
Abstract
Single-molecule localization microscopy (SMLM) has gained widespread use for visualizing the morphology of subcellular organelles and structures with nanoscale spatial resolution. However, analysis tools for automatically quantifying and classifying SMLM images have lagged behind. Here we introduce Enhanced Classification of Localized Point clouds by Shape Extraction (ECLiPSE), an automated machine learning analysis pipeline specifically designed to classify cellular structures captured through two-dimensional or three-dimensional SMLM. ECLiPSE leverages a comprehensive set of shape descriptors, the majority of which are directly extracted from the localizations to minimize bias during the characterization of individual structures. ECLiPSE has been validated using both unsupervised and supervised classification on datasets, including various cellular structures, achieving near-perfect accuracy. We apply two-dimensional ECLiPSE to classify morphologically distinct protein aggregates relevant for neurodegenerative diseases. Additionally, we employ three-dimensional ECLiPSE to identify relevant biological differences between healthy and depolarized mitochondria. ECLiPSE will enhance the way we study cellular structures across various biological contexts.
Similar content being viewed by others


Main
Cells are compartmentalized into various structural units including membrane-bound and membraneless subcellular organelles, cytoskeletal structures and supramolecular protein assemblies. Each of these organizational units possesses unique and complex structural and morphological properties that span a range of length scales to match their function. The distinct morphology of organelles help adapt them to specific functions, and can change in response to cellular needs as well as in disease states1. Similarly, aggregation of proteins into solid inclusions with specific morphological properties is a hallmark of several neurodegenerative diseases2. Therefore, techniques to characterize and classify subcellular compartments on the basis of their structural and morphological properties are invaluable in studying both cell physiology and pathology.
Recent advancements in super-resolution microscopy have revolutionized our ability to visualize the intricate morphological features of subcellular compartments and organelles at nanoscale spatial resolution3. Super-resolution microscopy can capture subtle changes in the morphology and structure of these subcellular components, which were once inaccessible due to the diffraction limit of light. For this purpose, single-molecule localization microscopy (SMLM) techniques have been widely adopted by the cell biology community as they do not require highly specialized microscope hardware4. However, the development of analysis tools to accurately classify individual subcellular structures into distinct categories on the basis of shape and morphology has not kept pace with advancements in super-resolution microscopy, particularly in the context of SMLM. This is because SMLM data consist of point clouds rather than pixelated intensity-based images, which is less compatible with traditional image processing and analysis techniques.
Current tools employ template-based or template-free strategies to classify and align super-resolution data of highly symmetric, self-similar and frequently simplistic structures, such as the nuclear pore complex (NPC), to facilitate single-particle averaging for the purpose of accurately describing the structure of interest5,6,7,8,9,10. However, these methods do not explicitly ascertain the morphological characteristics of unique structures, leading to the recent development of tools, such as SEgmentation and MORphological fingErprinting (SEMORE)11, which combines both geometric and kinetics-based descriptors in its analysis. Alternative tools, such as Localization Model Fit (LocMoFit), depend on model fitting with predefined and often basic geometric models to achieve the structure-matching and extract basic quantitative properties12. LocMoFit enables the extraction of a number of geometric features that are included in the model, such as size and symmetry angle, to determine the degree of variability among individual structures. Although this method is valuable, it can only quantify a small number of parameters and has not been utilized to classify structures into distinct categories as it relies on analyzing structures that are identical or highly similar. Recently, an automated structure analysis program (ASAP) was developed to quantify and classify structures based on a limited number of geometric shape descriptors13. ASAP was applied to SMLM images of NPCs, endocytic vesicles and Bax protein pores, all of which assemble into small (~100 nm) and simple structures resembling either rods, arcs or circles. Although ASAP represents an important improvement in the structural classification of super-resolution data, one of its drawbacks is that it requires rendering the SMLM point cloud data into pixelated images followed by thresholding and binarization, which may introduce artifacts and cause the loss of important information. Furthermore, the limited number of shape descriptors in ASAP makes it less applicable to structures with complex shapes and larger sizes. To fill the gaps in advanced classification tools, we developed an analysis pipeline called Enhanced Classification of Localized Point clouds by Shape Extraction (ECLiPSE) that expands the toolbox for classifying structures in two-dimensional (2D) and three-dimensional (3D) SMLM data. ECLiPSE does not require user input in its default settings, which are robust for most applications and can accurately describe and classify structures of different sizes and complexity. This includes organelles with distinct morphologies, cytoskeletal filaments and diverse protein aggregates.
Results
Two-dimensional ECLiPSE pipeline
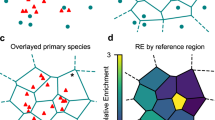
The workflow of ECLiPSE is shown in Fig. 1. After data acquisition and segmentation of super-resolution data, which can be done using existing techniques and packages (for example, Voronoi tessellation or DBScan14,15,16,17; Fig. 1a,b), the first step in ECLiPSE involves calculating 68 (2D data) or 69 (3D data) (see below) shape descriptors, of which the majority is extracted directly from the point cloud data (Fig. 1c, 2D descriptors). The shape descriptors include geometric properties, boundary properties, skeleton properties, texture properties, Hu moments and fractal properties (Supplementary Tables 1 and 2 for 2D and 3D properties, respectively). We note that for all visualization purposes in this manuscript (except Fig. 1c), the localizations were rendered into intensity-based images, but calculations were done on the raw, unprocessed localizations. It is important to note that not every shape descriptor is equally effective at distinguishing between various structures and the specific descriptors that provide the highest degree of separation can vary depending on each biological application. To address this, we have incorporated an automated variable selection step (Supplementary Note 1 and Supplementary Fig. 1), which can be used to automatically select the most informative descriptors that distinguish between different groups in the data without prior knowledge of what these descriptors are (Fig. 1d). If information on group identity is not available, data compression algorithms such as principal component analysis (PCA) could be used to extract such information. Once the shape descriptors are calculated and optionally undergo variable selection, the data can be explored in the PCA-space using these quantitative features (Fig. 1e). This approach offers a preliminary visual representation of the extent to which datasets are separated within the PCA space and can uncover subpopulations within the data. Additionally, information about distinct groups can be employed to color code the data points, revealing their degree of separation. The final step is the classification (Fig. 1f) using many machine learning models including supervised models (for example, K-nearest neighbors, random forest, partial least squares for discrimination, logistic regression for discrimination and so on) and unsupervised models (for example, partial or agglomerative hierarchical clustering). Model training and validation is performed independently from one another by selecting different subsets of the full dataset (Supplementary Note 2). The training and validation process can be performed multiple times and the best performing model(s) is (are) then automatically selected to predict the class membership of new data that the user provides. Detailed information on the unsupervised and supervised classification models and hyperparameters used in this work can be found in Supplementary Note 2, Supplementary Figs. 2–4 and Supplementary Tables 3 and 4.

a, A schematic representation of the SMLM data acquisition process (t is time). b, Segmentation of the localizations into individual clusters (applied to a region of interest of the lysosome data using a maximum Voronoi area of 684 nm2 and a minimum of 25 localizations and subsequent filtering of lysosomes with an area less than 0.123 µm2 or greater than 0.479 µm2; green: low density and blue: high density). c, Feature extraction from segmented point cloud clusters generate descriptors including geometric, boundary, skeleton and so on. d, Example distributions of features that adequately or poorly separate the different classes in the validation data, as determined by automatic variable selection (28/67 descriptors that provide clear class separation). e, Data exploration using PCA with and without variable selection. f, Optimized classification results for the validation data (97.1 ± 0.1% accuracy), obtained by the random forest classifier (100 best models out of 1,000 generated models). g, Difference confusion matrices between ECLiPSE (logistic regression, no variable selection) and ASAP (10 nm rendering precision, 1.5 × 105 threshold, discriminant classifier). Left: validation data (96.9% versus 93.5% accuracy for ECLiPSE and ASAP, respectively). Right: tau aggregation data (92.9% versus 80.6% accuracy for ECLiPSE and ASAP, respectively). The blue values represent superior results for ECLiPSE (that is, positive diagonal values and negative off-diagonal values), whereas red values represent inferior results for ECLiPSE (that is, negative diagonal values and positive off-diagonal values).

a, Example clusters of the four different tau aggregate species. b, Data exploration using PCA indicates the complexity of the data. c, Classification results obtained (89.8 ± 0.4% accuracy) using the logistic regression classifier on the variable selected tau aggregates data. d, Tau clearance, after removal of Dox, observed over a period of 10 days shows a visual reduction of tau aggregate cluster sizes over time. e, Tau aggregate species prediction on the total dataset demonstrates the rapid decrease in branched fibrils, pre-NFTs and NFTs after Dox removal, whereas linear fibrils show delayed degradation kinetics. FOV, field of view. f, Four representative 2D SMLM images of cells containing two different patient specific TDP-43 strains. g, Representative clusters of the two patient specific TDP-43 strains. h, Data exploration using PCA indicates that a nonnegligible subset of the data clusters are similar between the two patient-specific strains. i, Classification results obtained (89.9 ± 0.6% accuracy) using the partial least squares classifier on the nonvariable selected TDP-43 data. For classification in c and i, only the results obtained by the 100 best models out of 1,000 generated models are shown. For a–e: +Dox control, n = 29 cells; day 1 − Dox, n = 27 cells; day 2 − Dox, n = 27 cells; day − 3 Dox, n = 27 cells; day 4 − Dox, n = 29 cells; day 5 − Dox, n = 27 cells; day 10 − Dox, n = 28 cells; all three biological replicates. The bar plots in e represent mean ± s.d. of the prediction of the 100 best models as shown in c. For f–i strain A: n = 15 cells (three biological replicates) and strain B: n = 19 cells (three biological replicates).
Two-dimensional ECLiPSE validation and benchmarking
We first validated our approach using ground truth 2D SMLM datasets of five distinct structures including organelles (lysosomes and mitochondria), cytoskeletal filaments (microtubules), supramolecular assemblies (NPC, data that were reused from a previously published dataset)18 and aggregates of the tau protein (Supplementary Fig. 5 and Methods). Performing variable selection on this dataset largely reduced the variance between members of the same class, as shown by the exploratory PCA analysis, but did not substantially improve class separation (Fig. 1e and Supplementary Video 1). We then trained several types of machine learning models using a limited subset of training data (approximately 550 samples per group) and subsequently made predictions using ground truth data that had been excluded from the training dataset. The best results were achieved with the random forest classifier with an average prediction accuracy of 97.1 ± 0.1% across all categories (the prediction accuracy is the average percentage of correctly classified data over all classes; Fig. 1f). The error in the prediction represents the standard deviation over all prediction models, each trained on a different subset of training data, to quantify the robustness of the machine learning training step. The small standard deviation indicates that the results are robust regardless of the choice of the training dataset and given this robustness, the number of models can be drastically reduced in practice. Additionally, the low standard deviations across different classification methods (Supplementary Figs. 3 and 4) also further indicate the robustness of the developed descriptors for the biological quantification.
The largest confusion in the prediction was between lysosomes and mitochondria (94.0 ± 1.2% and 94.8 ± 0.9%, respectively) as these classes are morphologically more similar to each other than to the other classes. We thus selected these two classes for a side-by-side comparison of our approach to the previously developed ASAP (Supplementary Note 3, Supplementary Figs. 6 and 7 and Supplementary Tables 5 and 6). Since ASAP does not provide an automatic model selection, we used the classification method included as default setting (discriminant analysis). Moreover, given that ASAP requires pixelated images, we also tested how its performance depends on the image rendering parameters, in particular the width of the rendering point spread function (PSF) and binary image threshold (Supplementary Fig. 6). The accuracy of the ASAP prediction was dependent on both parameters as expected (Supplementary Fig. 7), and these parameters must therefore be manually optimized to achieve maximal results. Additionally, a full study on the influence of these parameters on ASAP classification accuracy for all available methods was performed. Surprisingly, it revealed that the relationship between rendering PSF width and prediction accuracy was model dependent. Sometimes, a larger rendering PSF size led to more accurate predictions, but for other classification methods, smaller PSF sizes resulted in superior performance (Supplementary Table 5). Moreover, upon comparing ASAP with ECLiPSE using their default settings, ECLiPSE demonstrated a superior average prediction accuracy by 6.1% over ASAP, and ECLiPSE also performed better than the best average prediction accuracy achieved in the optimized study presented in Supplementary Table 5. A similar result was also obtained when utilizing the validation dataset including all five classes (Fig. 1g, left), where the difference in average prediction accuracy is 3.4%, with a maximum difference in prediction accuracy of 13.5% for the more heterogeneous mitochondria class. Furthermore, with ASAP (Supplementary Table 5), a notable disparity in prediction accuracy was observed across various classification methods, whereas this inconsistency was not present when employing ECLiPSE (Supplementary Fig. 3). These results demonstrate several advantages of our approach over existing tools: the ability to use the unbiased raw point cloud data, automated variable selection and automated model selection. These collectively provide improved performance and robustness over previous state-of-the-art methods. Importantly, this improved performance does not come at the expense of computational load, as the speed of ECLiPSE and ASAP were comparable, with the descriptor calculation in ECLiPSE being roughly 10% faster than ASAP on the example data included in the manuscript (see Supplementary Note 3 for a step-by-step comparison between ECLiPSE and ASAP).
Distinct tau and TDP-43 aggregate classes quantified using 2D ECLiPSE
We next applied our approach to two biological applications, acquired using 2D SMLM: clearance of tau protein aggregates (Fig. 2a–e) and detection of TAR DNA-binding protein 43 (TDP-43) proteinopathy morphotypes (Fig. 2f–i). Both applications represent aggregation of proteins into insoluble inclusions that play a role in several neurodegenerative diseases19. Tau is a neuronal microtubule associated protein, which undergoes aberrant posttranslational modifications and aggregation in several tauopathies, including frontotemporal dementia with Parkinsonism linked to chromosome 17 (FTDP-17), Pick’s disease and Alzheimer’s disease20. It has been shown that these tau inclusions are morphologically diverse and disease specific (for example, neurofibrillary tangles (NFTs) in Alzheimer’s disease and Pick’s bodies in Pick’s disease)21. Recent work suggests that there are molecularly and structurally distinct disease-specific tau strains in which the tau protofilaments assume a distinct fold that leads to disease-specific tau aggregation21,22,23,24. However, the relationship between the molecular signatures (for example, posttranslational modifications) of tau proteins, the tau protofilament structure and the morphology of the resulting tau aggregates is not clearly understood. Using SMLM, we previously showed that tau forms morphologically diverse aggregates in an FTDP-17 engineered cell model25. These aggregates were broadly categorized into four classes based on visual inspection: linear fibrils, branched fibrils, preneurofibrillary tangles (pre-NFTs) and NFTs25 (Fig. 2a and Supplementary Fig. 8). Interestingly, these aggregate classes were enriched with hyperphosphorylation marks on distinct tau residues (phospho-Ser202/205 for linear fibrils and NFTs, and phospho-Thr231 for branched fibrils)25. An unbiased classification of these tau aggregates is crucial for obtaining insights into the progression of tau pathology. However, this task is particularly challenging due to the irregular and highly diverse morphological features of these aggregates. The automated, high-throughput and unbiased classification of these previously identified tau aggregates is crucial for obtaining insights into the progression of tau pathology. We elected to use supervised classification for this purpose given the morphological complexity and heterogeneity of tau aggregates (unsupervised classification results gave unsatisfactory results; Supplementary Fig. 2b). We therefore manually annotated a small subset of the data (~15% of the data used in the training/validation step) into the four classes mentioned above based on their morphology and size. We then trained and validated ECLiPSE on these preannotated data, which showed that ECLiPSE accurately discriminates between members of the four different tau morphological classes (Fig. 2b,c). The average prediction accuracy was 89.8 ± 0.4%, which is remarkably high given the high complexity and morphological similarity among the different tau aggregate structures. Additionally, we compared ECLiPSE and ASAP on this complex dataset and found that, at default settings for both methods, ECLiPSE yielded a 12.3% increase in overall prediction power relative to ASAP when accounting for all aggregate classes (Fig. 1g, right and Supplementary Table 6). Notably, ECLiPSE demonstrated an impressive 17.7% improvement in prediction accuracy for some of the most challenging comparisons (Supplementary Table 6). Once again, these results underscore the robustness of our approach in handling challenging biological data where the morphology of structures is complex and spans a broad size scale. To determine the contribution of the automated variable selection to the high performance, we repeated the prediction without variable selection and found that this step indeed improved the average prediction accuracy by 1.5% (Supplementary Fig. 9).
Following this validation, we next examined data in which we induced tau degradation. To do so, we inhibited the expression of soluble tau by removing doxycycline (Dox) in the QBI-293 (Clone 4.1) cells (Fig. 2d and Methods). We observed a decrease in total tau amounts and tau aggregates at days 1–10 after removing Dox (Fig. 2d and Supplementary Fig. 10), which is consistent with previous biochemical analysis26. Previous work had shown that this loss corresponds to tau degradation mediated by both the proteasome and autophagy pathways, but it remains unclear how the different tau aggregate classes are cleared over time. Using ECLiPSE, we predicted the number of the four morphological tau aggregate classes at different time points following Dox removal, which allowed us to determine the timing of degradation of these different classes (Fig. 2e). It is important to note that we used the classification models that were already trained above for predicting tau aggregate classes in this new dataset, without the need for further manual annotation and training. In general, the classification models are trained for a specific biological application and can be applied to new data acquired on a different microscope without retraining, as long as the underlying biology is similar and the acquired data is pointillist in nature. For the clearance of tau aggregates, interestingly, we found that while branched fibrils, pre-NFTs and NFTs showed a rapid and consistent decay starting at day 1 after Dox removal, linear fibrils persisted up to day 5 displaying more delayed degradation kinetics (Fig. 2e). These results suggest that it may be more challenging to clear linear fibrils compared with other morphological tau aggregate classes. Alternatively, it is possible that other aggregate classes are broken down into linear fibrils, resulting in their accumulation over time. Since both autophagy and proteasome pathways are involved in tau aggregate clearance26, in the future, this approach would be useful to determine whether specific pathways clear distinct classes of tau aggregates and the mechanisms of why the linear fibrils have a delayed degradation kinetics.
Finally, we used our approach to discriminate between brain-derived TDP-43 strains, obtained from two patients with frontotemporal lobular degeneration with TDP-43 immunoreactive pathology (FTLD-TDP). In normal conditions, TDP-43 is found in the nucleus and plays an important role in RNA regulation27. In pathology, changes in cleavage and posttranslational modifications of TDP-43 lead to its cytoplasmic accumulation and aggregation into inclusions, similar to tau27. Previous work has demonstrated that extracts derived from the postmortem brain samples of individuals with FTLD-TDP can seed morphologically distinct TDP-43 aggregates in both animal and cell models28. This finding supports the existence of distinct TDP-43 strains that possess unique seeding and spreading properties, which is highly relevant to understanding the pathophysiology of the disease. However, previous work has relied on low resolution images and simple geometric measurements (for example, circularity) to distinguish between ‘globular-like’ versus ‘wisp-like’ TDP-43 aggregates seeded by these different strains. Manual measurements and classification of low-resolution images can be a slow and subjective process. While this approach is useful, super-resolution information is needed to precisely visualize and quantify the morphology of TDP-43 aggregates and robustly classify them. We thus aimed to apply ECLiPSE to determine if this approach could detect morphologically distinct TDP-43 aggregates in cell models. TDP-43 extracted from two distinct postmortem FTLD-TDP brains was used to seed TDP-43 aggregates in cell models. The resulting aggregates were acquired using 2D SMLM (Fig. 2f,g, Supplementary Fig. 11 and Methods). Visual inspection confirmed that one strain led to the formation of more globular-like aggregates (Fig. 2f,g, strain A and Supplementary Fig. 11a), whereas the other strain predominantly seeded aggregated that resembled linear fibrils, previously described as wisps (Fig. 2f, g, strain B and Supplementary Fig. 11b). Extracting shape descriptors enabled us to further confirm these differences in morphology using PCA analysis (Fig. 2h). Finally, we applied the machine learning classification on the clustered localization data of aggregates from the two TDP-43 strains not included in the training data and showed that ECLiPSE predicts the distinct morphologies with very high accuracy (89.9 ± 0.6%) (Fig. 2i). Furthermore, upon examining the aggregates that were accurately or inaccurately classified (Supplementary Fig. 12a), it became evident that the ‘misidentified’ aggregates of one strain exhibited morphological features characteristic of the other strain, and vice versa. We can therefore conclude that although a strain primarily seeds aggregates with a specific morphological trait, a considerable proportion of the seeded aggregates still exhibits morphological similarities to the other strain even when the strains are derived from two distinct postmortem FTLD-TDP brains. In the future, application of ECLiPSE to classify the presence of morphologically distinct protein aggregates in postmortem brain tissue can enable linking aggregate morphology to patient-specific proteinopathy strains.
Three-dimensional ECLiPSE pipeline and validation
While 2D SMLM imaging is straightforward and suitable for most applications, organelles and other subcellular assemblies are often 3D in nature. The ability to use 3D morphological features to classify these structures can potentially improve classification and prediction accuracy. We therefore also extended ECLiPSE to the analysis of 3D SMLM data by extending the shape descriptors to 3D (Supplementary Table 2). To validate 3D ECLiPSE, we acquired 3D SMLM images of lysosomes and mitochondria (Fig. 3a). The mitochondria were measured either with or without prior treatment with antimycin and oligomycin A, which leads to depolarization of mitochondria and subsequent changes to mitochondrial morphology. In particular, mitochondria became rounded up and lost their elongated morphology in response to this treatment. We chose this application as mitochondrial membrane potential is crucial for energy storage during oxidative phosphorylation and loss of mitochondrial membrane potential is deleterious for mitochondrial function, which can be indicative of various pathologies29,30.

a, Representative images for 3D data color coded according to depth using the color scale bar (lysosomes (left), healthy mitochondria (middle) and depolarized mitochondria (right)). The insets show zoomed-in 3D views of the regions within the white boxes. b, Data exploration using PCA after feature extraction using ECLiPSE shows a clear separation between lysosomes and mitochondria (left), but a nonnegligent amount of similarities between healthy and depolarized mitochondria (right). c, The classification results using the partial least squares classifier on the variable selected lysosome versus mitochondria data (left; 98.6 ± 0.1% accuracy) and the random forest classifier on the variable selected healthy versus depolarized mitochondria (right; 75.8 ± 0.6% accuracy). d, The quantification of four biological properties of healthy and depolarized mitochondria indicating that two nonvariable selected properties do not show significant differences (number of localizations and boundary surface curvature (two left-most graphs)) and two variable selected properties show significant differences (major axis and sphericity (two right-most graphs)). For classification in c, only the results obtained by the 100 best models out of 1,000 generated models is shown. Lysosomes: n = 11 cells (three biological replicates); healthy mitochondria: n = 16 cells (four biological replicates); depolarized mitochondria: n = 9 cells (three biological replicates). In d the black line represents the median of the shown biological property and 1% upper and lower values were removed only for visualization purposes. P values were calculated using a two-sided Wilcoxon rank sum test.
Once the 3D descriptors were calculated on these organelles and variable selection was performed, PCA was used to explore the data, followed by supervised classification (Fig. 3b,c, left: lysosomes versus healthy mitochondria and right: healthy versus depolarized mitochondria, and Supplementary Videos 2 and 3). Whereas the 2D classification of the lysosome and mitochondria still carried a small degree of confusion between these organelles, the 3D quantitative features represented in the PCA space showed a much larger degree of separation, which was also reflected in the high classification accuracy obtained (98.6 ± 0.1%). This validates the robust quantification of 3D SMLM data using 3D ECLiPSE. Furthermore, when the mitochondria were compared with one another, there was a certain degree of overlap between the two groups in the PCA space as the treatment was relatively gentle. Nevertheless, an average classification accuracy of 75.8 ± 0.6% was obtained, further validating the use of ECLiPSE for 3D SMLM data. Representative images of wrongly classified mitochondria for either group are shown in Supplementary Fig. 12b. Additionally, using the automatic variable selection step of ECLiPSE (Supplementary Fig. 1c, right), biological properties were identified that showed clear differences between these mitochondria with or without treatment as well as properties that were conserved after treatment (Fig. 3d). For example, the number of localizations and mean surface boundary curvature did not change upon treatment whereas major axis and sphericity decreased as expected from mitochondria becoming rounded upon treatment.
To showcase an alternative approach for classifying these three organelles all together, the results of a hierarchical classification approach are reported in Supplementary Fig. 13, demonstrating the strength of the 3D ECLiPSE quantification to discriminate between morphologically similar lysosomes and depolarized mitochondria. Generally speaking, this hierarchical classification approach is a useful strategy when there is substantial overlap between some groups in the data but not others, which is the case for this combined dataset as shown by the exploratory PCA analysis (Supplementary Fig. 13a and Video 4). More detailed information can be found in Supplementary Note 2.
Discussion
We have developed a robust feature extraction and classification pipeline for structures and organelles in both 2D and 3D SMLM data. ECLiPSE works on point cloud data and is compatible with any SMLM modality. Importantly, ECLiPSE does not require user input and can be run using default settings giving satisfactory and robust results for most applications. Furthermore, it has comparable speed to existing methods for classifying structures in SMLM data, while providing superior performance. However, care should be taken to ensure the high quality of the individual organelles/structures used by ECLiPSE as issues, such as incomplete labeling, insufficient image acquisition or improper segmentation can affect classification performance, especially when working with small datasets. Moreover, when ECLiPSE is employed for supervised classification rather than as an exploratory tool (that is, unsupervised classification to discover groupings in the data), a training dataset is required to build the models, which may necessitate manual annotation of a limited subset of the available data.
We envision that ECLiPSE will be broadly applicable for classifying aggregation of proteins in neurodegenerative diseases to determine patient-specific aggregation prone protein strains, determining changes in organelle morphology in disease states or in response to drug treatment, classifying cell type-specific cytoskeletal architecture and other supramolecular assemblies. In the future, ECLiPSE can be expanded to include assigning pseudo-time stamps to protein aggregates or other biological structures based on their evolving morphological properties. Additionally, ECLiPSE can be adapted to generate pseudo-multicolor super-resolution images by color coding spatially distinct structures within single-color images. The classification step of ECLiPSE can be further expanded to include class modeling to provide a metric for assessing how well a new sample belongs to an existing class based on its similarity to the modeled classes. This addition will further increase robustness and allow identifying low-quality, nonrepresentative or novel samples. Finally, the descriptor calculation step of ECLiPSE can be expanded to enhance compatibility with cellular structures that encompass multiple scales of spatial information within the same structure (for example, chromatin structures).
Overall, we have developed a versatile toolbox for the structural and morphological characterization of super-resolution microscopy data, offering broad applicability and numerous potential applications and future directions.
Methods
A full list of the reagents, the supplier and the article number can be found in Supplementary Data 1.
Two-dimensional sample preparation
Aggregates of tau protein
Stable human embryonic kidney-derived QBI-293 cells (Clone 4.126; kindly provided by Virginia M.-Y. Lee, University of Pennsylvania) expressing full-length human tau T40 (2N4R) carrying the P301L mutation with a green fluorescent protein (GFP) tag were grown in Dulbecco’s modified Eagle medium (DMEM) supplemented with 10% (vol/vol) tetracycline-screened fetal bovine serum, 1% (vol/vol) sodium pyruvate (10 mM), 1% (vol/vol) antibiotic–antimycotic and 20 mM l-glutamine, 5 µg ml−1 blasticidin, 200 µg ml−1 zeocin and maintained in an incubator at 37 °C with 5% CO2. Clone 4.1 was continuously maintained in media containing 100 ng ml−1 Dox (Dox+), or Dox was removed from the culture media for several days to perform experiments (day 1 − Dox, day 2 − Dox and so on) and then fixed. Cells were then incubated with stabilizing buffer (modified tryptone soya broth: 50 mM of PIPES, 5 mM of egtazic acid, 5 mM of MgSO4. 7H2O and 90 mM of KOH in distilled water, pH 7) for 3 min and then methanol (ice cold) was added for 3 min. After that, cells were washed with modified tryptone soya broth twice, followed by blocking for 1 h using 4% (wt/vol) bovine serum albumin (BSA) in phosphate buffer saline (PBS). They were then immunostained with GFP VHH nanobody, recombinant binding protein conjugated with Alexa Fluor 647 in 4% (wt/vol) BSA and 0.2% (vol/vol) Triton X-100 in PBS.
NPC
U-2 OS genome-edited Nup96-mEGPF cells (clone 195, 300174, CLS Cell Lines Service) were grown at 37 °C with 5% CO2 in DMEM, to which MEM nonessential amino acid, GlutaMAX and 10% (vol/vol) PBS was added. The cells were then fixed in PBS containing 4% (vol/vol) paraformaldehyde for 25 min and blocked for 1 h using 3% BSA and 0.2% (vol/vol) Triton X-100 in PBS. The cells were then immunostained with GFP VHH nanobody, recombinant binding protein conjugated with Alexa Fluor 647, and then washed for four times using washing buffer consisting of 0.2% (vol/vol) blocking buffer and 0.05% (vol/vol) Triton X-100 in PBS for 10 min.
NPC data have previously been used by Bohrer et al.18.
Microtubules
BSC-1 cells (CCL-26, American Type Culture Collection (ATCC)) were permeabilized for 10–30 s in buffer containing 80 mM PIPES–KOH pH 7.1, 1 mM egtazic acid, 1 mM MgCl2, 0.5% (vol/vol) Triton X-100 and 10% (vol/vol) glycerol, followed by fixation in PBS containing 3% (vol/vol) paraformaldehyde and 0.1% (vol/vol) glutaraldehyde at 37 °C for 10 min. The fixed cells were washed twice with PBS before incubation with 0.1% (wt/vol) sodium borohydride for 7 min at 25 °C and washed again three times with PBS. Cells were incubated with blocking buffer consisting of PBS containing 10% (vol/vol) donkey serum, 0.2% (vol/vol) Triton X-100 and 0.05 mg ml−1 sonicated salmon sperm single-stranded DNA before incubation with mouse anti-acetylated α-tubulin antibody at 1:100 dilution in blocking buffer for 1 h at 25 °C or overnight at 4 °C. The excess antibody was removed by three washes in 1× wash buffer before incubating with docking-strand-conjugated secondary anti-mouse antibody (docking strand 1) at 1:100 dilution in antibody incubation buffer for 1 h at 25 °C. The excess secondary antibody was removed by three washes with 1× wash buffer and twice with PBS.
Lysosomes
HeLa cells (CRM-CCL-2, ATCC) were fixed in PBS containing 4% (vol/vol) paraformaldehyde warmed to 37 °C for 20 min at 25 °C. Fixed cells were washed three times with PBS, followed by permeabilization in 0.1% (vol/vol) saponin in PBS. Cells were then incubated in blocking buffer consisting of PBS containing 10% (vol/vol) donkey serum, 0.1% (vol/vol) saponin and 0.05 mg ml−1 sonicated salmon sperm single-stranded DNA for 1 h at 25 °C before incubation with mouse anti-LAMP2 at 1:100 dilution in blocking buffer for 1 h at 25 °C. The excess antibody was removed by three washes in 1× wash buffer before incubating with docking strand-conjugated secondary anti-mouse antibody (docking strand 1) at 1:100 dilution in antibody incubation buffer for 1 h at 25 °C. The excess secondary antibody was removed by three washes with 1× wash buffer and twice with PBS.
Mitochondria
Cos-7 cells (CRL-1651, ATCC) were fixed in PBS containing 4% (vol/vol) paraformaldehyde warmed to 37 °C for 20 min at 25 °C. Fixed cells were washed three times with PBS, followed by permeabilization in 0.2% (vol/vol) Triton X-100 in PBS. Cells were then incubated in blocking buffer consisting of PBS containing 10% (vol/vol) donkey serum, 0.2% (vol/vol) Triton X-100 and 0.05 mg ml−1 sonicated salmon sperm single-stranded DNA for 1 h at 25 °C before incubation with rabbit anti-Tom20 at 1:100 dilution in blocking buffer for 1 h at 25 °C or overnight at 4 °C. The excess antibody was removed by three washes in 1× wash buffer before incubating with docking strand-conjugated secondary anti-rabbit antibody (docking strand 2) at 1:25 or 1:100 dilution in antibody incubation buffer for 1 h at 25 °C. The excess secondary antibody was removed by three washes with 1× wash buffer and twice with PBS.
TDP-43
Brain seeds
Human postmortem brains were obtained from the University of Pennsylvania Center for Neurodegenerative Disease Research Brain Bank31. All necessary written informed consent forms were obtained from the patients or their next of kin and confirmed at the time of death. Sarkosyl-insoluble TDP-43 protein from the frozen frontal cortex of patients with FTLD-TDP was prepared as follows32: 1% (vol/vol) Triton X-100 high salt buffer was used to extract the gray matter from the frontal cortex (frozen) and then myelin was removed to create a pellet. This pellet was then treated with benzonase before being extracted with 2% (vol/vol) sarkosyl–high salt buffer32 and subsequently washed and resuspended using Dulbecco’s phosphate-buffered saline (dPBS) by sonication (QSonica). Finally, to remove large protein debris, the sarkosyl-insoluble fraction was spun at 5,000g for 5 min at 4 °C.
Brain-derived TDP-43 extracts from FTLD-TDP cases used in the present study have been characterized previously by Porta et al., 2021 (cases 4 and 12)28.
Cellular TDP-43 aggregation assay
iGFP-NLSm cells (clone #6.B7; generated from QBI-293A cells, source QBI-293A: #AES0506, Quantum) were plated on coated poly-d-lysine chambered coverglass Nunc Labtek II (16,000 cells per well) and transduced after 24 h with 0.5 µg brain-derived TDP-43 extracts (100–300 pg TDP-43 per well)28,32. Briefly, brain extracts were sonicated and diluted with dPBS and mixed with single-use tubes of BioPORTER as a protein delivery reagent28,32. Protein–bioporter complexes were added to the cells and incubated for 4 h. Cells were placed back on fresh medium in presence of 1.0 μg ml−1 Dox and cultured for 3 additional days.
Immunocytochemistry
To remove cytoplasmic soluble proteins and visualize the formation of phosphorylated TDP-43 aggregates, transduced iGFP-NLSm were fixed in 4% paraformaldehyde containing 1% (vol/vol) Triton X-100 for 15 min at room temperature. After blocking, cells were incubated with the mAb phospho-specific p409-410 antibody (1:5,000) overnight at 4 °C. After three washes with dPBS, cells were incubated with Alexa Fluor 405–Alexa Fluor 647 conjugated anti-rabbit secondary antibody at 1:100 dilution in 4% BSA in PBS for 1 h at room temperature in the dark. Stained cells were rinsed with PBS containing 2% BSA and 0.5% (vol/vol) Triton X-100 and then subsequently kept in PBS at 4 °C.
Three-dimensional sample preparation
Lysosomes
HeLa cells (#CRM-CCL-2, ATCC) were fixed and permeabilized as described above. This was followed by blocking in blocking buffer consisting of PBS containing 10% (vol/vol) donkey serum and 0.1% (vol/vol) saponin for 1 h at 25 °C before incubation with mouse anti-LAMP2 at 1:100 dilution in the same blocking buffer for 1 h at 25 °C. Then, the excess antibody was removed by three washes in 1× wash buffer before incubating with Alexa Fluor 405–Alexa Fluor 647 conjugated anti-mouse antibody at 1:100 dilution in antibody incubation buffer for 1 h at 25 °C. The excess secondary antibody was then finally removed by three washes with 1× wash buffer and twice with PBS.
Mitochondria
HeLa cells (#CRM-CCL-2, ATCC) were either treated with 10 µM antimycin A and 10 µM oligomycin A for the depolarized mitochondria, or with the vehicle control (0.02% (vol/vol) ethanol and 0.1% (vol/vol) DMSO) in DMEM media, supplemented with 10% (vol/vol) fetal bovine serum, 1× antibiotic–antimycotic, 1× GlutaMAX and 1 mM sodium pyruvate for 3 h at 37 °C. After, the cells were fixed and permeabilized as described above. This was followed by blocking in blocking buffer consisting of PBS containing 10% (vol/vol) donkey serum and 0.1% (vol/vol) saponin for 1 h at 25 °C before incubation with rabbit anti-Tom20 at 1:100 dilution in the same blocking buffer for 1 h at 25 °C. Then, the excess antibody was removed by three washes in 1 × wash buffer before incubating with Alexa Fluor 405–Alexa Fluor 647 conjugated anti-rabbit antibody at 1:100 dilution in antibody incubation buffer for 1 h at 25 °C. The excess secondary antibody was then finally removed by three washes with 1× wash buffer and twice with PBS.
Two-dimensional and 3D SMLM imaging
Microscope system
All data acquisitions were performed on the Oxford Nanoimager microscope equipped with a 100× oil immersion objective (numerical aperture 1.45) and 405, 488, 561 and 640 nm lasers, 498–551 nm and 576–620 nm band-pass filters in channel 1, 666–705–839 nm band-pass filters in channel 2 and a Hamamatsu Flash 4 V3 scientific complementary metal–oxide–semiconductor (sCMOS) camera.
For 3D image acquisition, the 3D astigmatism lens was engaged during acquisition. Before each measurement, TetraSpeck microspheres were imaged to establish a calibration curve of the z positions (in steps of 10 nm).
STORM imaging
The STORM imaging buffer used contained 50 mM Tris, 10 mM NaCl, 0.5 mg ml−1 glucose oxidase, 40 µg ml−1 catalase, 10% (wt/vol) glucose and 30 mM cysteamine (stock, 77 mg ml−1 of 360 mM HCl) at pH 7.5.
This buffer was used to image the 2D aggregates of tau protein data, the 2D NPC data, the 3D lysosome data and the 3D healthy and depolarized mitochondria data.
DNA-PAINT imaging
ATTO-655 (2D microtubules), Cy3B (2D lysosomes) or Cy3 (2D mitochondria) conjugated Imager 1(P) strands were added to the imaging chamber at 0.5 nM concentration in imaging buffer.
Image acquisition
The 2D aggregates of tau
Images were collected at HiLo illumination angle with a 15 ms exposure time for 50,000 frames at 27 °C with constant laser power.
Two-dimensional NPC
Images were collected at HiLo illumination angle with a 10 ms exposure time for 40,000 frames at 30 °C with constant laser power.
Two-dimensional microtubules
Images were collected at HiLo illumination angle with a 100 ms exposure time for 10,000 frames at 30 °C with constant laser power.
Two-dimensional lysosomes
Images were collected at HiLo illumination angle with a 100 ms exposure time for 25,000 frames at 30 °C with constant laser power.
Two-dimensional mitochondria
Images were collected at HiLo illumination angle with a 10 ms exposure time for 50,000 frames at 30 °C with constant laser power.
Two-dimensional TDP-43
Images were collected at HiLo illumination angle with a 15 ms exposure time for 25,000 frames at 30 °C with constant laser power.
Three-dimensional lysosomes
Images were collected in 3D mode with a 10 ms exposure time for 25,000 frames at 30 °C with constant 405 nm and 640 nm laser power.
Three-dimensional healthy and depolarized mitochondria
Images were collected in 3D mode with a 10 ms exposure time for 25,000 frames at 30 °C with constant 405 nm and 640 nm laser power.
Data analysis
ECLiPSE data requirements
ECLiPSE was implemented in MATLAB (MathWorks) and requires that the data is pointillistic in nature (that is, a list of x/y coordinates (2D data, in pixels or nm) or x/y/z coordinates (3D data, in nm)). Additionally, ECLiPSE requires that the data is already segmented into individual organelles (using your preferred method, such as Voronoi segmentation) and organized as a cell variable. An example of how the data should be organized is available as the example dataset (and accompanying example script).
For morphological and structural property quantification of the SMLM data, these are the only requirements. However, if ECLiPSE is used to classify the data into different groups, it requires the presence of structure (for example, different organelle types or different treatment types and so on) and that this structure can be captured by the ECLiPSE descriptors. Many descriptors are calculated by ECLiPSE, allowing to cover many possibilities, but ECLiPSE also allows to extract only the relevant descriptors using the included variable selection step.
Two-dimensional localization
The localizations of all 2D image acquisitions were generated and drift corrected using the Nanoimager operating and analysis software (Oxford Nanoimager v1.18.3.15066). No additional postprocessing was performed for the 2D data, except for the 2D microtubule data, where localizations beyond 30 nm precision (x and y) and outside the 10–150 nm PSF width (σx/y) range were removed from downstream analysis.
Three-dimensional localization
The localizations of the 3D images was done using the spline-PSF fitter (v201217)33, after establishing a calibration curve on the TetraSpeck microspheres for the z-position using the same spline–PSF fitter. Unreliable and noisy localizations were then removed from consideration by applying the following filters: z-position between −450 and 450 nm (determined to correspond to the reliable z-range according to the calibration curve), PSF width (σx/y) between 50 and 700 nm, photon intensity higher than 700 photons and localization precision (x and y) below 65 nm. Additionally, the localizations for which the algorithm did not converge (that is, exceeded the maximum number of iterations) and/or the fit of the PSF was not reliable (relative log-likelihood <−2) were removed.
Two-dimensional clustering with Voronoi segmentation
All Voronoi segmentation was performed using a custom-made MATLAB code (https://github.com/melikelakadamyali/StormAnalysisSoftware ref. 34).
Aggregates of tau
Tau aggregate localizations were Voronoi segmented and clustered based on a maximum Voronoi area of 410 nm2 and a minimum of five localizations. Clusters with less than 500 localizations were additionally filtered and removed from the analysis to investigate only the larger tau aggregate species (corresponding to larger linear fibrils, branched fibrils, pre-NFTs and NFTs).
NPC
NPC localizations were Voronoi segmented and clustered based on a maximum of 821 nm2 Voronoi area and a minimum of 75 localizations. Clusters with an area smaller than 0.011 µm2 and greater than 0.023 µm2 were discarded as nonspecific background clusters or clustering artifacts.
Microtubules
Microtubule localizations were Voronoi segmented and clustered based on a maximum Voronoi area of 5,475.6 nm2 and a minimum of five localizations. Clusters with an area smaller than 0.041 µm2 were discarded as nonspecific background clusters. Sections of microtubules were selected as regions of interest only from well-separated single microtubules.
Lysosomes
Lysosome localizations were Voronoi segmented and clustered based on a maximum Voronoi area ranging from 342 to 684 nm2 and a minimum of 25 localizations. Lysosome clusters with and area less than 0.123 µm2 and greater than 0.479 µm2 were discarded as nonspecific background clusters or clustering artifacts.
Mitochondria
Mitochondria localizations were Voronoi segmented and clustered based on a maximum Voronoi area ranging from 178 to 2,738 nm2 and a minimum of 25 localizations. Mitochondria clusters with an area less than 0.274 µm2 and greater than 1.03 µm2 were discarded as nonspecific background clusters or clustering artifacts.
TDP-43
The phosphorylated TDP-43 localizations were Voronoi segmented and clustered based on a maximum Voronoi area of 684 nm2 (strain A) and 1.37 µm2 (strain B) and a minimum of 10 localizations. Additionally, clusters with an area smaller than 0.05 µm2 were excluded from the data analysis.
Three-dimensional clustering with Voronoi segmentation
All 3D Voronoi segmentation was performed using Point Cloud Analyst (v0.10.0)17.
Lysosomes
Lysosome localizations were Voronoi segmented following the construction of 3D Voronoi polygons from the localizations. The cut distance was set at 60.0, with a minimum area of 10 and the minimum number of localizations was set at 500. Once the clusters were segmented, the data were filtered by only retaining the clusters with a volume between 106.5 and 108 nm3, to remove incompletely labeled lysosomes and multiple lysosomes that were spatially too close to be distinguishable from one another.
Healthy and depolarized mitochondria
Mitochondria localizations were Voronoi segmented following the construction of 3D Voronoi polygons from the localizations. The cut distance was set at 50.0 with a minimum area of 10 and the minimum number of localizations was set at 500. Once the clusters were segmented, the data were filtered by keeping only the clusters with a volume between 5.5 × 107 and 109 nm3 to remove incompletely labeled mitochondria and mitochondrial networks that were improperly segmented so that properties can be calculated of single mitochondria. Note that both healthy and depolarized mitochondria data were analyzed in the same way to ensure a fair assessment.
Datasets
Two-dimensional validation data
The validation dataset was constructed by combining 1,190 tau, 1,660 NPCs, 1,328 microtubules, 1,106 lysosome and 1,181 mitochondria structures. The tau structures were randomly selected from the manually annotated branched fibrils class of the tau aggregate data.
Two-dimensional aggregates of tau data
This dataset consists of data that originate from three biological replicates for each of the imaged days in the degradation process. The total number of data are as follows: 11,263 clusters for +Dox control (29 cells), 7,461 clusters for day 1 − Dox (27 cells), 7,517 clusters for day 2 − Dox (27 cells), 3,583 clusters for day 3 − Dox (27 cells), 1,981 clusters for day 4 − Dox (29 cells), 1,865 clusters for day 5 − Dox (27 cells) and 288 clusters for day 10 − Dox (28 cells). To construct the dataset for classification model training, a subset of the data (representing ~30% of the data from these days) from the +Dox control, day 1 − Dox and day 2 − Dox imaging days was manually annotated. To avoid bias in this process, the data were randomized and the person annotating was blinded from the origins of the data. After annotation, 963 linear fibrils, 5,218 branched fibrils, 1,098 pre-NFTs and 1,350 NFTs were identified.
TDP-43 data
The TDP-43 data were constructed by combining the data from the two different strains of TDP-43 (strain A and strain B). The total number of clusters was 900 (15 fields of view) and 694 (19 fields of view), respectively.
Three-dimensional lysosome versus mitochondria data
This dataset consists of segmented and clustered data of lysosomes and mitochondria, all imaged using 3D STORM. The total number of clusters was 5,424 clusters (11 cells) for the lysosomes and 1,939 clusters (16 cells) for the mitochondria.
Three-dimensional healthy versus depolarized mitochondria data
This dataset consists of segmented and clustered data of healthy or depolarized mitochondria (that is, treated with 10 µM antimycin A and 10 µM oligomycin A), all imaged using 3D STORM. The total number of clusters was 1,939 clusters (16 cells) for the healthy mitochondria and 3,413 clusters (9 cells) for the depolarized mitochondria.
Statistical analysis
The statistical test performed in Fig. 3d and Supplementary Fig. 10 is a two-sided Wilcoxon rank sum test.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
All datasets used to validate and showcase ECLiPSE are publicly available via figshare at https://doi.org/10.6084/m9.figshare.26499379 (ref. 35). Statistical source data for Fig. 3d are provided with this paper. Additionally, an example dataset of the 2D and 3D validation data on which the ECLiPSE can be tested is provided with the source code via https://github.com/LakGroup/ECLiPSE (ref. 36). Source data are provided with this paper.
Code availability
The source code, example scripts and documentation of ECLiPSE can be found at https://github.com/LakGroup/ECLiPSE (ref. 36).
References
Heald, R. & Cohen-Fix, O. Morphology and function of membrane-bound organelles. Curr. Opin. Cell Biol. 26, 79–86 (2014).
Soto, C. & Pritzkow, S. Protein misfolding, aggregation, and conformational strains in neurodegenerative diseases. Nat. Neurosci. 21, 1332–1340 (2018).
Bond, C., Santiago-Ruiz, A. N., Tang, Q. & Lakadamyali, M. Technological advances in super-resolution microscopy to study cellular processes. Mol. Cell 82, 315–332 (2022).
Lelek, M. et al. Single-molecule localization microscopy. Nat. Rev. Methods Primers 1, 39 (2021).
Broeken, J. et al. Resolution improvement by 3D particle averaging in localization microscopy. Methods Appl. Fluoresc. 3, 014003 (2015).
Heydarian, H. et al. 3D particle averaging and detection of macromolecular symmetry in localization microscopy. Nat. Commun. 12, 2847 (2021).
Heydarian, H. et al. Template-free 2D particle fusion in localization microscopy. Nat. Methods 15, 781–784 (2018).
Loschberger, A. et al. Super-resolution imaging visualizes the eightfold symmetry of gp210 proteins around the nuclear pore complex and resolves the central channel with nanometer resolution. J. Cell Sci. 125, 570–575 (2012).
Schnitzbauer, J. et al. Correlation analysis framework for localization-based superresolution microscopy. Proc. Natl Acad. Sci. USA 115, 3219–3224 (2018).
Huijben, T. A. P. M. et al. Detecting structural heterogeneity in single-molecule localization microscopy data. Nat. Commun. 12, 3791 (2021).
Bender, S. W. B., Dreisler, M. W., Zhang, M., Kæstel-Hansen, J. & Hatzakis, N. S. SEMORE: SEgmentation and MORphological fingErprinting by machine learning automates super-resolution data analysis. Nat. Commun. 15, 1763 (2024).
Wu, Y.-L. et al. Maximum-likelihood model fitting for quantitative analysis of SMLM data. Nat. Methods 20, 139–148 (2023).
Danial, J. S. H. & Garcia-Saez, A. J. Quantitative analysis of super-resolved structures using ASAP. Nat. Methods 16, 711–714 (2019).
Andronov, L., Orlov, I., Lutz, Y., Vonesch, J.-L. & Klaholz, B. P. ClusterViSu, a method for clustering of protein complexes by Voronoi tessellation in super-resolution microscopy. Sci. Rep. 6, 24084 (2016).
Lagache, T. et al. Mapping molecular assemblies with fluorescence microscopy and object-based spatial statistics. Nat. Commun. 9, 698 (2018).
Levet, F. et al. SR-Tesseler: a method to segment and quantify localization-based super-resolution microscopy data. Nat. Methods 12, 1065–1071 (2015).
Levet, F. & Sibarita, J.-B. PoCA: a software platform for point cloud data visualization and quantification. Nat. Methods https://doi.org/10.1038/s41592-023-01811-4 (2023).
Bohrer, C. H. et al. A pairwise distance distribution correction (DDC) algorithm to eliminate blinking-caused artifacts in SMLM. Nat. Methods 18, 669–677 (2021).
Ross, C. A. & Poirier, M. A. Protein aggregation and neurodegenerative disease. Nat. Med. 10, S10–S17 (2004).
Lee, V. M., Goedert, M. & Trojanowski, J. Q. Neurodegenerative tauopathies. Annu. Rev. Neurosci. 24, 1121–1159 (2001).
Chung, D.-E. C., Roemer, S., Petrucelli, L. & Dickson, D. W. Cellular and pathological heterogeneity of primary tauopathies. Mol. Neurodegener. 16, 57 (2021).
Falcon, B. et al. Structures of filaments from Pick’s disease reveal a novel tau protein fold. Nature 561, 137–140 (2018).
Fitzpatrick, A. W. P. et al. Cryo-EM structures of tau filaments from Alzheimer’s disease. Nature 547, 185–190 (2017).
Scheres, S. H., Zhang, W., Falcon, B. & Goedert, M. Cryo-EM structures of tau filaments. Curr. Opin. Struct. Biol. 64, 17–25 (2020).
Gyparaki, M. T. et al. Tau forms oligomeric complexes on microtubules that are distinct from tau aggregates. Proc. Natl Acad. Sci. USA 118, e2021461118 (2021).
Guo, J. L. et al. The dynamics and turnover of tau aggregates in cultured cells: insights into therapies for tauopathies. J. Biol. Chem. 291, 13175–13193 (2016).
de Boer, E. M. J. et al. TDP-43 proteinopathies: a new wave of neurodegenerative diseases. J. Neurol. Neurosurg. Psychiatry 92, 86–95 (2020).
Porta, S. et al. Distinct brain-derived TDP-43 strains from FTLD-TDP subtypes induce diverse morphological TDP-43 aggregates and spreading patterns in vitro and in vivo. Neuropathol. Appl. Neurobiol. 47, 1033–1049 (2021).
Wang, W., Zhao, F., Ma, X., Perry, G. & Zhu, X. Mitochondria dysfunction in the pathogenesis of Alzheimer’s disease: recent advances. Mol. Neurodegener. 15, 30 (2020).
Zorova, L. D. et al. Mitochondrial membrane potential. Anal. Biochem. 552, 50–59 (2018).
Toledo, J. B. et al. A platform for discovery: the University of Pennsylvania Integrated Neurodegenerative Disease Biobank. Alzheimer Dement. 10, 477 (2014).
Porta, S. et al. Patient-derived frontotemporal lobar degeneration brain extracts induce formation and spreading of TDP-43 pathology in vivo. Nat. Commun. 9, 4220 (2018).
Li, Y. et al. Real-time 3D single-molecule localization using experimental point spread functions. Nat. Methods 15, 367–369 (2018).
Hugelier, S. & Arab, A. STORM analysis software. GitHub https://github.com/melikelakadamyali/StormAnalysisSoftware (2023).
Hugelier, S. et al. ECLiPSE data. figshare https://doi.org/10.6084/m9.figshare.26499379 (2024).
Hugelier, S. & Kim, H. H.-S. ECLiPSE code. GitHub https://github.com/LakGroup/ECLiPSE (2024).
Acknowledgments
We thank B. Basak for providing the depolarization treatment drugs. We acknowledge funding from following grants: NIH RO1 GM133842 (M.L.), RM1 GM136511 (M.L.), UO1 DA052715 (M.L.) and RO1 AR079224 (M.L.).
Author information
Authors and Affiliations
Contributions
S.H. and M.L. conceived and designed the research study. S.H. and H.H.-S.K. designed the ECLiPSE algorithms and pipeline. M.T.G., C.B., Q.T., A.N.S.-R. and S.P. performed experimental work and provided data. S.H. performed the analysis with assistance from H.H.-S.K. S.H. assessed method performance. S.H. and M.L. wrote the manuscript with input from all other authors.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Methods thanks the anonymous reviewer(s) for their contribution to the peer review of this work. Primary Handling Editor: Rita Strack, in collaboration with the Nature Methods team.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Supplementary Information
Supplementary Notes 1–3, References, Figs. 1–13, Tables 1–6 and Videos 1–4 (captions only).
Supplementary Data 1
List of all reagents, antibodies and cells used in this manuscript.
Supplementary Video 1
PCA (three principal components are shown) applied to the validation data shows that the developed descriptors can discriminate well between five different classes. The left video shows PCA applied to the nonvariable selected data (using all 68 descriptors; 62.9% explained variance) in which classes are well separated but show a large within-class variance. The video on the right shows PCA applied to the variable selected data (using only 28 descriptors; 76.5% explained variance) demonstrating that the within-class variance is heavily reduced without compromising the separation between the different classes. Blue: tau aggregates; teal green: NPC; light green: microtubules; cyan: lysosomes; orange–yellow: mitochondria.
Supplementary Video 2
PCA (three principal components are shown) applied to the 3D data containing lysosomes and mitochondria shows that the developed descriptors can discriminate well between the lysosomes and the mitochondria. The left video shows PCA applied to the nonvariable selected data (using all 69 descriptors; 57.2% explained variance). The video on the right shows PCA applied to the variable selected data (using only 29 descriptors; 76.5% explained variance). Blue: lysosomes; orange–yellow: mitochondria.
Supplementary Video 3
PCA (three principal components are shown) applied to the 3D data containing healthy and depolarized mitochondria shows that a large percentage of the two different classes overlap, meaning that their biological properties are similar. The left video shows PCA applied to the nonvariable selected data (using all 69 descriptors; 56.6% explained variance). The video on the right shows PCA applied to the variable selected data (using only 29 descriptors; 69.1% explained variance). Orange–yellow: healthy mitochondria; cyan: depolarized mitochondria.
Supplementary Video 4
PCA (three principal components are shown) applied to the 3D data containing lysosomes versus healthy versus depolarized mitochondria shows that the developed descriptors can discriminate well between the lysosomes and the two types of mitochondria, but there is more confusion between the healthy and depolarized mitochondria. The left video shows PCA applied to the nonvariable selected data (using all 69 descriptors; 57.4% explained variance) in which lysosomes and mitochondria are well separated but show a large within-class variance. The video on the right shows PCA applied to the variable selected data (using only 33 descriptors; 72.2% explained variance).
Source data
Source Data Fig. 3d.
Statistical source data for Fig. 3d.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Hugelier, S., Tang, Q., Kim, H.HS. et al. ECLiPSE: a versatile classification technique for structural and morphological analysis of 2D and 3D single-molecule localization microscopy data. Nat Methods 21, 1909–1915 (2024). https://doi.org/10.1038/s41592-024-02414-3
Received:
Accepted:
Published:
Version of record:
Issue date:
DOI: https://doi.org/10.1038/s41592-024-02414-3
This article is cited by
-
Super-resolution microscopy for structural biology
Nature Methods (2025)
