
Abstract
Triple-negative breast cancer (TNBC) is a highly aggressive subtype of breast cancer with poor prognosis. The current first-line treatment for advanced TNBC (aTNBC) is determined by the expression of programmed cell death-ligand 1 (PD-L1). In the ATRACTIB trial—a multicenter, single-arm, phase 2 study—we evaluated the combination of atezolizumab, paclitaxel and bevacizumab as first-line treatment for patients with aTNBC, independently of PD-L1 status. The primary endpoint was investigator-assessed progression-free survival. One hundred female patients were enrolled, with most evaluable tumors being PD-L1-negative (97.6%). The primary endpoint was met, with a median progression-free survival of 11.0 months (95% confidence interval (CI): 9.0–13.4; P < 0.001). The objective response rate was 63.0% (95% CI: 52.8–72.4) and median overall survival was 27.4 months (95% CI: 23.4–37.4). No treatment-related deaths or new safety signals were observed. This combination demonstrated significant antitumor activity as first-line therapy for aTNBC patients and merits further investigation. ClinicalTrials.gov Identifier: NCT04408118.
Similar content being viewed by others

Main
Triple-negative breast cancer (TNBC) is characterized by the lack of expression of estrogen and progesterone receptors and no overexpression or amplification of human epidermal growth factor receptor 2 (HER2)1,2. TNBC represents approximately 10 to 15% of all breast cancer (BC) diagnoses1,3 and is typically very aggressive, with a shorter time to recurrence and poorer prognosis than the other subtypes in both the early and advanced settings4,5.
Programmed cell death-ligand 1 (PD-L1) and its receptor, programmed cell death 1 (PD-1), are targets for immune checkpoint inhibitors (ICIs). Moreover, PD-L1 has been validated as both a prognostic and predictive biomarker for response to immunotherapy across different cancers6. TNBC is considered the most immunogenic among all BC subtypes, characterized by a higher tumor mutational burden and increased infiltration of T cells. Approximately 40% of patients enrolled in immunotherapy trials for advanced TNBC (aTNBC) have PD-L1-positive tumors7,8,9. However, PD-L1 positivity is influenced by the assay used, the algorithm and defined cutoff point used to determine expression, and the metastatic site where PD-L1 status is evaluated10,11,12,13,14.
Current first-line treatment for aTNBC is determined by the expression of PD-L1 (refs. 15,16). The IMpassion130 (ref. 8) and KEYNOTE-355 (refs. 7,17) trials demonstrated that patients with PD-L1-positive aTNBC have a substantial improvement in progression-free survival (PFS) with the addition of ICIs, respectively atezolizumab or pembrolizumab, to standard chemotherapy. In KEYNOTE-355, a statistically significant improvement in overall survival (OS) was also observed7. Conversely, these two pivotal trials did not show a benefit in PFS or OS in patients with PD-L1-negative aTNBC. Consequently, while several clinical trials testing new approaches are ongoing, chemotherapy remains the standard first-line treatment for this population.
Despite treatment advancements, the median OS for aTNBC remains poor, ranging from 16 months in patients with PD-L1-negative tumors to 24 months in those with PD-L1-positive tumors7,8. This underscores the urgent need of developing additional therapeutic strategies for patients with aTNBC. Although PD-L1 expression is used to predict responses to ICI-based regimens in aTNBC, it is not a definitive biomarker, and some patients with PD-L1 negative tumors have exhibited responses to these therapies18,19,20. Therefore, identifying novel predictive biomarkers beyond PD-L1 expression is essential to enhance the efficacy of PD-1/PD-L1-targeted immunotherapies and to broaden their use beyond PD-L1-positive tumors. Additionally, there is also a critical need to explore the potential synergism of these immunotherapies in combination with other targeted or cytotoxic agents, not only to extend the benefits of immunotherapy to patients who have not demonstrated favorable outcomes in previously reported trials but also to enhance therapeutic efficacy in those who do respond, with the goal of delaying disease progression and improving overall outcomes.
Bevacizumab is an antibody directed against vascular endothelial growth factor (VEGF) that reduces the angiogenic and oncogenic activity of VEGF21. When combined with chemotherapy, bevacizumab has demonstrated a synergistic effect in HER2-negative advanced BC, notably improving both PFS and response rates22,23,24,25,26. Beyond its impact on angiogenesis, VEGF can also exert immunosuppressive effects within the tumor microenvironment by impairing antigen presentation by tumor cells. Antiangiogenic agents can counteract this, potentially increasing the efficacy of concurrent treatments27,28. Preclinical studies in TNBC models have shown that combining anti-VEGF therapies with ICIs can improve antitumor activity of these therapies by normalizing tumor vasculature and promoting immune cells infiltration29,30. Indeed, the immunomodulatory properties of bevacizumab make it a promising candidate to use in combination with other immunotherapeutic approaches31,32,33,34,35. A study assessing sequential treatment with bevacizumab and durvalumab showed efficacy in HER2-negative advanced BC, with bevacizumab enhancing the overall response to durvalumab through immune-priming effects36. Moreover, combining bevacizumab with immunotherapy and chemotherapy has also demonstrated clinical benefit in multiple tumors, including HER2-negative advanced BC37.
In this study, we hypothesized that combining atezolizumab plus paclitaxel and bevacizumab could improve outcomes in patients with previously untreated aTNBC, regardless of PD-L1 status. Here, we present the efficacy and safety results of this combination from the ATRACTIB phase 2 study.
Results
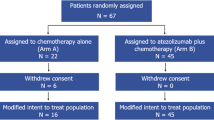
From 2 October 2020 to 4 May 2022, a total of 100 patients were recruited at 22 sites in four countries. All enrolled patients were female, with a median age of 55 years (range, 32 to 84), and 75% of them had an Eastern Cooperative Oncology Group (ECOG) performance status of 0 (Table 1). Overall, 29% patients had de novo metastatic BC and 56% presented visceral involvement, including lung, liver, brain, adrenal gland, kidney, spleen and other thoracic and abdominal regions. Additionally, 46% of patients had three or more metastatic sites. Among the 70 patients who had undergone prior (neo)adjuvant therapy, 61 were treated with a taxane-based regimen and none received prior PD-1 or PD-L1 inhibitors or bevacizumab. A tumor sample was available for PD-L1 evaluation in 85 patients (31.8% from a metastatic site), of whom 83 had PD-L1-negative tumors (97.6%, 83 out of 85) (Table 1). For the remaining 15 patients, PD-L1 expression could not be evaluated or samples were not available.
At the time of data cutoff (17 April 2024), all patients had discontinued study treatment, with a median follow-up duration of 19.4 months (range, 0.9 to 37.4). The main reason for treatment discontinuation was disease progression, which occurred in 66% of patients. Nineteen (19%) patients had completed study treatment and transitioned to post-trial access. Other reasons for treatment discontinuation were unacceptable toxicity (7%), the need for surgical intervention related to the disease under study (4%) and withdrawal of consent (4%) (Fig. 1). One patient who discontinued treatment due to unacceptable toxicity simultaneously reported progressive disease.

a, Toxicity prompted the decision to discontinue treatment in four patients, but treatment discontinuations were documented in the context of disease progression. b, Patients discontinued treatment in the framework of the study, but were offered to continue receiving treatment outside the trial, as per treating physician decision and in accordance with the marketing authorization holders’ policy. c, Three discontinuations due to AEs (vomiting and cough, digestive toxicity, pulmonary thromboembolism and autoimmune-mediated hepatitis) and four discontinuations due to serious AEs (cerebral accident, sepsis, fatigue and thrombocytopenia, and ejection fraction decreased). n, number of patients.
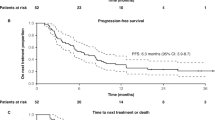
The primary endpoint of this study was met, with a median PFS of 11.0 months (95% CI: 9.0–13.4; P < 0.001) (Fig. 2). Objective response rate (ORR) was 63% (95% CI: 52.8–72.4), 14% of patients had complete responses (3% of them unconfirmed) and 49% had partial responses (6% of them unconfirmed) (Table 2). The median best percentage change from baseline was −59.8% (range, −100.0 to 94.9) (Fig. 3a). Clinical benefit rate (CBR) was 80% (95% confidence interval (CI): 70.8–87.3). Median duration of response (DoR) was 10.1 months (95% CI: 7.3–13.9) and median time to treatment response (TTR) was 1.9 months (95% CI: 1.9–3.7). Median OS was 27.4 months (95% CI: 23.4–37.4); the 12-month OS rate was 81.3% (95% CI: 71.9–87.8), and the 24-month OS was 59.8% (95% CI: 46.3–71.0) (Fig. 3b). A post hoc analysis of the treatment effect on PFS, OS and ORR across key subgroups is presented in Extended Data Figs. 1–3. These analyses suggested a worse PFS in patients with bone (hazard ratio (HR) 1.90; 95% CI 1.18–3.07; P = 0.008) and liver metastasis (HR 1.69; 95% CI 1.02–2.79; P = 0.041), as well as in those with HER2-0 (immunohistochemistry (IHC) 0) status (HR 1.67; 95% CI 1.00–2.78; P = 0.048). In patients with confirmed PD-L1-negative tumors (83 out of 100), median PFS was 9.3 months (95% CI: 7.8–13.2) and median OS was 24.5 months (95% CI: 22.9–not reached) (Extended Data Figs. 1 and 2 and Extended Data Table 1). Additional detailed data on clinical outcomes in patients with PD-L1-negative tumors are provided in Extended Data Table 1.

Kaplan–Meier curve of investigator-assessed PFS in the full analysis set. KM, Kaplan–Meier.

a, Waterfall plot of BOR per RECIST v.1.1. Dotted lines mark the thresholds for partial response (≥30% decrease) and disease progression (≥20% increase). b, OS. Reasons for censoring: reaching the end of the study as per protocol (48%), loss to follow up (7%), patient decision (7%) and investigator decision (1%). aPatients with only non-target lesions.bThree patients discontinued before post baseline assessment, one due to progressive disease and two due to withdrawal of consent.
All patients received at least one dose of each study drug. The median relative dose intensities (RDIs) were 93.8% for atezolizumab, 60.7% for paclitaxel and 91.7% for bevacizumab. The median treatment durations were 9.3 months for atezolizumab, 5.6 months for paclitaxel and 9.5 months for bevacizumab.
All patients experienced treatment emergent adverse events (TEAEs), with 61% presenting grade (G) 3 or 4 TEAEs (Extended Data Table 2). Discontinuations of any of the study treatments due to treatment-related TEAEs occurred in 48% of patients, with paclitaxel discontinuations being the most frequently reported (41%) (Extended Data Table 2). There were no treatment-related deaths in this study. The most common non-hematological TEAEs of any grade included fatigue (63%; 7% G3), neurotoxicity (46%; 10% G3 or G4), diarrhea (44%; 4% G3 or G4) and alopecia (41%). Neutropenia (27%; 12% G3 or G4) and anemia (24%) were the most frequent hematologic TEAEs (Table 3). Individual evaluation of safety by study drug is detailed in Supplementary Tables 1–3. Of note, 26% of patients experienced hypertension (6% G3) related to the treatment with bevacizumab, with one patient developing a G4 hypertensive crisis (Supplementary Table 3). Serious TEAEs occurred in 34% of patients (28% G3 or G4) and events of clinical interest (ECIs) were reported in 51% of patients (15% G3 or G4) (Supplementary Tables 4 and 5).
Immune-related adverse events (irAEs) of any grade occurred in 32% of patients, with 8% of them experiencing G3 events (Supplementary Table 6). No G4 irAEs were reported. The most frequently reported irAEs of any grade were thyroid disorders (21%; 1% G3), then immune-mediated hepatitis (4%; 3% G3), gastrointestinal disorders (3%; 3% G3), nephritis (3%; 3% G3) and Addison’s disease (3%; 0% G3).
Discussion
Here, we describe the primary results of the ATRACTIB phase 2 study, which demonstrated encouraging antitumor activity and a manageable safety profile of the atezolizumab, paclitaxel and bevacizumab triplet combination as first-line therapy for patients with aTNBC, enrolled irrespectively of PD-L1 status. The primary endpoint was met, with a median PFS of 11.0 months. Moreover, ORR was 63%, DoR was 10.1 months and the median OS was 27.4 months.
The randomized phase 3 IMpassion130, IMpassion131 and KEYNOTE-355 studies evaluated the role of chemo-immunotherapy as frontline treatment for aTNBC, both in the intention-to-treat and in the PD-L1-positive populations (~40%). The median PFS of 11.0 months observed in ATRACTIB was superior to the median PFS of 7.2, 5.7 and 7.5 months reported among the intention-to-treat populations of the IMpassion130, IMpassion131 and KEYNOTE-355, respectively7,8,9. ATRACTIB was an open-label study with no predefined selection by PD-L1 status, but highly enriched in patients with PD-L1-negative disease, as tumors of 83 of the 85 patients with evaluable basal samples classified as PD-L1-negative (97.6%) by central review. The median PFS and OS in this group were 9.3 and 24.5 months, respectively. Interestingly, in the IMpassion130 study, patients with PD-L1-negative aTNBC treated with atezolizumab plus nab-paclitaxel—a population that closely resembles that of ATRACTIB—had a median PFS of 5.6 months8 and a median OS of 19.7 months38, results considerably lower than those in ATRACTIB. Irrespective of PD-L1 status, our results also surpass those obtained with bevacizumab in combination with paclitaxel in aTNBC, with a median PFS of 8.1 months and a median OS of 18.9 months26. While these comparisons are noteworthy, differences in trial design and patient characteristics should be considered when interpreting efficacy results, particularly given the known variability of immunotherapy outcomes between phase 2 and phase 3 trials39.
In our study, OS was not a primary endpoint; therefore, these data are purely descriptive. The median OS of 27.4 months may have been influenced by the high censoring rate, as 48% of patients were censored due to study completion, limiting its ability to accurately reflect the true survival benefit. However, early survival estimates, before most censures, indicates a 12-month OS rate of 81.3%, which compares favorably with the observed 12-month OS rates of ~72% in the IMpassion130 study38 and ~65.0% in the KEYNOTE-355 study7. Moreover, the OS benefit of bevacizumab treatment in aTNBC has been challenging to demonstrate22,23,24,26, but our findings underscore its potential in combination strategies.
The toxicity profile observed in ATRACTIB was manageable, with G3 or G4 TEAEs occurring in 61% of patients and 48% discontinuing at least one of the treatments due to treatment-related TEAEs, primarily attributed to paclitaxel toxicity (41%). In comparison, IMpassion130 reported a lower incidence of G3 and G4 TEAEs (48.7%) and a discontinuation rate due to TEAEs of 15.9% (ref. 8). However, in the E2100 trial evaluating paclitaxel plus bevacizumab, there was an increased incidence of G3 and G4 adverse events (AEs), including hypertension (14.8%), fatigue (9.1%) and proteinuria (3.6%). Notably, the treatment discontinuation rate due to TEAEs was 51.3%, largely attributed to cumulative toxic effects23. In a phase 2 study evaluating the triple combination of nivolumab, bevacizumab and paclitaxel, the safety profile was comparable to that observed in ATRACTIB, with 58% incidence of G3 and G4 AEs37. Overall, no new safety signals9,23 or treatment-related deaths were reported.
As previously mentioned, ATRACTIB predominantly included patients with PD-L1-negative tumors, for which there is an unmet clinical need. This selection may have resulted from concurrent trials selectively enrolling PD-L1-positive patients, as well as from investigators opting against selecting patients with PD-L1-positive disease because of the lack of clinical benefit observed for atezolizumab plus paclitaxel in IMpassion131 (ref. 9). Moreover, atezolizumab and pembrolizumab were approved as first-line treatments for aTNBC during ATRACTIB recruitment, becoming the standard of care for patients with PD-L1-positive tumors and reducing the need for their clinical trial participation. Finally, differences in PD-L1 assessment methods should also be acknowledged, as the VENTANA SP142 assay—used in ATRACTIB—may classify some tumors as PD-L1-negative that could potentially meet combined positive score positivity thresholds. However, the use of SP142 maintains consistency with prior pivotal trials of atezolizumab in this population8,9, providing support in the methodology and potential comparability of the results.
Other new combinations in aTNBC further reinforce chemo-immunotherapy plus antiangiogenic strategies. The FUTURE-C-PLUS study, evaluating famitinib (VEGF receptor inhibitor), camrelizumab (anti-PD-1) and nab-paclitaxel as first-line treatment for immunomodulatory aTNBC, reported a median PFS of 13.6 months and an ORR of 81.3%, with efficacy in both PD-L1-positive (35.4%) and PD-L1-negative (27.1%) patients40. In comparison, the ATRACTIB trial achieved a median PFS of 11.0 months and an ORR of 63.0% in a predominantly PD-L1-negative population, with 14% of complete responses. Similarly, combining nivolumab, bevacizumab and paclitaxel in a phase 2 study of TNBC has shown an ORR of 59% and a median PFS of 7.8 months, with no correlation between PD-L1 expression and efficacy37. Moreover, other studies evaluating bispecific antibodies, like ivonescimab (targeting VEGF and PD-1)41 and PM8002/BNT327 (targeting VEGF and PD-L1)42, as first-line treatments for aTNBC have also demonstrated promising activity in PD-L1-negative aTNBC. These findings suggest that antiangiogenic agents may be key to paving the way for the use of immunotherapy in PD-L1-negative aTNBC.
Further studies are still needed to identify biomarkers for optimal patient selection. In our study, subgroup analyses were conducted to explore potential predictors of response, and ongoing evaluations—including proteomic plasma profiling and tumor biopsy assessments—may help identify novel biomarkers. The FUTURE-C-PLUS study described PD-L1/CD31 double positivity as a potential biomarker for improved outcomes and revealed other genetic alterations associated with treatment response40,43, warranting further research into new biomarker-driven approaches beyond PD-L1 status.
The ATRACTIB study has two main strengths: a large sample size of 100 patients, which is substantial for a phase 2 trial, and the inclusion of a predominantly PD-L1-negative population, a challenging-to-treat subgroup. However, the predominance of PD-L1-negative tumors also represents a limitation, as it restricts the extrapolation of results to PD-L1-positive patients. Another limitation is the single-arm design, which precludes a formal assessment of the contribution of each agent in the regimen, as well as the potential synergy between bevacizumab and the atezolizumab–paclitaxel combination. Additionally, the high censoring rate for OS data limits its interpretability, rendering the OS results primarily descriptive.
Two important clinical questions arise from our findings. First, whether this regimen may be effective in patients previously treated with immunotherapy, a population not included in the ATRACTIB. Although data on this scenario is limited, the synergy between antiangiogenic agents and immunotherapy (± chemotherapy) has been demonstrated18,44,45,46, suggesting that combining bevacizumab with immunotherapy and chemotherapy may offer additional benefits for this population. Second, the feasibility of including patients with estrogen receptor (ER)-low tumors in future trials evaluating treatment strategies for TNBC, as retrospective analyses suggest biological and clinical similarities between tumors with ER expression levels between 1 to 9% and those with ER expression <1% (refs. 47,48,49,50), including comparable expression of immune biomarkers associated with immunotherapy response. Based on this evidence, it may be reasonable to extrapolate the potential use of our triplet to the ER-low population.
Beyond antiangiogenic combinations, emerging PD-1/PD-L1-targeted immunotherapy plus antibody-drug conjugates (ADCs) strategies are reshaping the first-line treatment landscape in aTNBC. The MORPHEUS-panBC trial (atezolizumab plus sacituzumab govitecan) reported an ORR of 76.7% and a PFS of 12.2 months in patients with PD-L1-positive disease51, while the BEGONIA trial (datopotamab deruxtecan plus durvalumab) showed an ORR of 79% and a PFS of 13.8 months in a predominantly PD-L1-low population (87.1%)52,53. These findings, together with those from ATRACTIB, suggest that immunotherapy benefits may extend beyond PD-L1-positive tumors, particularly in combination with antiangiogenics or ADCs.
In conclusion, the ATRACTIB study demonstrated that the combination of atezolizumab plus paclitaxel and bevacizumab exhibits encouraging antitumor activity as a first-line treatment for patients with aTNBC, irrespective of PD-L1 expression. Importantly, bevacizumab appears to improve the efficacy of immunotherapy in this patient population. In the context of the evolving treatment landscape for aTNBC and the potential introduction of Trop-2-directed ADCs as first-line therapy, our findings warrant further investigation by evaluating the role of bevacizumab in combination with emerging ADCs and immunotherapies, regardless of PD-L1 status.
Methods
Study design and participants
The ATRACTIB study was a single-arm, open-label, multicenter phase 2 trial conducted across 24 sites in Spain, Germany, Italy and France. The trial protocol is available in the Supplementary Information.
Eligible patients were aged 18 years or older with unresectable locally advanced or metastatic TNBC, confirmed by local assessment and defined as <1% expression for ER and progesterone receptor by IHC, and negative for HER2 (0 to 1+ by IHC or 2+ and negative by in situ hybridization test) according to American Society of Clinical Oncology/College of American Pathologists criteria54,55. Patients were included regardless of their PD-L1 status. No prior therapy for advanced disease was allowed and patients who received prior (neo)adjuvant therapies (taxane-based chemotherapy, immunotherapy and/or antiangiogenic agents) were required to have a disease-free interval of at least 12 months, or at least 6 months in the case of non-taxane-based chemotherapy. Patients with measurable or non-measurable disease as per RECIST v.1.1 were included (patients with bone-only lesions were eligible), and availability of recently or newly obtained tumor sample from either primary breast tumor or the metastatic site was required, unless contraindicated due to site inaccessibility and/or participant safety concerns. In those cases, patient eligibility was evaluated by a Sponsor’s qualified designee. Patients with known active uncontrolled or symptomatic central nervous system metastases, or a history of leptomeningeal disease were excluded, as well as patients with active or a history of autoimmune disease, or other clinical situations that could entail higher risk for patients. Patients were enrolled regardless of sex, which was collected according to the identity information provided by the patients. Complete inclusion and exclusion criteria are provided in the Supplementary Methods.
This study was conducted in compliance with the Declaration of Helsinki and was approved by the institutional review boards or independent ethics committees at each site: Comité de Ética de la Investigación con medicamentos del Hospital Universitari Arnau de Vilanova de la Gerencia Territorial de Lleida (Spain), Comité de Protection des Personnes TOURS – Région Center – Ouest 1 (France), Ethik-Kommission II der Universität Heidelberg (Medizinische Fakultät Mannheim) and Ethik-Kommission an der Universität Duisburg-Essen (Germany), and Comitatio Etico IRCCS Pascale and Comitato Etico Territoriale CAMPANIA 1 (Italy). All patients provided written informed consent. This trial is registered at ClinicalTrials.gov, NCT04408118.
Procedures
In treatment cycles of 28 days, patients received an intravenous infusion of atezolizumab (840 mg) on days 1 and 15; if the first infusion was tolerated over 60 min, subsequent infusions were delivered over 30 min. Paclitaxel (90 mg per m2) was administered on days 1, 8 and 15 as an intravenous infusion over 60 min. Bevacizumab (10 mg per kg) was administered as an intravenous infusion over 30 to 90 min on days 1 and 15. Dose reductions were allowed for paclitaxel as defined by prespecified protocol guidelines but not allowed to atezolizumab and bevacizumab per their respective labels. If one of the study drugs was permanently discontinued, the remaining treatments, either as monotherapy or in combination, could continue to be administered if there was no contraindication. Treatment was given until progressive disease, unacceptable toxicity, death, withdrawal or discontinuation from the study for any other reason, whichever occurred first. End of study was defined as 12 months following the enrollment of the last patient or upon the occurrence of at least 67 PFS events with 100 enrolled patients, whichever occurred later. For patients with ongoing treatment at the time of end of study, who were required to discontinue study treatment as part of the study protocol, transition to post-trial access was offered. This decision was made in consultation with the treating physician and in accordance with the marketing authorization holders’ policy.
Tumor assessments were conducted by computed tomography or magnetic resonance imaging according to RECIST v.1.1 at baseline and every 8 weeks up to 12 months. Thereafter, disease assessment was performed every 12 weeks until the end of treatment visit. Bone scans were conducted at baseline and repeated every 24 weeks for patients with bone lesions identified at baseline, unless clinical or biochemical bone progression was suspected.
PD-L1 expression was assessed with the VENTANA SP142 immunohistochemistry assay, and PD-L1 positivity was defined as ≥1% PD-L1 expression on tumor-infiltrating immune cells.
Outcomes
The primary endpoint of this study was investigator-assessed PFS as per RECIST v.1.1. Secondary endpoints were ORR and CBR as per RECIST v.1.1; best percentage change from baseline; DoR; TTR; OS; and safety, including AEs, TEAEs, serious AEs, ECI (the definition of ECI is detailed in the Supplementary Methods), irAEs and rates of discontinuations and dose reductions because of AEs as per Common Terminology Criteria for Adverse Events v.5.0. The definition of all endpoints is provided in the Supplementary Methods.
Exploratory endpoints were immune-related PFS; immune-related ORR; association of tumor and/or immune-related biomarkers (for example, PD-L1, tumor-infiltrating lymphocytes, tumor mutational burden or cytokine profile) with treatment efficacy; and changes in mutation and copy number in oncogenes, tumor suppressor genes and/or genes associated with disease progression, as evaluated in liquid biopsy. Exploratory endpoints are under evaluation and not reported here.
Statistical analysis
We conducted the efficacy and safety analyses in the full analysis set, comprising all enrolled patients who received at least one dose of study drugs. The median PFS was calculated using the Kaplan–Meier method. The maximum likelihood estimation for the exponential distribution test was used to compare differences between observed and historical rates56,57. The PFS analysis was designed to test the null hypothesis that the true median PFS was 7 months or less11. The alternative hypothesis was that the true median PFS was greater than or equal to 9.5 months (HR 0.74). We estimated that enrolling 100 patients would provide 80% power at a nominal level of one-sided α of 0.05. This primary objective would be met with a median PFS of 8.6 months and 67 PFS events observed. The R code used for sample size calculation and primary analysis is provided in the Supplementary Methods. The 95% CI for median PFS, OS, TTR and DoR were estimated using the Greenwood method. ORR and CBR were estimated with the 95% Clopper–Pearson CI based on an exact binomial test. The best percentage change from baseline was described for each patient with a waterfall plot. For forest plot analysis, HR and associated 95% CI for PFS and OS were estimated using Cox proportional hazards models with Breslow’s method of tie handling. The odd ratios and associated 95% CI for ORR were calculated using logistic regression models. RDI for each study drug was calculated from treatment initiation to discontinuation of all study drugs; RDI was defined as the actual dose of the study drug administered to each patient divided by the scheduled dose per study protocol58,59. We used descriptive statistics to summarize baseline characteristics and safety data. Patients without any post-baseline assessment for primary PFS or secondary endpoints OS were censored after one day of treatment. For best overall response, patients lacking post-baseline assessments were considered as non-responders or without clinical benefit. Statistical analyses were performed with SAS software (v.9.4) and with R software (v.4.2.2). Detailed statistical analyses are described in the statistical analysis plan in the Supplementary Information.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
Data collected within this study will be made available to researchers after contacting the corresponding author and upon revision and approval based on scientific merit by the ATRACTIB trial management group (which includes a qualified statistician) of a detailed proposal for their use. The data required for the approved, specified purposes will be provided after the completion of a data sharing agreement that will be set up by the study sponsor, beginning 1 month and ending 5 years after article publication. All data provided will be anonymized to respect the privacy of patients who have participated in the trial in line with applicable laws and regulations. The estimated time frame for response will be within 30 days. Please address requests for data to the corresponding author. The study protocol and statistical analysis plan (SAP) is provided as Supplementary Information.
Code availability
The code for the calculation of sample size and the analysis of the primary endpoint of this manuscript is reported in Supplementary Methods, ʽR code for sample size calculation and endpoint analysisʼ.
References
Zagami, P. & Carey, L. A. Triple negative breast cancer: Pitfalls and progress. NPJ Breast Cancer 8, 95 (2022).
Gennari, A. et al. ESMO Clinical Practice Guideline for the diagnosis, staging and treatment of patients with metastatic breast cancer. Ann. Oncol. 32, 1475–1495 (2021).
Surveillance Research Program, National Cancer Institute. SEER*Explorer: An Interactive Website for SEER Cancer Statistics (SEER Incidence Data, November 2024, accessed 16 May 2025); https://seer.cancer.gov/statistics-network/explorer
Courtney, D. et al. Breast cancer recurrence: factors impacting occurrence and survival. Ir. J. Med Sci. 191, 2501–2510 (2022).
Dent, R. et al. Triple-negative breast cancer: clinical features and patterns of recurrence. Clin. Cancer Res. 13, 4429–4434 (2007).
Munari, E. et al. PD-1/PD-L1 in cancer: pathophysiological, diagnostic and therapeutic aspects. IJMS 22, 5123 (2021).
Cortes, J. et al. Pembrolizumab plus chemotherapy in advanced triple-negative breast cancer. N. Engl. J. Med. 387, 217–226 (2022).
Schmid, P. et al. Atezolizumab and nab-paclitaxel in advanced triple-negative breast cancer. N. Engl. J. Med. 379, 2108–2121 (2018).
Miles, D. et al. Primary results from IMpassion131, a double-blind, placebo-controlled, randomised phase III trial of first-line paclitaxel with or without atezolizumab for unresectable locally advanced/metastatic triple-negative breast cancer. Ann. Oncol. 32, 994–1004 (2021).
Noske, A. et al. Comparison of assessment of programmed death-ligand 1 (PD-L1) status in triple-negative breast cancer biopsies and surgical specimens. J. Clin. Pathol. 77, 239–245 (2024).
Rugo, H. S. et al. Performance of PD-L1 immunohistochemistry (IHC) assays in unresectable locally advanced or metastatic triple-negative breast cancer (mTNBC): post-hoc analysis of IMpassion130. Ann. Oncol. 30, v858–v859 (2019).
Vlajnic, T. et al. PD-L1 expression in triple-negative breast cancer—a comparative study of 3 different antibodies. Appl. Immunohistochem. Mol. Morphol. 30, 726–730 (2022).
Sigurjonsdottir, G. et al. Comparison of SP142 and 22C3 PD-L1 assays in a population-based cohort of triple-negative breast cancer patients in the context of their clinically established scoring algorithms. Breast Cancer Res. 25, 123 (2023).
Prince, E. A., Sanzari, J. K., Pandya, D., Huron, D. & Edwards, R. Analytical concordance of PD-L1 assays utilizing antibodies from FDA-approved diagnostics in advanced cancers: a systematic literature review. JCO Precis. Oncol. 5, 953–973 (2021).
ESMO triple-negative breast cancer living guidelines. ESMO www.esmo.org/living-guidelines/esmo-metastatic-breast-cancer-living-guideline/triple-negative-breast-cancer (2023).
National Comprehensive Cancer Network. NCCN Guidelines: breast cancer version 2.2024. https://www.nccn.org/patients/guidelines/content/PDF/stage_iv_breast-patient.pdf (2024).
Cortes, J. et al. Pembrolizumab plus chemotherapy versus placebo plus chemotherapy for previously untreated locally recurrent inoperable or metastatic triple-negative breast cancer (KEYNOTE-355): a randomised, placebo-controlled, double-blind, phase 3 clinical trial. Lancet 396, 1817–1828 (2020).
Socinski, M. A. et al. Atezolizumab for first-line treatment of metastatic nonsquamous NSCLC. N. Engl. J. Med. 378, 2288–2301 (2018).
Mok, T. S. K. et al. Pembrolizumab versus chemotherapy for previously untreated, PD-L1-expressing, locally advanced or metastatic non-small-cell lung cancer (KEYNOTE-042): a randomised, open-label, controlled, phase 3 trial. Lancet 393, 1819–1830 (2019).
Hellmann, M. D. et al. Nivolumab plus Ipilimumab in advanced non–small-cell lung cancer. N. Engl. J. Med. 381, 2020–2031 (2019).
Garcia, J. et al. Bevacizumab (Avastin®) in cancer treatment: a review of 15 years of clinical experience and future outlook. Cancer Treat. Rev. 86, 102017 (2020).
Miles, D. W. et al. Phase III study of bevacizumab plus docetaxel compared with placebo plus docetaxel for the first-line treatment of human epidermal growth factor receptor 2–negative metastatic breast cancer. J. Clin. Oncol 28, 3239–3247 (2010).
Miller, K. et al. Paclitaxel plus bevacizumab versus paclitaxel alone for metastatic breast cancer. N. Engl. J. Med. 357, 2666–2676 (2007).
Robert, N. J. et al. RIBBON-1: randomized, double-blind, placebo-controlled, phase III trial of chemotherapy with or without bevacizumab for first-line treatment of human epidermal growth factor receptor 2-negative, locally recurrent or metastatic breast cancer. J. Clin. Oncol. 29, 1252–1260 (2011).
Brufsky, A. et al. Progression-free survival (PFS) in patient subgroups in RIBBON-2, a phase III trial of chemotherapy (chemo) plus or minus bevacizumab (BV) for second-line treatment of HER2-negative, locally recurrent or metastatic breast cancer (MBC). J. Clin. Oncol. 28, 1021 (2010).
Miles, D. W. et al. First-line bevacizumab in combination with chemotherapy for HER2-negative metastatic breast cancer: pooled and subgroup analyses of data from 2447 patients. Ann. Oncol. 24, 2773–2780 (2013).
Fukumura, D., Kloepper, J., Amoozgar, Z., Duda, D. G. & Jain, R. K. Enhancing cancer immunotherapy using antiangiogenics: opportunities and challenges. Nat. Rev. Clin. Oncol. 15, 325–340 (2018).
Hack, S. P., Zhu, A. X. & Wang, Y. Augmenting anticancer immunity through combined targeting of angiogenic and PD-1/PD-L1 pathways: challenges and opportunities. Front. Immunol. 11, 598877 (2020).
Reguera-Nuñez, E. et al. Therapeutic impact of Nintedanib with paclitaxel and/or a PD-L1 antibody in preclinical models of orthotopic primary or metastatic triple negative breast cancer. J. Exp. Clin. Cancer Res. 38, 16 (2019).
Li, Q. et al. Low-dose anti-angiogenic therapy sensitizes breast cancer to PD-1 blockade. Clin. Cancer Res. 26, 1712–1724 (2020).
Oyama, T. et al. Vascular endothelial growth factor affects dendritic cell maturation through the inhibition of nuclear factor-kappa B activation in hemopoietic progenitor cells. J. Immunol. 160, 1224–1232 (1998).
Goel, S. et al. Normalization of the vasculature for treatment of cancer and other diseases. Physiol. Rev. 91, 1071–1121 (2011).
Wallin, J. J. et al. Atezolizumab in combination with bevacizumab enhances antigen-specific T-cell migration in metastatic renal cell carcinoma. Nat. Commun. 7, 12624 (2016).
Roland, C. L. et al. Cytokine levels correlate with immune cell infiltration after anti-VEGF therapy in preclinical mouse models of breast cancer. PLoS ONE 4, e7669 (2009).
Hegde, P. S., Wallin, J. J. & Mancao, C. Predictive markers of anti-VEGF and emerging role of angiogenesis inhibitors as immunotherapeutics. Semin. Cancer Biol. 52, 117–124 (2018).
Quintela-Fandino, M. et al. Immuno-priming durvalumab with bevacizumab in HER2-negative advanced breast cancer: a pilot clinical trial. Breast Cancer Res. 22, 124 (2020).
Ozaki, Y. et al. Safety and efficacy of nivolumab plus bevacizumab, paclitaxel for HER2-negative metastatic breast cancer: Primary results and biomarker data from a phase 2 trial (WJOG9917B). Eur. J. Cancer 171, 193–202 (2022).
Emens, L. A. et al. First-line atezolizumab plus nab-paclitaxel for unresectable, locally advanced, or metastatic triple-negative breast cancer: IMpassion130 final overall survival analysis. Ann. Oncol. 32, 983–993 (2021).
Li, X. et al. Comparison of efficacy discrepancy between early-phase clinical trials and phase III trials of PD-1/PD-L1 inhibitors. J. Immunother. Cancer 12, e007959 (2024).
Chen, L. et al. Famitinib with camrelizumab and nab-paclitaxel for advanced immunomodulatory triple-negative breast cancer (FUTURE-C-Plus): An open-label, single-arm, phase II trial. Clin. Cancer Res. 28, 2807–2817 (2022).
Ouyang, Q. et al. 347MO The safety and efficacy of ivonescimab in combination with chemotherapy as first-line (1L) treatment for triple-negative breast cancer (TNBC). Ann. Oncol. 35, S360–S361 (2024).
Wu, J. et al. 348MO A phase Ib/II study to assess the safety and efficacy of PM8002/BNT327 in combination with nab-paclitaxel for first-line treatment of locally advanced or metastatic triple-negative breast cancer. Ann. Oncol. 35, S361 (2024).
Wu, S.-Y. et al. Combined angiogenesis and PD-1 inhibition for immunomodulatory TNBC: concept exploration and biomarker analysis in the FUTURE-C-Plus trial. Mol. Cancer 21, 84 (2022).
Finn, R. S. et al. Atezolizumab plus bevacizumab in unresectable hepatocellular carcinoma. N. Engl. J. Med. 382, 1894–1905 (2020).
McDermott, D. F. et al. Clinical activity and molecular correlates of response to atezolizumab alone or in combination with bevacizumab versus sunitinib in renal cell carcinoma. Nat. Med. 24, 749–757 (2018).
Oaknin, A. et al. Atezolizumab plus bevacizumab and chemotherapy for metastatic, persistent, or recurrent cervical cancer (BEATcc): a randomised, open-label, phase 3 trial. Lancet https://doi.org/10.1016/S0140-6736(23)02405-4 (2023).
Chen, H., Huang, F., Chen, Q. & Deng, Y. Impact of estrogen receptor expression level on response to neoadjuvant chemotherapy and prognosis in HER2-negative breast cancers. BMC Cancer 23, 841 (2023).
Loi, S. et al. Neoadjuvant nivolumab and chemotherapy in early estrogen receptor-positive breast cancer: a randomized phase 3 trial. Nat. Med. 31, 433–441 (2025).
Cardoso, F. et al. Pembrolizumab and chemotherapy in high-risk, early-stage, ER+/HER2− breast cancer: a randomized phase 3 trial. Nat. Med. 31, 442–448 (2025).
Yoder, R. et al. Impact of low versus negative estrogen/progesterone receptor status on clinico-pathologic characteristics and survival outcomes in HER2-negative breast cancer. NPJ Breast Cancer 8, 80 (2022).
Schmid, P. 181O – Interim analysis (IA) of the atezolizumab (atezo) + sacituzumab govitecan (SG) arm in patients (pts) with triple-negative breast cancer (TNBC) in MORPHEUS-pan BC: a phase Ib/II study of multiple treatment (tx) combinations in pts with locally advanced/metastatic BC (LA/mBC). Ann. Oncol. 9, 1–47 (2024).
Schmid, P. et al. 379MO Datopotamab deruxtecan (Dato-DXd) + durvalumab (D) as first-line (1L) treatment for unresectable locally advanced/metastatic triple-negative breast cancer (a/mTNBC): Updated results from BEGONIA, a phase Ib/II study. Ann. Oncol. 34, S337 (2023).
Schmid, P. et al. 166MO Datopotamab deruxtecan (Dato-DXd) + durvalumab (D) as first-line (1L) treatment for unresectable locally advanced/metastatic triple-negative breast cancer (a/mTNBC): initial results from BEGONIA, a phase Ib/II study. Ann. Oncol. 33, S199 (2022).
Allison, K. H. et al. Estrogen and progesterone receptor testing in breast cancer: ASCO/CAP Guideline update. J. Clin. Oncol. 38, 1346–1366 (2020).
Wolff, A. C. et al. Human epidermal growth factor receptor 2 testing in breast cancer: ASCO-College of American Pathologists Guideline update. J. Clin. Oncol. https://doi.org/10.1200/JCO.22.02864 (2023).
Sampayo-Cordero, M. et al. A single-arm study design with non-inferiority and superiority time-to-event endpoints: a tool for proof-of-concept and de-intensification strategies in breast cancer. Front. Oncol. 13, 1048242 (2023).
Jung, S.-H. Randomized Phase II Cancer Clinical Trials (Chapman and Hall/CRC, 2013).
Hryniuk, W. M. & Goodyear, M. The calculation of received dose intensity. J. Clin. Oncol. 8, 1935–1937 (1990).
Wildiers, H. & Reiser, M. Relative dose intensity of chemotherapy and its impact on outcomes in patients with early breast cancer or aggressive lymphoma. Crit. Rev. Oncol. Hematol. 77, 221–240 (2011).
Acknowledgements
We thank the patients and their caregivers for participating in this study, as well as the trial teams at the participating sites and the trial unit at Medica Scientia Innovation Research (MEDSIR). We thank A. Rynne Vidal and V. Di Giacomo from TPM Science, for providing writing support, funded by MEDSIR. This study was sponsored by MEDSIR. This study was funded by Hoffmann-La Roche, who did not participate in data collection, data analysis, data interpretation or the writing of this report.
Author information
Authors and Affiliations
Consortia
Contributions
M.G., J.M.P.-G., J.C. and A.L.-C. were responsible for the conception and methodology. Formal analysis, data curation and visualization were provided by J.A.G. and M.S.-C. All authors were responsible for data interpretation, data validation and resources. Administrative support was provided by O.B., J.R.-M. and S.G.-V. The original draft was written by O.B., G.A., J.M.P.-G. and M.G. while the final writing, review, editing and approval of the manuscript was carried out by all authors. Supervision was given by M.G., J.M.P.-G., J.C. and A.L.-C. Project administration was the responsibility of MEDSIR as the study sponsor.
Corresponding authors
Ethics declarations
Competing interests
M.G. reports having received honoraria from Novartis, Gilead, AstraZeneca and Pfizer; having personal support for attending meetings and/or travel from Roche, Pfizer, AstraZeneca and Gilead; and having received honoraria for advisory board participation from Gilead, Novartis, AstraZeneca and Pfizer. I.B. reports having received honoraria as Medical Monitor from MEDSIR; having received honoraria for advisory board participation from AstraZeneca, Bristol-Myers Squibb, Celgene, Daiichi Sankyo, Eisai, Gilead, Grünenthal, GSK, Jazz Pharmaceutical, Lilly, MSD, Novartis, Pfizer, Pierre-Fabre, Roche, Seagen and Veracyte; having personal support for attending meetings and/or travel from AstraZeneca, Bristol-Myers Squibb, Daiichi Sankyo, Gilead, Lilly, Novartis, Pfizer, Pierre-Fabre and Roche; and having received institutional financial support from Agendia, AstraZeneca, Lilly, Pfizer and Roche. A.C.-S. reports having received personal honoraria for advisory board participation from GlaxoSmithKline and AstraZeneca; having personal honoraria for speakers’ bureaus from GlaxoSmithKline, AstraZeneca, MSD, Eisai, Accord Healthcare, Pfizer, and Pharma&; having personal support for attending meetings and/or travel from Pfizer, GlaxoSmithKline and MSD. They are also founder of ONCARE Madrid. F.M. reports having consulting fees from AstraZeneca, Clovis, Daiichi Sankyo, EISAI, Gilead, GlaxoSmithKline, Novartis, Myriad Genetic, Seagen, Stemline Menarini, Lilly, MSD, Pfizer, Roche, Bionteck and Nerviano; having personal honoraria for lectures, presentations, speakers bureaus, manuscript writing or educational events for AstraZeneca, Clovis, Daiichi Sankyo, EISAI, Gilead, GlaxoSmithKline, Myriad Genetic, Seagen, Stemline Menarini, Lilly, MSD, Pfizer and Roche; having personal support for attending meetings and/or travel from AstraZeneca, Daiichi Sankyo, Gilead, GlaxoSmithKline, Stemline Menarini, Lilly, Pfizer and Roche; and having received honoraria for advisory board participation from Immutep, Amgen and Palleos. S.M. has received personal support for attending meetings and/or travel from AstraZeneca, Daiichi Sankyo, Gilead, Lilly, Novartis, MSD and Pfizer; and has received institutional financial support from Lilly. I.C.-P. reports having received personal honoraria as consultant, advisor and speaker from Gilead, MSD, Roche and AstraZeneca; having received personal honoraria for educational events from Roche and Daiichi Sankyo; and having received institutional financial support from Roche. S.R. reports having personal honoraria for speakers’ bureaus from Roche; and having personal support for attending meetings and/or travel from Accord Healthcare and Lilly. A.M.-B. reports having personal support for attending meetings and/or travel from GlaxoSmithKline, MSD and Roche; having received personal honoraria for speaker participation from AstraZeneca, GlaxoSmithKline and Seagen; and has received institutional financial support from GlaxoSmithKline. E.L. reports having received personal honoraria from Pfizer, Lilly, Novartis, Gilead, Daichii and AstraZeneca; and having received personal honoraria for advisory board participation from Gilead and Daichii. M.T.T. reports having received honoraria for local meetings from AstraZeneca, Bristol-Myers Squibb, Celgene, Daiichi Sankyo, Eisai, Gilead, Grünenthal, GlaxoSmithKline, Lilly, MSD, Novartis, Pfizer, Pierre-Fabre and Roche; and having personal support for attending meetings and/or travel from Merck, Pzifer and Servier. M. de L. reports having received honoraria for lectures, presentations, speakers bureaus, manuscript writing and educational events from Eli Lilly, Novartis, Seagen, Takeda, Roche, Daiichi Sankyo, Tomalab, Gilead, Genetic, Menarini, Sophos and Istituto Gentili; having received economical support for attending meetings and/or travel from Gilead, Novartis, Roche and AstraZeneca; and having participated on a Data Safety Monitoring Board or Advisory Board for Pfizer, AstraZeneca, Sanofi, Seagen, Novartis, Ipsen, Roche, Pierre-Fabre, Daiichi Sankyo, GSK, MSD and Menarini. S.G.-V., J.A.G., O.B., J.R.-M. and M.S.-C. are full-time employees at MEDSIR. G.A. reports having personal support for attending meetings and/or travel along with personal honoraria from MEDSIR. J.M.P.-G. reports having received personal honoraria for advisory board participation from Lilly, Roche, Eisai, Daichii Sankyo, AstraZeneca, Seattle Genetics, MSD and Gilead; having personal support for attending meetings and/or travel from Roche; and being an employee at MEDSIR. J.C. reports being a consultant/advisor for Roche, AstraZeneca, Seattle Genetics, Daiichi Sankyo, Lilly, Merck Sharp & Dohme, Leuko, Bioasis, Clovis Oncology, Boehringer Ingelheim, Ellipses, Hibercell, BioInvent, Gemoab, Gilead, Menarini, Zymeworks, Reveal Genomics, Scorpion Therapeutics, Expres2ion Biotechnologies, Jazz Pharmaceuticals, Abbvie, BridgeBio, Biontech, Biocon, Circle Pharma, Delcath Systems, Inc., Hexagon Bio; receiving honoraria from Roche, Novartis, Eisai, Pfizer, Lilly, Merck Sharp & Dohme, Astrazeneca, Gilead, Steamline Therapeutics and Daiichi Sankyo; has stock of MAJ3 Capital and Leuko (relative); received travel, accommodation and expenses from Roche, Novartis, Eisai, Pfizer, Daiichi Sankyo, Astrazeneca, Gilead Merck Sharp & Dhome, Steamline Therapeutics; and has the following patents: Pharmaceutical Combinations of A Pi3k Inhibitor and a Microtubule Destabilizing Agent. Javier Cortés Castán, Alejandro Piris Giménez, Violeta Serra Elizalde. WO 2014/199294A—Issued, Her2 as a predictor of response to dual HER2 blockade in the absence of cytotoxic therapy. Aleix Prat, Antonio Llombart, Javier Cortés.US 2019/ 0338368 A1—Licensed. J.C. also reports their institution received research funding from Roche, Ariad Pharmaceuticals, AstraZeneca, Baxalta GMBH/Servier Affaires, Bayer Healthcare, Eisai, F. Hoffman-La Roche, Guardanth Health, Merck Sharp & Dohme, Pfizer, Piqur Therapeutics, Puma C and Queen Mary University of London. A.L.-C. reports receiving research support from Roche, Agendia, Lilly, Pfizer, Novartis, Merck Sharp & Dohme, Gilead and Daichii-Sanyo; consulting or advisory role for Lilly, Roche, Pfizer and Novartis; speakers’ bureaus from Lilly, AstraZeneca, Pfizer, Novartis and Merck Sharp & Dohme; travel support from Roche, Pfizer, Steamline Therapeutics, Merck Sharp & Dhome and AstraZeneca; and stock or other ownership of MAJ3 Capital and Initia-Research. The other authors declare no competing interests.
Peer review
Peer review information
Nature Medicine thanks Paolo Tarantino, Masakazu Toi, Hui Zhang and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Primary Handling Editor: Ulrike Harjes, in collaboration with the Nature Medicine team.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data
Extended Data Fig. 1 Forest plot of subgroup analysis for progression-free survival according to baseline characteristics and selected prognostic factors.
HR and associated 95% CI were estimated using Cox proportional-hazards models with Breslow’s method of tie handling. CI, confidence interval; ECOG, Eastern Cooperative Oncology Group; HER2, human epidermal growth factor receptor 2; HR, hazard ratio; IHC, immunohistochemistry; OS, overall survival; PD-L1, programmed death-ligand 1; ref, reference subgroup.
Extended Data Fig. 2 Forest plot of subgroup analysis for overall survival according to baseline characteristics and selected prognostic factors.
HR and associated 95% CI were estimated using Cox proportional-hazards models with Breslow’s method of tie handling. CI, confidence interval; ECOG, Eastern Cooperative Oncology Group; HER2, human epidermal growth factor receptor 2; HR, hazard ratio; IHC, immunohistochemistry; OS, overall survival; PD-L1, programmed death-ligand 1; ref, reference subgroup.
Extended Data Fig. 3 Forest plot of subgroup analysis for objective response rate according to baseline characteristics and selected prognostic factors.
The odd ratios and associated 95% CI for ORR were calculated using logistic regression models. CI, confidence interval; ECOG, Eastern Cooperative Oncology Group; HER2, human epidermal growth factor receptor 2; IHC, immunohistochemistry; OR, Odds ratio, ORR, objective response rate; PD-L1, programmed death-ligand 1; ref, reference subgroup.
Supplementary information
Supplementary Information
Supplementary Methods, Study protocol and Statistical analysis plan.
Supplementary Table 1
Treatment emergent adverse events (TEAEs) related to atezolizumab. Supplementary Table 2 Treatment emergent adverse events (TEAEs) related to paclitaxel. Supplementary Table 3 Treatment emergent adverse events (TEAEs) related to bevacizumab. Supplementary Table 4 Serious treatment emergent adverse events (TEAEs) by maximum severity. Supplementary Table 5 Adverse events of clinical interest (ECIs) by maximum severity. Supplementary Table 6 Immune-related adverse events (irAEs).
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Gion, M., Blancas, I., Cortez-Castedo, P. et al. Atezolizumab plus paclitaxel and bevacizumab as first-line treatment of advanced triple-negative breast cancer: the ATRACTIB phase 2 trial. Nat Med 31, 2746–2754 (2025). https://doi.org/10.1038/s41591-025-03734-3
Received:
Accepted:
Published:
Version of record:
Issue date:
DOI: https://doi.org/10.1038/s41591-025-03734-3
This article is cited by
-
Harnessing albumin’s natural tumor-targeting properties: nanoplatform strategies for triple-negative breast cancer therapy
Discover Nano (2026)
-
Conserved sphingolipid metabolism under transcriptomic diversity: a prognostic and therapeutic target in triple-negative breast cancer
Journal of Translational Medicine (2025)
-
Advances in nanoparticle-based doxorubicin delivery: precision strategies for targeted treatment of triple-negative breast cancer
Discover Nano (2025)
