
Abstract
Myalgic encephalomyelitis/chronic fatigue syndrome (ME/CFS) is a chronic illness with a multifactorial etiology and heterogeneous symptomatology, posing major challenges for diagnosis and treatment. Here we present BioMapAI, a supervised deep neural network trained on a 4-year, longitudinal, multi-omics dataset from 249 participants, which integrates gut metagenomics, plasma metabolomics, immune cell profiling, blood laboratory data and detailed clinical symptoms. By simultaneously modeling these diverse data types to predict clinical severity, BioMapAI identifies disease- and symptom-specific biomarkers and classifies ME/CFS in both held-out and independent external cohorts. Using an explainable AI approach, we construct a unique connectivity map spanning the microbiome, immune system and plasma metabolome in health and ME/CFS adjusted for age, gender and additional clinical factors. This map uncovers altered associations between microbial metabolism (for example, short-chain fatty acids, branched-chain amino acids, tryptophan, benzoate), plasma lipids and bile acids, and heightened inflammatory responses in mucosal and inflammatory T cell subsets (MAIT, γδT) secreting IFN-γ and GzA. Overall, BioMapAI provides unprecedented systems-level insights into ME/CFS, refining existing hypotheses and hypothesizing unique mechanisms—specifically, how multi-omics dynamics are associated to the disease’s heterogeneous symptoms.
This is a preview of subscription content, access via your institution
Access options
Access Nature and 54 other Nature Portfolio journals
Get Nature+, our best-value online-access subscription
$32.99 / 30 days
cancel any time
Subscribe to this journal
Receive 12 print issues and online access
$259.00 per year
only $21.58 per issue
Buy this article
- Purchase on SpringerLink
- Instant access to the full article PDF.
USD 39.95
Prices may be subject to local taxes which are calculated during checkout





Similar content being viewed by others


Data availability
All individual-level data supporting the findings of this study are publicly available. The complete dataset used in this study, including clinical metadata (including demographic information), immune profiles, plasma metabolomics, Quest Diagnostics blood measurements, and processed microbiome data, is available on GitHub: https://github.com/ohlab/BioMapAI/blob/main/data/Omics_Dataset.zip. Raw gut metagenomic sequencing data have been deposited in BioProject: https://www.ncbi.nlm.nih.gov/bioproject/PRJNA1125469.
Code availability
BioMapAI framework and user guide is available at https://github.com/ohlab/BioMapAI. All original code, processed data and trained model of this study has been deposited at Github.
References
Ruiz-Pablos, M., Paiva, B., Montero-Mateo, R., Garcia, N. & Zabaleta, A. Epstein-Barr virus and the origin of myalgic encephalomyelitis or chronic fatigue syndrome. Front. Immunol. 12, 656797 (2021).
Su, R. et al. The TLR3/IRF1/type III IFN axis facilitates antiviral responses against enterovirus infections in the intestine. mBio 11, e02540-20 (2020).
Anderson, D. E. et al. Lack of cross-neutralization by SARS patient sera towards SARS-CoV-2. Emerg Microbes Infect. 9, 900–902 (2020).
Cairns, R. & Hotopf, M. A systematic review describing the prognosis of chronic fatigue syndrome. Occup. Med. Oxf. Engl. 55, 20–31 (2005).
Cortes Rivera, M., Mastronardi, C., Silva-Aldana, C. T., Arcos-Burgos, M. & Lidbury, B. A. Myalgic encephalomyelitis/chronic fatigue syndrome: a comprehensive review. Diagnostics 9, 91 (2019).
Hare, P. J., LaGree, T. J., Byrd, B. A., DeMarco, A. M. & Mok, W. W. K. Single-cell technologies to study phenotypic heterogeneity and bacterial persisters. Microorganisms 9, 2277 (2021).
Zeeshan, S., Xiong, R., Liang, B. T. & Ahmed, Z. 100 years of evolving gene–disease complexities and scientific debutants. Brief. Bioinform. 21, 885–905 (2020).
Poenaru, S., Abdallah, S. J., Corrales-Medina, V. & Cowan, J. COVID-19 and post-infectious myalgic encephalomyelitis/chronic fatigue syndrome: a narrative review. Ther. Adv. Infect. Dis. 8, 20499361211009385 (2021).
Reuken, P. A. et al. Longterm course of neuropsychological symptoms and ME/CFS after SARS-CoV-2-infection: a prospective registry study. Eur. Arch. Psychiatry Clin. Neurosci. 274, 1903–1910 (2024).
Bretherick, A. D. et al. Typing myalgic encephalomyelitis by infection at onset: a DecodeME study. NIHR Open Res. 3, 20 (2023).
Bae, J. & Lin, J.-M. S. Healthcare utilization in myalgic encephalomyelitis/chronic fatigue syndrome (ME/CFS): analysis of US ambulatory healthcare data, 2000–2009. Front. Pediatr. 7, 185 (2019).
Zheng, Y. & Zhu, Z. Editorial: retrieving meaningful patterns from big biomedical data with machine learning approaches. Front. Genet. 14, 1177996 (2023).
Leelatian, N. et al. Unsupervised machine learning reveals risk stratifying glioblastoma tumor cells. eLife 9, e56879 (2020).
Su, Q. et al. The gut microbiome associates with phenotypic manifestations of post-acute COVID-19 syndrome. Cell Host Microbe 32, 651–660 (2024).
Bourgonje, A. R., van Goor, H., Faber, K. N. & Dijkstra, G. Clinical value of multiomics-based biomarker signatures in inflammatory bowel diseases: challenges and opportunities. Clin. Transl. Gastroenterol. 14, e00579 (2023).
Marcos-Zambrano, L. J. et al. Applications of machine learning in human microbiome studies: a review on feature selection, biomarker identification, disease prediction and treatment. Front. Microbiol. 12, 634511 (2021).
Guo, C. et al. Deficient butyrate-producing capacity in the gut microbiome is associated with bacterial network disturbances and fatigue symptoms in ME/CFS. Cell Host Microbe 31, 288–304 (2023).
Raijmakers, R. P. H. et al. Multi-omics examination of Q fever fatigue syndrome identifies similarities with chronic fatigue syndrome. J. Transl. Med. 18, 448 (2020).
Germain, A. et al. Plasma metabolomics reveals disrupted response and recovery following maximal exercise in myalgic encephalomyelitis/chronic fatigue syndrome. JCI Insight 7, e157621 (2023).
Che, X. et al. Metabolomic evidence for peroxisomal dysfunction in myalgic encephalomyelitis/chronic fatigue syndrome. Int. J. Mol. Sci. 23, 7906 (2022).
Liñares-Blanco, J., Fernandez-Lozano, C., Seoane, J. A. & López-Campos, G. Machine learning based microbiome signature to predict inflammatory bowel disease subtypes. Front. Microbiol. 13, 872671 (2022).
He, F. et al. Development and external validation of machine learning models for diabetic microvascular complications: cross-sectional study with metabolites. J. Med. Internet Res. 26, e41065 (2024).
Mora-Ortiz, M., Trichard, M., Oregioni, A. & Claus, S. P. Thanatometabolomics: introducing NMR-based metabolomics to identify metabolic biomarkers of the time of death. Metabolomics 15, 37 (2019).
Balasubramanian, R. et al. Metabolomic profiles associated with all-cause mortality in the Women’s Health Initiative. Int. J. Epidemiol. 49, 289–300 (2020).
Li, H., Ren, M. & Li, Q. 1H NMR-based metabolomics reveals the intrinsic interaction of age, plasma signature metabolites, and nutrient intake in the longevity population in Guangxi, China. Nutrients 14, 2539 (2022).
Kondoh, H. & Kameda, M. Metabolites in aging and aging-relevant diseases: frailty, sarcopenia and cognitive decline. Geriatr. Gerontol. Int. 24, 44–48 (2024).
Peng, S., Shen, Y., Wang, M. & Zhang, J. Serum and CSF metabolites in stroke-free patients are associated with vascular risk factors and cognitive performance. Front. Aging Neurosci. 12, 193 (2020).
Duerler, P., Vollenweider, F. X. & Preller, K. H. A neurobiological perspective on social influence: serotonin and social adaptation. J. Neurochem. 162, 60–79 (2022).
Pomrenze, M. B., Paliarin, F. & Maiya, R. Friend of the Devil: negative social influences driving substance use disorders. Front. Behav. Neurosci. 16, 836996 (2022).
Laslett, A.-M. Empirical and conceptual paradigms for studying secondary impacts of a person’s substance use. Addiction 117, 3148–3149 (2022).
Carco, C. et al. Increasing evidence that irritable bowel syndrome and functional gastrointestinal disorders have a microbial pathogenesis. Front. Cell. Infect. Microbiol. 10, 468 (2020).
Saffouri, G. B. et al. Small intestinal microbial dysbiosis underlies symptoms associated with functional gastrointestinal disorders. Nat. Commun. 10, 2012 (2019).
Liang, S., Wu, X., Hu, X., Wang, T. & Jin, F. Recognizing depression from the microbiota–gut–brain axis. Int. J. Mol. Sci. 19, 1592 (2018).
Zhu, F., Tu, H. & Chen, T. The microbiota–gut–brain axis in depression: the potential pathophysiological mechanisms and microbiota combined antidepression effect. Nutrients 14, 2081 (2022).
Topan, R. & Scott, S. M. Sleep: an overlooked lifestyle factor in disorders of gut-brain interaction. Curr. Treat. Options Gastroenterol. 21, 435–446 (2023).
Moens de Hase, E. et al. Impact of metformin and Dysosmobacter welbionis on diet-induced obesity and diabetes: from clinical observation to preclinical intervention. Diabetologia 67, 333–345 (2024).
Amabebe, E., Robert, F. O., Agbalalah, T. & Orubu, E. S. F. Microbial dysbiosis-induced obesity: role of gut microbiota in homoeostasis of energy metabolism. Br. J. Nutr. 123, 1127–1137 (2020).
Kavanagh, P. et al. Tentative identification of the phase I and II metabolites of two synthetic cathinones, MDPHP and α-PBP, in human urine. Drug Test. Anal. 12, 1442–1451 (2020).
Lenoir, M. et al. Butyrate mediates anti-inflammatory effects of Faecalibacterium prausnitzii in intestinal epithelial cells through Dact3. Gut Microbes 12, 1–16 (2020).
Sokol, H. et al. Faecalibacterium prausnitzii is an anti-inflammatory commensal bacterium identified by gut microbiota analysis of Crohn disease patients. Proc. Natl Acad. Sci. 105, 16731–16736 (2008).
Monteiro, C. R. A. V. et al. In vitro antimicrobial activity and probiotic potential of Bifidobacterium and Lactobacillus against species of Clostridium. Nutrients 11, 448 (2019).
Nguyen-Lefebvre, A. T., Selzner, N., Wrana, J. L. & Bhat, M. The hippo pathway: a master regulator of liver metabolism, regeneration, and disease. FASEB J. 35, e21570 (2021).
Kim, C.-S. Roles of diet-associated gut microbial metabolites on brain health: cell-to-cell interactions between gut bacteria and the central nervous system. Adv. Nutr. 15, 100136 (2024).
Rebeaud, J., Peter, B. & Pot, C. How microbiota-derived metabolites link the gut to the brain during neuroinflammation. Int. J. Mol. Sci. 23, 10128 (2022).
Ahmad, S. et al. Gut microbiome-related metabolites in plasma are associated with general cognition. Alzheimers Dement. 17, e056142 (2021).
Ahmed, Z., Zeeshan, S., Xiong, R. & Liang, B. T. Debutant iOS app and gene‐disease complexities in clinical genomics and precision medicine. Clin. Transl. Med. 8, e26 (2019).
Xiong, R. et al. Multi-omics of gut microbiome-host interactions in short- and long-term myalgic encephalomyelitis/chronic fatigue syndrome patients. Cell Host Microbe 31, 273–287 (2023).
Sweetman, E. et al. Current research provides insight into the biological basis and diagnostic potential for myalgic encephalomyelitis/chronic fatigue syndrome (ME/CFS). Diagnostics 9, 73 (2019).
Hamine, S., Gerth-Guyette, E., Faulx, D., Green, B. B. & Ginsburg, A. S. Impact of health chronic disease management on treatment adherence and patient outcomes: a systematic review. J. Med. Internet Res. 17, e3951 (2015).
Derman, I. D. et al. High-throughput bioprinting of the nasal epithelium using patient-derived nasal epithelial cells. Biofabrication 15, 044103 (2023).
Fleming, E. et al. Cultivation of common bacterial species and strains from human skin, oral, and gut microbiota. BMC Microbiol. 21, 278 (2021).
Trivedi, M. S. et al. Identification of myalgic encephalomyelitis/chronic fatigue syndrome-associated DNA methylation patterns. PLoS ONE 13, e0201066 (2018).
Bouquet, J. et al. Whole blood human transcriptome and virome analysis of ME/CFS patients experiencing post-exertional malaise following cardiopulmonary exercise testing. PLoS ONE 14, e0212193 (2019).
Caruana, E. J., Roman, M., Hernández-Sánchez, J. & Solli, P. Longitudinal studies. J. Thorac. Dis. 7, E537–E540 (2015).
Aurora, C., Cecilia, A. & Adina, H. The role of diet in the treatment of chronic diseases case study. ARS Med. Tomitana 27, 153–156 (2021).
Therrien, R. & Doyle, S. Role of training data variability on classifier performance and generalizability. In Medical Imaging 2018: Digital Pathology Vol. 10581, 58–70 (SPIE, 2018).
Krumina, A. et al. Clinical profile and aspects of differential diagnosis in patients with ME/CFS from Latvia. Med. (Mex.) 57, 958 (2021).
Zubcevik, N. et al. Symptom clusters and functional impairment in individuals treated for Lyme borreliosis. Front. Med. 7, 464 (2020).
Lathan, C., Spira, J. L., Bleiberg, J., Vice, J. & Tsao, J. W. Defense automated neurobehavioral assessment (DANA)-psychometric properties of a new field-deployable neurocognitive assessment tool. Mil. Med. 178, 365–371 (2013).
Resnick, H. E. & Lathan, C. E. From battlefield to home: a mobile platform for assessing brain health. mHealth 2, 30 (2016).
Lee, J. et al. Hemodynamics during the 10-minute NASA Lean Test: evidence of circulatory decompensation in a subset of ME/CFS patients. J. Transl. Med. 18, 314 (2020).
Committee on the Diagnostic Criteria for Myalgic Encephalomyelitis/ChronicFatigue Syndrome, Board on the Health of Select Populations, & Institute of Medicine. Beyond Myalgic Encephalomyelitis/Chronic Fatigue Syndrome: Redefining an Illness (National Academies Press, 2015).
36-Item Short Form Survey (SF-36) Scoring Instructions. RAND Health Care https://www.rand.org/health-care/surveys_tools/mos/36-item-short-form/scoring.html (1992).
Blanco-Míguez, A. et al. Extending and improving metagenomic taxonomic profiling with uncharacterized species using MetaPhlAn 4. Nat. Biotechnol. 41, 1633–1644 (2023).
Edgar, R. C. Search and clustering orders of magnitude faster than BLAST. Bioinformatics 26, 2460–2461 (2010).
Love, M. I., Huber, W. & Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550 (2014).
Saripuddin, M., Suliman, A., Syarmila Sameon, S. & Jorgensen, B. N. Random undersampling on imbalance time series data for anomaly detection. In Proc. 2021 4th International Conference on Machine Learning and Machine Intelligence 151–156 (Association for Computing Machinery, 2022).
Lundberg, S. & Lee, S.-I. A Unified approach to interpreting model predictions. Preprint at https://doi.org/10.48550/arXiv.1705.07874 (2017).
Friedman, J. H., Hastie, T. & Tibshirani, R. Regularization paths for generalized linear nodels via coordinate descent. J. Stat. Softw. 33, 1–22 (2010).
Kuhn, M. Building predictive models in R using the caret package. J. Stat. Softw. 28, 1–26 (2008).
Langfelder, P. & Horvath, S. WGCNA: an R package for weighted correlation network analysis. BMC Bioinformatics 9, 559 (2008).
Antonov, M., et al. igraph enables fast and robust network analysis across programming languages. Preprint at https://doi.org/10.48550/arXiv.2311.10260 (2023).
Koo, T. K. & Li, M. Y. A guideline of selecting and reporting intraclass correlation coefficients for reliability research. J. Chiropr. Med. 15, 155–163 (2016).
Nelder, J. A. & Wedderburn, R. W. M. Generalized linear models. J. R. Stat. Soc. Ser. Gen. 135, 370–384 (1972).
Bakdash, J. Z. & Marusich, L. R. Repeated measures correlation. Front. Psychol. 8, 456 (2017).
Gu, C. Smoothing Spline ANOVA Models: R Package gss (Springer, 2013).
Acknowledgements
We are thankful to the Oh, Unutmaz and Li laboratories for inspiring discussions and acknowledge the contribution of the Genome Technologies Service at The Jackson Laboratory for expert assistance with sample sequencing for the work described in this publication. We also thank the clinical support team at the Bateman Horne Center and all the individuals who participated in this study. This work was funded by 1U54NS105539. R.X., E.A., R.C., S.D.V., L.K., C.G., L.B., D.U. and J.O. were funded by 1U54NS105539. J.O. is additionally supported by the NIH (1 R01 AR078634-01, DP2 GM126893-01, 1 U19 AI142733, 1 R21 AR075174).
Author information
Authors and Affiliations
Contributions
Conceptualization was carried out by D.U., J.O., S.D.V., L.B. and R.X. Data curation was performed by R.X., C.G., S.D.V. and L.B. Formal analysis was conducted by R.X. Funding acquisition was secured by D.U., J.O., S.D.V. and L.B. Clinical sample design and collection were conducted by S.D.V. and L.B. Investigation was carried out by R.X., C.G., E.A., S.D.V., L.B., R.C. and L.K. Project administration was handled by J.O., D.U., L.B., S.D.V. and C.G. Resources were provided by D.U., J.O., S.D.V. and L.B. Supervision was provided by J.O. Visualization and writing were completed by R.X. and J.O. Writing, review and editing were performed by R.X., C.G., S.D.V., L.B., D.U. and J.O.
Corresponding author
Ethics declarations
Competing interests
S.D.V. is affiliated and has a financial interest with The BioCollective, a company that provided the BioCollector, the collection kit used for at-home stool collection discussed in this paper. The other authors declare no competing interests.
Peer review
Peer review information
Nature Medicine thanks Carmen Scheibenbogen and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Primary Handling Editors: Lorenzo Righetto and Saheli Sadanand, in collaboration with the Nature Medicine team.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data
Extended Data Fig. 1 Data Pairedness Overview and Heterogeneity in Healthy and Patients.
a, Cohort Composition and Data Collection. Over four years, 515 time points were collected: baseline year from all 249 donors (Healthy N=96, ME/CFS N=153); second year from 168 individuals (Healthy N=58, ME/CFS N=110); third year from 94 individuals (Healthy N=13, ME/CFS N=81); fourth year from N=4 ME/CFS patients. Clinical metadata and blood measures were collected at all 515 points. Immune profiles from PBMCs were recorded at 489 points, microbiome data from stool samples at 479 points, and plasma metabolome data at 414 points. A total of 1,471 biosamples were collected. b-c, Heterogeneity of b, Healthy Controls and c, All Patients in Symptom Severity and omics Profiles. Supplemental information for Fig. 1b, which shows examples from 20 patients. Variability in symptom severity (top) and omics profiles (bottom) for all healthy controls and all patients with 3-4 time points. The top x-axis numbers represent 12 symptoms, arranged in the same order as Extended Data Fig. 1f, g (left to right, top to bottom). d, Distribution of 12 Clinical Symptoms in ME/CFS and Control. Density plots compare the distributions of 12 clinical scores between control (blue) and ME/CFS patients (orange) with the x-axis represents the values of symptom severity (scaled from 0%, no symptom, to 100%, most severe) and the y-axis represents the frequency (count) of data points. e, Principal Coordinates Analysis (PCoA) of each ‘Omics. PCoA based on Bray-Curtis distance. Control samples (blue) and ME/CFS patients (red) show distinct clustering. Here, except for the clinical scores, controls are indistinguishable from patients, highlighting the difficulty of building classification models. f-g, Symptom Progression Over Time in f, Healthy vs. g, ME/CFS Patients. Symptom progression for each individual (represented by different colors) is shown using line plots of symptom severity (y-axis) over time points (years 1–4). Compared to healthy controls, ME/CFS patients exhibit higher severity, greater heterogeneity, and inconsistent or nonlinear progression (indicated by substantial variation over time without a consistent pattern) in clinical symptoms. Related to: Figs. 1–2.
Extended Data Fig. 2 BioMapAI’s Performance at Clinical Score Reconstruction and Disease Classification.
a, Density map of True vs. Predicted Clinical Scores. Supplemental information for Fig. 2b, which shows three examples. Here, the full set of 12 clinical scores compares the true score, \(y\) (Column 1), against BioMapAI’s predictions generated from different omics profiles – \({\hat{y}}_{{immune}}\), \({\hat{y}}_{{species}}\), \({\hat{y}}_{{KEGG}}\), \({\hat{y}}_{{metabolome}}\), \({\hat{y}}_{{quest}}\), \({\hat{y}}_{{omics}}\) (Columns 2–7). b, Scatter Plot of True vs. Predicted Clinical Scores. Scatter plots display the relationship between true clinical scores (x-axis) and predicted clinical scores (y-axis) for six different models: Omics, Immune, Species, KEGG, Metabolome, and Quest Labs. Each plot demonstrates the clinical score prediction accuracy for each model. c, ROC Curve for Disease Classification with Original Clinical Scores. The Receiver Operating Characteristic (ROC) curve evaluates the performance of disease classification using the original 12 clinical scores. The mean Area Under the Curve (AUC) is 0.99, indicating high prediction accuracy, which aligns with the clinical diagnosis of ME/CFS based on key symptoms. d, 3D t-SNE Visualization of Hidden Layers. 3D t-SNE plots show how BioMapAI progressively distinguishes disease from control across hidden layers for five trained ‘omics models: Immune, KEGG, Species, Metabolome, and Quest Labs. Each plot uses the first three principal components to show the spatial distribution of control samples (blue) and ME/CFS patients (red). The progression from the input layer (mixed groups) to Hidden Layer 3 (fully separated groups) illustrates how BioMapAI progressively learns to separate ME/CFS from healthy controls. Related to: Fig. 2.
Extended Data Fig. 3 Disease-Specific Biomarkers - Top 10 Biomarkers Shared across Clinical Symptoms and Multiple Models.
Through the top 30 high-ranking features for each score, we discovered that the most critical features for all 12 symptoms were largely shared and consistently validated across ML and DL models, particularly the foremost 10. Here, this multi-panel figure presents the top 10 most significant features identified by BioMapAI across five omics profiles, highlighting their importance in predicting clinical symptoms and diagnostic outcomes across BioMapAI, DNN, and GBDT models, along with their data prevalence. Each vertical section represents one omics profile, with columns of biomarkers ordered by average feature importance from right to left. From top to bottom: 1. Heatmap of SHAP Values from BioMapAI. This heatmap shows averaged SHAP values with the 12 scores on the rows and the top 10 features in the columns. Darker colors indicate a stronger impact on the model’s output; Consistency among the top 5 features suggests they are shared disease biomarkers crucial for all clinical symptoms; 2. Swarm Plot of SHAP Values from DNN. This plot represents the distribution of feature contributions from DNN, which is structurally similar to BioMapAI but omits the third hidden layer (\({Z}^{3}\)). SHAP values are plotted vertically, ranging from negative to positive, showing each feature’s influence on prediction outcomes. Points represent individual samples, with color gradients denoting actual feature values. For instance, Dysosmobacteria welbionis, identified as the most critical species, shows that greater species relative abundance correlates with a higher likelihood of disease prediction; 3. Bar Graphs of Feature Importance in GBDT. GBDT is another machine learning model used for comparison. Each bar’s height indicates a feature’s significance within the GBDT model, providing another perspective on the predictive relevance of each biomarker; 4. Heatmap of Normalized Raw Abundance Data. This heatmap compares biomarker prevalence between healthy and disease states, with colors representing z-scored abundance values, highlighting biomarker differences between groups. Supporting Materials: Extended Data Table 3. Related to: Fig. 3.
Extended Data Fig. 4 Symptom-Specific Biomarkers - Immune, KEGG and Metabolome Models.
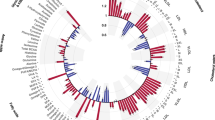
By linking omics profiles to clinical symptoms, BioMapAI identified unique symptom-specific biomarkers in addition to disease-specific biomarkers (Extended Data Fig. 3). Each omics has a circularized diagram (Fig. 3a, Extended Data Fig. 4b–d) to display how BioMapAI use this omics profile to predict 12 clinical symptoms and to discuss the contribution of disease- and symptom-specific biomarkers. Detailed correlation between symptom-specific biomarkers and their corresponding symptoms is in Extended Data Fig. 5. a, Examples of Sleeping Problem-Specific Species’ and Gastrointestinal-Specific Species’ Contributions. Supplemental information for Fig. 3d, which shows the contribution of pain-specific species. b-d, Circularized Diagram for Immune, KEGG and Metabolome Models. Supplemental information for Fig. 3a, which shows the species model. e-f, Zoomed Segment for Pain in KEGG and Metabolome Model. Supplemental information for Fig. 3b, which shows the zoomed segment for pain in the species and immune models. *Note, the reported biomarkers were calculated using the entire dataset and were not validated on held-out data. Abbreviations and Supporting Materials: Extended Data Fig. 5. Related to: Fig. 3.
Extended Data Fig. 5 Symptom-Specific Biomarkers - Different Correlation Patterns of Biomarkers to Symptom.
Supplemental information for Fig. 3c, which shows six pain biomarkers from multiple models. Here for each omics (a-d, Immune, Species, KEGG, Metabolome), we plotted the correlation of symptom-specific biomarkers (x-axis) to its related symptom (y-axis), colored by SHAP value (contribution to the symptom). P value by two-sided spearman correlation, FDR adjusted (Detailed statistics in Supplementary Table 5). Abbreviations: CD4, Cluster of Differentiation 4; CD8, Cluster of Differentiation 8; IFNg, Interferon Gamma; DC, Dendritic Cells; MAIT, Mucosal-Associated Invariant T; Th17, T helper 17 cells; CD4+ TCM, CD4+ Central Memory T cells; DC CD1c+ mBtp+, Dendritic Cells expressing CD1c+ and myelin basic protein; DC CD1c+ mHsp, Dendritic Cells expressing CD1c+ and heat shock protein; CD4+ TEM, CD4+ Effector Memory T cells; CD4+ Th17 rfx4+, CD4+ T helper 17 cells expressing RFX4; F. prausnitzii, Faecalibacterium prausnitzii; A. communis, Akkermansia communis; NAD, Nicotinamide Adenine Dinucleotide. Related to: Fig. 3.
Extended Data Fig. 6 omics WGCNA Modules and Host-Microbiome Network.
a, Correlation of WGCNA Modules with Clinical Metadata. Weighted Gene Co-expression Network Analysis (WGCNA) was used to identify co-expression modules for each omics layer: species, KEGG, immune, and metabolome. The top dendrograms show hierarchical clustering of ‘omics features, with modules identified. The bottom heatmap shows the relationship of module eigengenes (colored as per dendrogram) with clinical metadata – including demographic information and environmental factors - and 12 clinical scores. General linear models were used to determine the primary clinical drivers for each module, with the color gradient representing the coefficients (red = positive, blue = negative). Microbial modules were influenced by disease presence and energy-fatigue levels, while metabolome and immune modules correlated with age and gender. b-c, Microbiome-Immune-Metabolome Network in b, Patient and c, Healthy Subgroups. Supplemental information for Fig. 4a (Healthy Network) and 4b (Patient Subgroups). Figure 4a is the healthy network; here, Extended Data Fig. 6b presented the shifted correlations in all patients. Figure 4b represented the network in patient subgroups; here, Extended Data Fig. 6c is the corresponding healthy counterpart, for example, female patients were compared with female controls to exclude gender influences. d, Differences in Host-Microbiome Correlations between Healthy and Patient Subgroups. Selected host–microbiome module pairs are grouped on the x-axis (for example, pyruvate to blood modules, steroids to gut microbiome). Significant positive and negative correlations (top and bottom y-axis) of module members pairs are shown as dots for each subgroup (blue = healthy, orange = patient) (Spearman, adjusted p < 0.05), from left to right: Young, Elder, Female, Male, NormalWeight, OverWeight Healthy and Young, Elder, Female, Male, NormalWeight, OverWeight Patient. The middle bars represent the total count of associations. This panel highlights the shifts in host–microbiome networks from health to disease, for example, in patients, the loss of pyruvate to host blood modules correlation and the increase of INFg+ CD4 memory correlation with gut microbiome. Related to: Fig. 4.
Supplementary information
Supplementary Information
Supplementary Figs. 1 and 2.
Supplementary Tables
Supplemental Tables 1–8.
Supplementary Software 1
BioMapAI model software with example data and tutorials. See full details in our Github: https://github.com/ohlab/BioMapAI.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Xiong, R., Aiken, E., Caldwell, R. et al. AI-driven multi-omics modeling of myalgic encephalomyelitis/chronic fatigue syndrome. Nat Med 31, 2991–3001 (2025). https://doi.org/10.1038/s41591-025-03788-3
Received:
Accepted:
Published:
Version of record:
Issue date:
DOI: https://doi.org/10.1038/s41591-025-03788-3
This article is cited by
-
Neuro-immune Crosstalk in Atopic Dermatitis: A Multiaxial Regulatory Network and Novel Therapeutic Perspectives
Clinical Reviews in Allergy & Immunology (2026)
