study, key secondary and other functional endpoints appeared to be more sensitive measures for this age group and study duration, with the ability to detect functional decline earlier, as previously shown39. Based on the broad scoring intervals for each functional assessment, NSAA scores of 1 (performance of tasks with difficulty or compensation) can only decline to 0 if functions are completely lost and can only improve to 2 if compensations are eliminated39. A score of 1 represents a broad range of abilities: a patient performing a task with slight difficulty may score a 1, whereas a patient performing a task with great difficulty but still able to complete the task may also score a 1. Therefore, in this early ambulatory patient population, the NSAA may not have been sensitive enough to detect a difference that was statistically significant at 52 weeks. First, there was a greater proportion of patients in the placebo group who progressed past the key prognostic threshold of 5 s on the TTR, which represents an earlier loss of ambulation. This indicates that delandistrogene moxeparvovec may reduce the odds of progressing to a TTR of more than 5 s by up to 91% and has the potential to modify the course of the disease. Second, the functional endpoint SV95C is a novel digital objective measure of ambulatory performance of daily activities in patients’ normal daily environment that is qualified for use by the European Medicines Agency as a primary endpoint in clinical trials of DMD44,45,46. Finally, although the primary endpoint did not show a statistically significant difference at week 52 versus placebo, the global statistical test, a composite measure of efficacy, supported the totality of evidence of treatment effect with delandistrogene moxeparvovec and indicated the presence of a functional treatment effect after accounting for multiple hypotheses tested across the primary and secondary study endpoints. The pre-specified global statistical test combines information from multiple endpoints and reduces multiple testing problems into a single test against the global null hypothesis of no treatment effect on all endpoints. Although the TTR and 10MWR assessments are included in the NSAA as ‘rise from floor’ and ‘run’, the assessment of these items in the NSAA is categorical and scored with a 0, 1 or 2 based on the patient’s ability to complete the task. The TTR and 10MWR key secondary endpoints are quantitative and assess the time it takes for the patient to complete the assessment.
No significant differences were observed between delandistrogene moxeparvovec and placebo groups for either PROMIS measure at week 52. This may have been related to a ceiling effect, as evidenced by the high baseline scores, rendering the overall score insensitive to capturing potential improvements or differences over 52 weeks. Furthermore, given that the studied population was still in the maturational phase, this likely reduced the potential to observe differences between groups over the 52-week timeframe.
In earlier studies with long-term follow-up, ambulatory patients treated with delandistrogene moxeparvovec at age 5.1 years (mean) showed stabilization of NSAA total scores over 4 years in four males with DMD22. Notably, the mean patient age at 4 years after treatment (9.2 years) surpassed the mean age at which NSAA total score has been shown to peak and subsequently decline (6.3 years) by approximately 3 years22,39. Furthermore, delandistrogene moxeparvovec may confer benefit to patients at various stages of disease progression by resulting in greater improvements versus natural history in the maturational phase of the disease and prevention of decline in older patients39. Delandistrogene moxeparvovec aims to protect muscles against further damage and stabilize or slow the decline of function; therefore, treatment may result in a higher natural peak of motor function for patients treated in the maturational phase compared to stabilization of motor function in those treated in the plateau/early-decline phase, with divergence from the natural disease course expected to widen over time.
Potential study limitations include the placebo group being limited to 1 year, due to ethical concerns of withholding disease-modifying treatment from patients in need of treatment. Although the study was blinded, because vomiting and nausea were the most common TR-TEAEs shortly after infusion, patients or caregivers may have become aware of treatment allocation. In addition, TTR is assessed in the primary endpoint of NSAA total score as the item ‘rise from floor’ and separately as a key secondary endpoint, which may be perceived as an overlap of outcome measures. However, it is important to note that the assessment of items in the NSAA is categorical and scored with a 0, 1 or 2 based on the patient’s ability to complete the task, whereas the TTR endpoint is quantitative and evaluates the time it takes for the patient to complete the assessment.
In conclusion, delandistrogene moxeparvovec did not show a statistically significant difference compared to placebo in the primary endpoint at week 52. Key secondary endpoints and other functional endpoints numerically favored delandistrogene moxeparvovec in the overall population and age subgroups, although no statistical significance can be claimed. This is consistent with long-term results from earlier delandistrogene moxeparvovec trials and the essential myoprotective role of functional dystrophin5,20,22,47. No new safety signals were identified in EMBARK, supporting a manageable safety profile of delandistrogene moxeparvovec. Among the TR-SAEs, no life-threatening events, deaths or study discontinuations were reported, and all have resolved. Collectively, the delandistrogene moxeparvovec safety profile observed in EMBARK was consistent with that observed in other trials in the clinical development program19,20,21,22. Of note, no clinically significant complement-mediated AEs were observed in this study, consistent with other clinical studies that have used the rAAVrh74 vector. Other clinical studies of this gene therapy in patients younger and older than those studied in EMBARK are ongoing48,49,50, and open-label extension data from studies in process should provide a better understanding of the long-term effects of delandistrogene moxeparvovec51,52.
Methods
Trial oversight
This trial was conducted in accordance with the provisions of the Declaration of Helsinki and Good Clinical Practice guidelines23. The trial protocol and all amendments were approved by an institutional review board and ethics committee at each site. The full list of institutional review boards and ethics committees is available in the Supplementary Information. The protocol is available upon reasonable request. Here we report results from a planned analysis, per protocol, of Part 1 (52 weeks) of EMBARK (SRP-9001-301; ClinicalTrials.gov: NCT05096221), a large, phase 3, two-part, multinational, randomized, double-blind, placebo-controlled trial assessing delandistrogene moxeparvovec safety and efficacy in patients with DMD aged ≥4 years to <8 years23. The primary analysis of the study was performed after all patients completed Part 1. No interim analysis was planned before the completion of Part 1. EMBARK was conducted at 42 sites in the United States, Europe and Asia23. The first patient was enrolled on 8 November 2021, and the last patient was enrolled on 14 September 2022. Informed consent was obtained from parent(s)/legal guardian(s), and patients’ assent was obtained when indicated. No compensation was offered for participation in the study other than covering for meals and travel-related expenses. All authors contributed to the design of the study, data collection, analyses, interpretation, manuscript writing, reviewing and approval and the decision to publish. The sponsor had final responsibility for the design of the trial, protocol, database maintenance, trial conduct, data analyses and confirmation of the accuracy of the data. All authors gathered the data, had access to the data and vouch for its accuracy and completeness for fidelity to the trial protocol. All authors contributed to data analysis and interpretation as well as manuscript writing, reviewing and approval. All authors jointly decided to publish the manuscript. An independent data monitoring committee continues to monitor safety, efficacy, data quality and study integrity.
Trial design
For all patients, the day before infusion, and, in addition to baseline stable oral corticosteroids, standard-of-care corticosteroid dosage was continued, and prednisone 1 mg kg−1 d−1 was added for suppression of potential AEs caused by immune response to the AAV vector, continuing for at least 60 d to a maximum total dose of 60 mg d−1 and then tapered to pre-infusion dosing. Patients were randomized (1:1 ratio) by interactive response technology to either a single IV administration of commercial process delandistrogene moxeparvovec material (1.33 × 1014 vector genomes per kilogram (vg/kg), linear standard quantitative polymerase chain reaction (PCR)), or placebo (0.9% sodium chloride solution) through a peripheral limb vein and stratified by age group (≥4 years to <6 years or ≥6 years to <8 years) at randomization and by NSAA total score (≤22 or >22) at screening. The random allocation sequence was saved in the interactive response technology system, which automatically assigned treatment based on sequence. All patients, parents/caregivers, investigators and site staff were blinded, except for the unblinded site pharmacist.
The crossover study consists of Part 1 (52 weeks (complete)) and Part 2 (52 weeks) followed by an open-label, follow-up study of at least 5 years (Supplementary Fig. 3). In Part 2, patients who received placebo in Part 1 received delandistrogene moxeparvovec, whereas those treated with delandistrogene moxeparvovec in Part 1 received placebo.
Between November 2020 and August 2022, the protocol was updated three times. The updates to the protocol are summarized below.
-
Version 1 (17 November 2020) to Version 2 (2 August 2021)
-
The primary reasons necessitating updates to the protocol were to add a blinded crossover design, so that patients randomized to placebo in Part 1 of the study had the opportunity to receive delandistrogene moxeparvovec in Part 2, and patients randomized to delandistrogene moxeparvovec in Part 1 received placebo in Part 2 to maintain the blind; to further clarify and refine the inclusion and exclusion criteria as well as stratification factors; to adjust the sample size; and to add a transgene ELISA endpoint.
-
-
Version 2 (2 August 2021) to Version 3 (30 August 2021)
-
The primary reasons necessitating updates to the protocol were to update exon language for inclusion criterion 2 and to update safety monitoring and AESI language.
-
-
Version 3 (August 2021) to Version 4 (August 2022)
-
The primary reasons necessitating updates to the protocol were to update the randomization language to allow approximately 50% of patients to be randomized in the ≥4-year to <6-year age group and to update safety monitoring language.
-
Gene therapy description
Delandistrogene moxeparvovec uses the rAAVrh74 vector due to its transduction efficiency and relatively low seroprevalence in patients with DMD compared to other AAV serotypes. The muscle-specific MHCK7 promoter and cardiac enhancer region drives expression in cardiac and skeletal muscles, including the diaphragm, with minimal off-target expression, and the delandistrogene moxeparvovec transgene encodes the key functional domains of full-length dystrophin, including anchor regions at the N-terminus and cysteine-rich (CR) region for actin and the dystrophin-associated protein complex, respectively, spectrin repeats 1–3 and 24 and hinge domains 1, 2 and 4 to maintain molecular flexibility53. The delandistrogene moxeparvovec inverted terminal repeat (ITR) to ITR sequence is available in the Supplementary Information.
Patients
A patient must meet all of the following criteria to be eligible to participate in this study:
-
1.
Is male at birth (self-reported), ambulatory and ≥4 years to <8 years of age at the time of randomization.
-
2.
Has a definitive diagnosis of DMD before screening based on documentation of clinical findings and prior confirmatory genetic testing using a clinical diagnostic genetic test. The genetic report must describe a frameshift deletion, frameshift duplication, premature stop (‘nonsense’), canonical splice site mutation or other pathogenic variant in the DMD gene fully contained between exons 18 and 79 (inclusive) that is expected to lead to absence of dystrophin protein.
-
3.
Is able to cooperate with motor assessment testing.
-
4.
Has an NSAA total score >16 and <29 at the screening visit.
-
5.
Has a TTR from the floor of <5 s at the screening visit.
-
6.
Stable daily dose of oral corticosteroids for at least 12 weeks before screening, and the dose and regimen are expected to remain constant (except for modifications to accommodate changes in weight) throughout the study.
-
7.
Has rAAVrh74 antibody titers of less than 1:400 (that is, not elevated) as determined by an ELISA.
-
8.
Patients who are sexually active must agree to use, for the entire duration of the study, a condom, and the female sexual partner must also use a medically acceptable form of birth control (for example, oral contraceptive).
-
9.
Has (a) parent(s) or legal guardian(s) who is/are able to understand and comply with the study visit schedule and all other protocol requirements.
-
10.
Is willing to provide informed assent (if applicable) and has (a) parent(s) or legal guardian(s) who is/are willing to provide informed consent for the patient to participate in the study.
A patient who met any of the following criteria was excluded from this study:
-
1.
Has DMD gene:
-
a.
Pathogenic variants between or including exons 1–17
-
b.
In-frame deletions, in-frame duplications and variants of uncertain significance
-
c.
Pathogenic variants fully contained within exon 45 (inclusive).
-
a.
-
2.
Has a left ventricular ejection fraction of less than 40% on the screening echocardiogram or clinical signs and/or symptoms of cardiomyopathy.
-
3.
Major surgery within 3 months before day 1 or planned surgery or procedures that would interfere with the conduct of the study for any time during this study.
-
4.
Presence of any other clinically significant illness (including cardiac, pulmonary, hepatic, renal, hematologic and immunologic), behavioral disease, infection, malignancy, concomitant illness or requirement for chronic drug treatment that, in the opinion of the investigator, creates unnecessary risks for gene transfer, medical condition or extenuating circumstance that, in the opinion of the investigator, might compromise the patient’s ability to comply with the protocol-required testing or procedures or compromise the patient’s well-being, safety or clinical interpretability.
-
5.
Has serologic evidence of current, chronic or active HIV, hepatitis C or hepatitis B infection.
-
6.
Has a symptomatic infection (for example, upper respiratory tract infection, pneumonia, pyelonephritis and meningitis) within 4 weeks before day 1.
-
7.
Demonstrates cognitive delay or impairment that could confound motor development in the opinion of the investigator.
-
8.
Treatment with any of the following therapies according to the timeframes specified:
-
a.
Any time:
-
Gene therapy
-
Cell-based therapy (for example, stem cell transplantation)
-
CRISPR–Cas9 or any other form of gene editing
-
-
b.
Within 12 weeks of day 1 and any time during the study:
-
Use of human growth factor or vamorolone
-
-
c.
Within 6 months of day 1 and any time during the study:
-
Any investigational medication
-
Any treatment designed to increase dystrophin expression (for example, Translarna, EXONDYS 51, VILTEPSO, VYONDYS 53 and AMONDYS 45)
-
-
a.
-
9.
Has received a live virus vaccine within 4 weeks or inactive vaccine within 2 weeks of the day 1 visit or expects to receive a vaccination during the first 3 months after day 1.
-
10.
Has abnormal laboratory values considered clinically significant, including, but not limited to:
-
a.
Gamma-glutamyl transferase >2× the upper limit of normal
-
b.
Glutamate dehydrogenase >15 U L−1
-
c.
Total bilirubin > upper limit of normal (elevations in total bilirubin confirmed to be due to Gilbert’s syndrome are not exclusionary)
-
d.
White blood cell count >18,500 per microliter
-
e.
Platelets ≤150,000 per microliter
-
a.
-
11.
Family does not want to disclose patient’s study participation with general practitioner/primary care physician and other medical providers.
-
12.
In the opinion of the investigator, the patient is not likely to be compliant with the study protocol.
Race and ethnicity were self-reported, determined by a two-question format and categories consistent with US Food and Drug Administration guidance54. Sex was self-reported by the patient or the parent/guardian. Per disease etiology, only males were enrolled.
Patient withdrawal criteria
A patient can withdraw from study participation at any time for any reason. A patient who withdraws before dosing may be replaced at the discretion of the sponsor. In addition, the sponsor may decide to stop the study participation of any patient as deemed necessary. The investigator may also stop the study participation of any patient at any time. Reasons for withdrawal from the study include, but are not limited to:
-
The patient or parent(s)/legal guardian(s) withdraw(s) consent.
-
Before randomization and dosing, it is determined that the patient was erroneously included in the study (that is, was found to not have met the eligibility criteria).
The investigator or study staff will document the reason(s) for withdrawal on the electronic case report form. If withdrawn patients received the study drug, every effort should be made to request that the patient allows follow-up for safety purposes.
Patients who withdraw from the study must return the wearable device.
Patients who have been dosed and withdraw from the study but do not withdraw consent will be asked to continue telephone calls to collect AEs and concomitant medication information and have blood collected for laboratory assessments per protocol every week for the first 12 weeks (±3 d) after infusion (if patients withdraw within this window) and then for safety laboratory assessments approximately every 6 months (±1 month) starting from the date of the last safety laboratory assessment before withdrawal. For this study, safety laboratory assessments include the following: electrolytes, troponin, liver function, hematology, high-sensitivity C-reactive protein and complement, renal function and urinalysis.
Assessments and endpoints
Patients were monitored weekly for 12 weeks after infusion and at weeks 24, 36 and 52. The NSAA and TFTs (TTR, time to ascend 4 steps, 10MWR and 100MWR)21 were performed at baseline and at weeks 12, 24, 36 and 52. The NSAA is a categorical assessment, and items are scored with a 0, 1 or 2 based on the patient’s ability to complete the task. The TFTs are quantitative and assess the time it takes for the patient to complete the assessment. For SV95C assessments, a wearable device (Syde) was worn daily for 3 weeks before infusion and then for 3 weeks before week 12, 24, 36 and 52 clinic visits. Week 12 biopsies from the medial gastrocnemius muscle in a subset of patients (n = 31), performed at sites pre-selected based on experience in performing muscle biopsies as routine in their diagnostic repertoire, were collected using open or core biopsies; each patient had two samples of biopsies taken, and all samples were processed for western blot20,21. Baseline biopsy data were not available for comparison as muscle biopsies were performed only at week 12. AE reporting was continuous, beginning at informed consent/assent.
The modified intent-to-treat population (all randomized patients who received study treatment (excluding one patient enrolled under a regional addendum), N = 125) was the analysis population for efficacy endpoints (Supplementary Table 4). The primary endpoint was change from baseline to week 52 in NSAA total score. The three pre-specified key secondary endpoints (in rank order) were quantity of delandistrogene moxeparvovec micro-dystrophin expression at week 12 (western blot) and change from baseline to week 52 in TTR and 10MWR. Other secondary endpoints were change from baseline to week 52 in: SV95C44, 100MWR and time to ascend 4 steps; change from baseline to week 52 in PROMIS scores in the Mobility and Upper Extremity Function domains; and number of skills gained or improved at week 52 as measured by the NSAA.
Safety assessments in the safety population (all patients who received study treatment (excluding one patient enrolled under a regional addendum)) included TEAEs, SAEs, AEs of special interest, clinically significant changes in vital signs and physical examination findings and clinically relevant changes in safety laboratory assessments, electrocardiograms and echocardiograms. The exploratory endpoint in Part 1 included change in CK levels in blood.
Methodology for processing and analyzing biologic samples
Week 12 biopsies collected at study sites were from the lower extremities of the medial gastrocnemius muscle, or alternatively allowed muscle groups, in a subset of patients using open or probe biopsies in accordance with allocation protocols and as previously described20,21. Samples were mounted, frozen in 2-methylbutane (isopentane) cooled in liquid nitrogen, stored at −80 °C and transferred in dry ice to the sponsor laboratory and transferred frozen to −80 °C freezer storage.
Western blot analyses were performed following Good Clinical Laboratory Practice standards, in accordance with validated methodology adapted from Charleston et al.55. Homogenized biopsy samples were assayed for total protein. Negative controls and total protein samples (20 μg per sample) as well as a five-point standard curve (recombinant micro-dystrophin (Curia) ranging from 21.85 to 349.58 fmol mg−1 protein) were resolved using SDS-PAGE (Invitrogen). Membranes with transferred proteins were probed using an anti-dystrophin primary antibody (DYS3, 1:20; Leica Biosystems) and then anti-mouse immunoglobulin G-conjugated horseradish peroxidase (Amersham ECL anti-mouse immunoglobulin G peroxidase-linked species-specific whole antibody (from sheep)) (NA931V, 1:1,000; Cytiva). A chemiluminescence imaging system (Alliance Q9 Advanced Imager, UVITEC) was used to visualize bound enzyme activity, and ImageQuant TL version 8.2 software (Cytiva) was used to analyze the bands. Contrast was automatically adjusted in the entire image by ImageQuant TL software; quantitative value remained the same as the original untuned image. For the loading control, membranes were probed with anti-alpha actinin antibody (A7811, 1:100,000; Sigma-Aldrich) and then the same secondary antibody and imaging procedure as described above. In each sample, delandistrogene moxeparvovec micro-dystrophin was quantified using data that were normalized to each patient’s muscle content. Control samples used in western blot assays were kindly provided by Steven A. Moore (Wellstone Center, University of Iowa). As the muscle biopsy samples being tested are from patients with varying conditions of muscle degeneration, it is necessary to normalize delandistrogene moxeparvovec micro-dystrophin expression data generated by western blot to muscle content. Protein expression data generated by western blot are expressed as percent of normal control samples derived from a pool of normal control muscle biopsied. Muscle content is then determined using Masson’s trichrome histological stain paired with digital image analysis on a serial section within the same biopsy. The algorithm quantifies the area of muscle as a percentage of total area, generating percent muscle content. The adjusted values represent the percent normal delandistrogene moxeparvovec micro-dystrophin expression normalized to the percent muscle content. Thus, the resulting muscle content adjusted expression values provide meaningful measurement of micro-dystrophin expression in tissues with progressive muscle degeneration, as present in the DMD patient population.
Statistical analysis
Assuming an s.d. of 3.5 estimated based on previous delandistrogene moxeparvovec clinical studies19,20,21,22 and a 10% dropout rate at week 52, with a type 1 error of 0.05 (two-sided), a sample size of 120 with 1:1 randomization provided approximately 90% power to detect a mean difference of 2.2 in change in NSAA total score from baseline to week 52 between the delandistrogene moxeparvovec and placebo groups under the two-sample t-test. Estimate of effect size for difference between mean was equal to the ratio of expected difference and s.d. (2.2/3.5).
A restricted maximum likelihood-based mixed model for repeated measures (MMRM) analysis was used to compare delandistrogene moxeparvovec with placebo from baseline to week 52, with 95% CIs for the difference in LSM between treatment groups. SAS software version 9.4 was used to perform the statistical analysis for the primary endpoint. In this model, the response vector consisted of the change from baseline in NSAA total score at each post-baseline visit in Part 1. The model included the covariates of treatment group (categorical), visit (categorical), treatment group by visit interaction, age group at the time of randomization (categorical), baseline NSAA total score, age group at the time of randomization by visit interaction and baseline NSAA total score by visit interaction. All covariates were fixed effects in this analysis. An unstructured covariance matrix was used to model the within-patient variance–covariance errors. If the unstructured covariance structure resulted in a lack of convergence, the heterogeneous first-order autoregressive covariance structure was used. The Kenward–Roger approximation was used to estimate the denominator degrees of freedom. In the primary analysis, missing data were assumed to be missing at random. An MMRM analysis similar to the one for the primary endpoint was performed to compare the two treatment groups for each of the secondary endpoints, with baseline NSAA raw total score replaced with the corresponding baseline for the secondary endpoint, as well as NSAA group at the time of screening (≤22 versus >22) added as a covariate.
For the primary endpoint, a subgroup analysis was conducted with respect to all subgroup variables (≥4 years to <6 years or ≥6 years to <8 years) and NSAA total scores (≤22 versus >22). For each category of a subgroup variable, an MMRM similar to the primary analysis model was fitted using subset data. For age group subgroup analysis, age group and age group by visit interaction were removed from the MMRM model as a covariate. For the secondary endpoints, subgroup analysis was conducted with respect to the age and NSAA group (at the time of screening), using an analysis method similar to the subgroup analyses for the primary endpoint (with baseline NSAA total score being replaced with the baseline value for the corresponding endpoint in the MMRM model, as well as NSAA group at the time of screening (≤22 versus >22) added as a covariate, if applicable).
Because the primary endpoint did not meet statistical significance, and because the statistical analysis plan did not include a provision for correcting for multiplicity beyond the planned hierarchical testing procedure, results are reported as point estimates with between-group differences in LSM changes and 95% CIs. The widths of the CIs have not been adjusted for multiplicity and should not be used to infer definitive treatment effects for secondary outcomes or in subgroups.
To assess the totality of evidence wholistically and address the concern of multiple hypothesis testing, an additional pre-specified efficacy exploratory analysis that was not controlled for multiplicity within the hierarchical testing procedure was performed using the Wei–Lachin procedure56. The test was performed as a global statistical test on a composite of multiple endpoints (as pre-specified as a sensitivity analysis), assessing overall treatment effects among the primary, key secondary and other functional efficacy endpoints (NSAA total score, TTR, 10MWR, SV95C, 100MWR and time to ascend 4 steps). The global statistical test combines information from multiple endpoints and reduces multiple testing problems into a single test against the global null hypothesis of no treatment effect on all endpoints. The global statistical test was implemented by comparing the sum of observed t-statistics from multiple endpoints against the null distribution induced by 10,000 permutations57.
Hierarchical statistical testing (at completion of Part 1)
This analysis included the analyses of all data through the completion of Part 1 for the following endpoints:
-
Change in NSAA total score from baseline to week 52 (Part 1)
-
Quantity of delandistrogene moxeparvovec micro-dystrophin expression at week 12 (Part 1) as measured by western blota
-
Change in TTR from the floor from baseline to week 52 (Part 1)a
-
Change in time of 10MWR from baseline to week 52 (Part 1)a
-
Change in SV95C from baseline to week 52 (Part 1)
-
Change in time of 100MWR from baseline to week 52 (Part 1)
-
Change in time to ascend 4 steps from baseline to week 52 (Part 1)
-
Change in PROMIS Mobility score from baseline to week 52 (Part 1)
-
Change in PROMIS Upper Extremity score from baseline to week 52 (Part 1)
-
Number of skills gained or improved at week 52 (Part 1) as measured by the NSAA
aKey secondary efficacy endpoints.
Additional statistical considerations
Analyses of exploratory endpoints defined for Part 1 of the study were performed as follow-on analyses of the above endpoints. The Part 1 analysis also included disposition, demographics and baseline characteristics, medical history, concomitant medications, treatment exposure and compliance, baseline and post-baseline corticosteroids and protocol deviations.
The initial power analysis relied on data from the phase 1 study22. Subsequent adjustments to the power analysis assumptions were made in response to new findings from the additional phase 2 and phase 1b studies20,21.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The datasets presented in this article are not readily available as this study is ongoing, and access to the data is limited to those that support the findings of this study. De-identified patient-level data cannot be disclosed due to confidentiality agreements and the risk of re-identification. Qualified researchers may request access to the data that support the findings of Part 1 of this study and clinical study documents from Sarepta Therapeutics, Inc. by contacting medinfo@sarepta.com, subject to review by the study sponsors on a case-by-case basis. Data requests will be fulfilled within 90 d, and a data transfer agreement may be required.
References
Duan, D., Goemans, N., Takeda, S., Mercuri, E. & Aartsma-Rus, A. Duchenne muscular dystrophy. Nat. Rev. Dis. Prim. 7, 13 (2021).
Rodino-Klapac, L. R., Mendell, J. R. & Sahenk, Z. Update on the treatment of Duchenne muscular dystrophy. Curr. Neurol. Neurosci. Rep. 13, 332 (2013).
McDonald, C. M. et al. Long-term effects of glucocorticoids on function, quality of life, and survival in patients with Duchenne muscular dystrophy: a prospective cohort study. Lancet 391, 451–461 (2018).
Birnkrant, D. J. et al. Diagnosis and management of Duchenne muscular dystrophy, part 1: diagnosis, and neuromuscular, rehabilitation, endocrine, and gastrointestinal and nutritional management. Lancet Neurol. 17, 251–267 (2018).
McDonald, C. M. et al. Open-label evaluation of eteplirsen in patients with Duchenne muscular dystrophy amenable to exon 51 skipping: PROMOVI trial. J. Neuromuscul. Dis. 8, 989–1001 (2021).
Aartsma-Rus, A. et al. Theoretic applicability of antisense-mediated exon skipping for Duchenne muscular dystrophy mutations. Hum. Mutat. 30, 293–299 (2009).
Mendell, J. R. et al. Eteplirsen for the treatment of Duchenne muscular dystrophy. Ann. Neurol. 74, 637–647 (2013).
Bushby, K. et al. Diagnosis and management of Duchenne muscular dystrophy, part 1: diagnosis, and pharmacological and psychosocial management. Lancet Neurol. 9, 77–93 (2010).
Harper, S. et al. Modular flexibility of dystrophin: implications for gene therapy of Duchenne muscular dystrophy. Nat. Med. 8, 253–261 (2002).
Salva, M. Z. et al. Design of tissue-specific regulatory cassettes for high-level rAAV-mediated expression in skeletal and cardiac muscle. Mol. Ther. 15, 320–329 (2007).
US Food and Drug Administration. ELEVIDYS (delandistrogene moxeparvovec-rokl). Highlights of prescribing information. https://www.fda.gov/media/169679/download
US Food and Drug Administration. FDA expands approval of gene therapy for patients with Duchenne muscular dystrophy. https://www.fda.gov/news-events/press-announcements/fda-expands-approval-gene-therapy-patients-duchenne-muscular-dystrophy (2024).
UAE Ministry of Health and Prevention. Registered Medical Product Directory. https://mohap.gov.ae/en/more/registered-medical-product-directory (2024).
Qatar Ministry of Public Health. Qatar National Formulary. https://www.moph.gov.qa/english/OurServices/advancedsearch/Pages/servicesdetails.aspx?serviceId=234 (2024).
NHRA Bahrain. Pharmacy & Pharmaceutical Products Regulation (PPR). https://www.nhra.bh/Departments/PPR/ (2024).
Oman Ministry of Health. List of registered pharmaceutical manufacturers and products. https://www.moh.gov.om/en/hospitals-directorates/directorates-and-centers-at-hq/drug-safety-center/#Resources (2024).
Kuwait Ministry of Health. Drug and Dietary Supplement Price List. https://e.gov.kw/sites/kgoenglish/Pages/eServices/MOH/DrugFoodSupplementPrices.aspx (2024).
Ministry of Health Israel. The Israeli Drug Registry. https://israeldrugs.health.gov.il/#!/byDrug (2024).
Mendell, J. et al. Assessment of systemic delivery of rAAVrh74.MHCK7.micro-dystrophin in children with Duchenne muscular dystrophy: a nonrandomized controlled trial. JAMA Neurol. 77, 1121–1131 (2020).
Mendell, J. R. et al. Expression of SRP-9001 dystrophin and stabilization of motor function up to 2 years post-treatment with delandistrogene moxeparvovec gene therapy in individuals with Duchenne muscular dystrophy. Front. Cell Dev. Biol. 11, 1167762 (2023).
Zaidman, C. M. et al. Delandistrogene moxeparvovec gene therapy in ambulatory patients (aged ≥4 to <8 years) with Duchenne muscular dystrophy: 1-year interim results from study SRP-9001-103 (ENDEAVOR). Ann. Neurol. 94, 955–968 (2023).
Mendell, J. R. et al. Long-term safety and functional outcomes of delandistrogene moxeparvovec gene therapy in patients with Duchenne muscular dystrophy: a phase 1/2a nonrandomized trial. Muscle Nerve 69, 93–98 (2024).
A gene transfer therapy study to evaluate the safety and efficacy of SRP-9001 (delandistrogene moxeparvovec) in participants with Duchenne muscular dystrophy (DMD) (EMBARK). https://clinicaltrials.gov/ct2/show//NCT05096221
Khan, S. et al. T-cell response to micro-dystrophin in a patient treated with delandistrogene moxeparvovec gene therapy: a case of immune-mediated myositis. In 28th International Annual Congress of the World Muscle Society (2023); https://investorrelations.sarepta.com/static-files/51d79e1a-7ef7-46c6-93e3-c63b7be11238
Zambon, A. A. et al. Peak functional ability and age at loss of ambulation in Duchenne muscular dystrophy. Dev. Med. Child Neurol. 64, 979–988 (2022).
Ertl, H. C. J. Immunogenicity and toxicity of AAV gene therapy. Front. Immunol. 13, 975803 (2022).
Salmon, F., Grosios, K. & Petry, H. Safety profile of recombinant adeno-associated viral vectors: focus on alipogene tiparvovec (Glybera®). Expert Rev. Clin. Pharm. 7, 53–65 (2014).
Chan, C., Harris, K. K., Zolotukhin, S. & Keeler, G. D. Rational design of AAV-rh74, AAV3B, and AAV8 with limited liver targeting. Viruses 15, 2168 (2023).
Ogbonmide, T. et al. Gene therapy for spinal muscular atrophy (SMA): a review of current challenges and safety considerations for onasemnogene abeparvovec (zolgensma). Cureus 15, e36197 (2023).
US Food and Drug Administration. ZOLGENSMA (onasemnogene abeparvovec-xioi). Highlights of prescribing information. https://www.fda.gov/media/126109/download?attachment
Goedeker, N. L. et al. Evaluation of rAAVrh74 gene therapy vector seroprevalence by measurement of total binding antibodies in patients with Duchenne muscular dystrophy. Ther. Adv. Neurol. Disord. 16, 17562864221149781 (2023).
Gao, G. et al. Clades of adeno-associated viruses are widely disseminated in human tissues. J. Virol. 78, 6381–6388 (2004).
Kropf, E., Markusic, D. M., Majowicz, A., Mingozzi, F. & Kuranda, K. Complement system response to adeno-associated virus vector gene therapy. Hum. Gene Ther. 35, 425–438 (2024).
Potter, R. A. et al. Use of plasmapheresis to lower anti-AAV antibodies in nonhuman primates with pre-existing immunity to AAVrh74. Mol. Ther. Methods Clin. Dev. 32, 101195 (2024).
Mendell, J. R. et al. Practical considerations for delandistrogene moxeparvovec gene therapy in patients with Duchenne muscular dystrophy. Pediatr. Neurol. 153, 11–18 (2024).
Verma, S. et al. Seroprevalence of adeno-associated virus neutralizing antibodies in males with Duchenne muscular dystrophy. Hum. Gene Ther. 34, 430–438 (2023).
A gene transfer therapy study to evaluate the safety and efficacy of delandistrogene moxeparvovec (SRP-9001) following imlifidase infusion in participants with Duchenne muscular dystrophy (DMD) determined to have pre-existing antibodies to recombinant adeno-associated virus serotype (rAAVrh74). https://classic.clinicaltrials.gov/ct2/show/NCT06241950
Goemans, N. et al. Prognostic factors for changes in the timed 4-stair climb in patients with Duchenne muscular dystrophy, and implications for measuring drug efficacy: a multi-institutional collaboration. PLoS ONE 15, e0232870 (2020).
Muntoni, F. et al. Categorising trajectories and individual item changes of the North Star Ambulatory Assessment in patients with Duchenne muscular dystrophy. PLoS ONE 14, e0221097 (2019).
Matthews, E., Brassington, R., Kuntzer, T., Jichi, F. & Manzur, A. Corticosteroids for the treatment of Duchenne muscular dystrophy. Cochrane Database Syst. Rev. 2016, CD003725 (2016).
Ferizovic, N. et al. Prognostic indicators of disease progression in Duchenne muscular dystrophy: a literature review and evidence synthesis. PLoS ONE 17, e0265879 (2022).
Arora, H. et al. Longitudinal timed function tests in Duchenne muscular dystrophy: imagingDMD cohort natural history. Muscle Nerve 58, 631–638 (2018).
Merlini, L. & Sabatelli, P. Improving clinical trial design for Duchenne muscular dystrophy. BMC Neurol. 15, 153 (2015).
Servais, L., Yen, K., Guridi, M. & Lukawy, J. Stride velocity 95th centile: insights into gaining regulatory qualification of the first wearable-derived digital endpoint for use in Duchenne muscular dystrophy trials. J. Neuromuscul. Dis. 9, 335–346 (2022).
European Medicines Agency. Qualification opinion on stride velocity 95th centile as a secondary endpoint in Duchenne muscular dystrophy measured by a valid and suitable wearable device. https://www.ema.europa.eu/en/documents/scientific-guideline/qualification-opinion-stride-velocity-95th-centile-secondary-endpoint-duchenne-muscular-dystrophy_en.pdf (2019).
European Medicines Agency. Qualification opinion for stride velocity 95th centile as primary endpoint in studies in ambulatory Duchenne muscular dystrophy studies. https://www.ema.europa.eu/en/documents/scientific-guideline/qualification-opinion-stride-velocity-95th-centile-primary-endpoint-studies-ambulatory-duchenne_en.pdf (2023).
Mendell, J. R. et al. Longitudinal effect of eteplirsen versus historical control on ambulation in Duchenne muscular dystrophy. Ann. Neurol. 79, 257–271 (2016).
A gene transfer therapy study to evaluate the safety and efficacy of SRP-9001 (delandistrogene moxeparvovec) in non-ambulatory and ambulatory participants with Duchenne muscular dystrophy (DMD) (ENVISION). https://clinicaltrials.gov/study/NCT05881408
A two-part, open-label systemic gene delivery study to evaluate the safety and expression of RO7494222 (SRP-9001) in subjects under the age of four with Duchenne muscular dystrophy (ENVOL) (2022-000691-19). https://www.clinicaltrialsregister.eu/ctr-search/trial/2022-000691-19/FR
A gene delivery study to evaluate the safety of and expression from SRP-9001 in Duchenne muscular dystrophy (DMD) (ENDEAVOR). https://clinicaltrials.gov/ct2/show/NCT04626674
A gene transfer therapy study to evaluate the safety of delandistrogene moxeparvovec (SRP-9001) in participants with Duchenne muscular dystrophy (DMD). https://classic.clinicaltrials.gov/ct2/show/NCT03375164
A randomized, double-blind, placebo-controlled study of SRP-9001 for Duchenne muscular dystrophy (DMD). https://classic.clinicaltrials.gov/ct2/show/NCT03769116
Asher, D. et al. Clinical development on the frontier: gene therapy for duchenne muscular dystrophy. Expert Opin. Biol. Ther. 20, 263–274 (2020).
US Food and Drug Administration. Collection of race and ethnicity data in clinical trials and clinical studies for FDA-regulated medical products. https://www.fda.gov/regulatory-information/search-fda-guidance-documents/collection-race-and-ethnicity-data-clinical-trials-and-clinical-studies-fda-regulated-medical (2024).
Charleston, J. S. et al. Eteplirsen treatment for Duchenne muscular dystrophy: exon skipping and dystrophin production. Neurology 90, e2146–e2154 (2018).
Wei, L. J. & Lachin, J. M. Two-sample asymptotically distribution-free tests for incomplete multivariate observations. J. Am. Stat. Assoc. 79, 653–661 (1984).
Li, D. et al. Assessment of treatment effect with multiple outcomes in 2 clinical trials of patients with Duchenne muscular dystrophy. JAMA Netw. Open 3, e1921306 (2020).
Acknowledgements
J.R.M.’s affiliation at the time of Part 1 of EMBARK was Center for Gene Therapy, Nationwide Children’s Hospital, Columbus, OH, USA (currently employed by Sarepta Therapeutics, Inc.). This trial was sponsored by Sarepta Therapeutics, Inc. and funded by Sarepta Therapeutics, Inc. and F. Hoffmann-La Roche, Ltd. The sponsor had final responsibility for the design of the trial, protocol, database maintenance, trial conduct, data analyses and confirmation of the accuracy of the data.
We thank the patients who participated in this trial and their families and caregivers; the members of the data monitoring committee; the staff who assisted with the trial at each site; the EMBARK Study Group (see full list in the Supplementary Information); the study sponsor teams that supported this trial; and C. Chen and B. Ravishankar of Sarepta Therapeutics, Inc. and A. Wong of F. Hoffmann-La Roche, Ltd. for manuscript development support.
The first draft of the manuscript was written by the lead and senior authors. All the authors participated in revisions. Medical writing and editorial assistance were provided by E. Turner of Nucleus Global in accordance with Good Publication Practice 2022 guidelines (https://www.ismpp.org/gpp-2022), funded by Sarepta Therapeutics, Inc. and F. Hoffmann-La Roche, Ltd.
Author information
Authors and Affiliations
Contributions
J.R.M., F.M., E.M.M., H.K., U.S.-S.: conception or design of the work; acquisition of data; interpretation of data; drafted the work or critically reviewed it; approved the submitted version; and agreed to be accountable for the accuracy and integrity of all aspects of the work. C.M.M.: acquisition of data; analysis of data; interpretation of data; drafted the work or critically reviewed it; approved the submitted version; and agreed to be accountable for the accuracy and integrity of all aspects of the work. E.C., C.L.-A., A.N., C.P., A.V. and C.M.Z.: acquisition of data; interpretation of data; drafted the work or critically reviewed it; approved the submitted version; and agreed to be accountable for the accuracy and integrity of all aspects of the work. M.G., A.P.M., C.R., C.W., D.R.A., E.D., S.M., R.A.P., W.Z. and P.F.: analysis of data; interpretation of data; drafted the work or critically reviewed it; approved the submitted version; and agreed to be accountable for the accuracy and integrity of all aspects of the work. T.S., J.S.E. and L.R.R.-K.: conception or design of the work; analysis of data; interpretation of data; drafted the work or critically reviewed it; approved the submitted version; and agreed to be accountable for the accuracy and integrity of all aspects of the work.
Corresponding author
Ethics declarations
Competing interests
J.R.M. received study funding from Sarepta Therapeutics while at Nationwide Children’s Hospital at the time of the study and is currently an employee of Sarepta Therapeutics. J.R.M. is a co-inventor of AAVrh74.MHCK7.micro-dys technology. F.M. has received honoraria and grants from Sarepta Therapeutics for participating at symposia and advisory boards and is involved as an investigator in Sarepta Therapeutics clinical trials. He reports participation in advisory boards for Novartis, F. Hoffmann-La Roche, Ltd., Edgewise Therapeutics, Dyne Therapeutics, Pfizer, PTC Therapeutics and Italfarmaco. C.M.M. reports grants from Capricor Therapeutics, Catabasis, Edgewise Therapeutics, Epirium Bio, Italfarmaco, Pfizer, PTC Therapeutics, Santhera Pharmaceuticals and Sarepta Therapeutics and has a consultancy/advisory role with Biomarin, Capricor Therapeutics, Catalyst, Edgewise Therapeutics, Italfarmaco, PTC Therapeutics, F. Hoffmann-La Roche, Ltd., Santhera Pharmaceuticals and Sarepta Therapeutics. He has received honoraria from PTC Therapeutics and Sarepta Therapeutics. E.M.M. has received fees from AveXis, Biogen and F. Hoffmann-La Roche, Ltd. E.C. has received honoraria from Sarepta Therapeutics for participating in advisory boards and research and/or grant support from the Centers for Disease Control and Prevention, CureSMA, the Muscular Dystrophy Association, the National Institutes of Health, Orphazyme, the Patient-Centered Outcomes Research Institute, Parent Project Muscular Dystrophy, PTC Therapeutics, Santhera, Sarepta Therapeutics and the US Food and Drug Administration. H.K. has received grants from Sarepta Therapeutics, Pfizer, PTC Therapeutics, Taiho Pharmaceutical Co., Ltd., Chugai Pharmaceutical Co., Nippon Shinyaku Co., Ltd. and Kaneka Corporation. H.K. has received fees from Sarepta Therapeutics, Pfizer, PTC Therapeutics, Chugai Pharmaceutical Co., Nippon Shinyaku Co. and Kaneka Corporation. C.L.-A. is an investigator in Sarepta Therapeutics clinical trials and a sub-investigator in studies sponsored by Pfizer, SolidBioSciences, Edgewise Therapeutics, Italfarmaco and Genentech/Roche. A.N. has received fees from AveXis, Biogen and F. Hoffmann-La Roche, Ltd. C.P. participates on an advisory board and is a consultant for Biogen, Sarepta Therapeutics, AveXis/Novartis Gene Therapies, Genentech/Roche and Scholar Rock; serves as a speaker for Biogen; and is a principal investigator of studies sponsored by AveXis/Novartis Gene Therapies, AMO Pharma, Astellas, Biogen, CSL Behring, Fibrogen, PTC Therapeutics, Pfizer, Sarepta Therapeutics and Scholar Rock. U.S.-S. has received honoraria for counseling and participating in invited talks from Sarepta Therapeutics and F. Hoffmann-La Roche, Ltd. A.V. has a consultancy/advisory role with AMO Pharma, AveXis, Biogen, Edgewise Therapeutics, FibroGen, Novartis, Pfizer, PTC Therapeutics, Sarepta Therapeutics, UCB Pharma, Catalyst and Scholar Rock; has received research funding from AMO Pharma, Capricor Therapeutics, Edgewise Therapeutics, FibroGen, the Muscular Dystrophy Association, Novartis, Parent Project Muscular Dystrophy, Pfizer, RegenxBio and Sarepta Therapeutics; and has other relationship(s) with MedLink Neurology for editorial services. C.M.Z. has received research support from Biogen and Novartis and has served on an advisory board for Sarepta Therapeutics. M.G., C.W. and P.F. are employees of F. Hoffmann-La Roche, Ltd. and may have stock options. A.P.M. and C.R. are employees of Roche Products, Ltd. and may have stock options in F. Hoffmann-La Roche, Ltd. D.R.A., E.D., S.M., R.A.P., T.S., W.Z. and J.S.E. are employees of Sarepta Therapeutics and may have stock options. L.R.R.-K. is an employee of Sarepta Therapeutics and may have stock options. In addition, she is a co-inventor of AAVrh74.MHCK7.micro-dys technology.
Peer review
Peer review information
Nature Medicine thanks James Dowling, Nicholas Freemantle and Ian Woodcock for their contribution to the peer review of this work. Primary Handling Editor: Anna Maria Ranzoni, in collaboration with the Nature Medicine team.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data
Supplementary information
Supplementary Information
Study group; investigator and recruitment sites; institutional review boards and ethics committees list; delandistrogene moxeparvovec ITR to ITR sequence; Supplementary Tables 1–4; and Supplementary Figs. 1–3.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Mendell, J.R., Muntoni, F., McDonald, C.M. et al. AAV gene therapy for Duchenne muscular dystrophy: the EMBARK phase 3 randomized trial. Nat Med 31, 332–341 (2025). https://doi.org/10.1038/s41591-024-03304-z
Received:
Accepted:
Published:
Version of record:
Issue date:
DOI: https://doi.org/10.1038/s41591-024-03304-z
This article is cited by
-
Evaluation of protein expression and oxidative stress index in Duchenne muscular dystrophy
Pediatric Research (2026)
-
Study Designs and Crafting Endpoints for Gene Therapy Development Programs in Rare Disease: A Narrative Review
Advances in Therapy (2026)
-
Emerging therapeutic strategies in muscular dystrophy: an updated review on pathogenesis and treatment advances
Molecular Biology Reports (2026)
-
Caregiver-reported Patient Experiences with Duchenne Muscular Dystrophy: Qualitative In-trial Interviews 1 Year After Delandistrogene Moxeparvovec in the Pivotal EMBARK Trial
Neurology and Therapy (2026)
-
Two-Year Outcomes Following Delandistrogene Moxeparvovec Treatment in Ambulatory Patients with Duchenne Muscular Dystrophy: Phase 3 EMBARK Trial
Neurology and Therapy (2026)
