
Abstract
Emerging spatial multiomics technologies provide an increasingly large amount of information content at multiple scales. However, it remains challenging to efficiently represent and harmonize diverse spatial datasets. Here we present Giotto Suite, a suite of modular packages that provides scalable and extensible end-to-end solutions for multiscale and multiomic data analysis, integration and visualization. At its core, Giotto Suite is centered around an innovative data framework, allowing the representation and integration of spatial omics data in a technology-agnostic manner. Giotto Suite integrates molecular, morphology, spatial and annotated feature information to create a responsive and flexible workflow, as demonstrated by applications to several state-of-the-art spatial technologies. Furthermore, Giotto Suite builds upon interoperable interfaces and data structures that bridge the established fields of genomics and spatial data science in R, thereby enabling independent developers to create custom-engineered pipelines. As such, Giotto Suite creates an immersive and multiscale ecosystem for spatial multiomic data analysis.
Similar content being viewed by others


Main
Biological tissues are organized in a hierarchical and structured manner that dictates their specific functions and occur at various scales. Examples are abundant, including the subcellular localization of transcripts across the polarized axis of intestinal enterocytes1; the different roles for individual liver cells dictated by hepatocyte zonation2; multicellular niches comprising macrophages, endothelial and cancer cells that promote metastasis3; or the layered organization of the brain4. As a result, large-scale efforts to systematically create spatial maps from various tissues5 and disease states6 are occurring with increased frequency. With the introduction and advancement of spatial omics technologies, including spatial transcriptomics7,8,9, proteomics10,11,12,13 and others14, researchers have the ability to visualize and analyze various molecular analytes within their tissue context and across different levels of the biological organization. Each of these techniques provides a unique level of resolution and data modality, creating an intricate, multidimensional map of a biological tissue. Regardless of the scale, the individual units of a tissue are single cells whose phenotypes are defined by the interplay of multiple regulatory layers, such as variations in the (epi-)genome, transcriptome, proteome and metabolome. Emerging research highlights the importance of integrating both multiomics data within single cells and multiscale patterns within the tissue. Connecting both intrinsic and extrinsic layers of variation will provide the foundation to understand how the activities of single cells jointly coordinate tissue function and organization in a systems biology manner.
For instance, although spatial transcriptomics allows the exploration of gene expression in a spatial context, spatial proteomics on the same, or immediately adjacent, tissue slice provides complementary insights into the spatial distribution of proteins. Furthermore, most technologies can be combined with imaging, which captures tissue and cellular morphology. Similarly, multiple serial sections from a tissue of interest can be profiled to create a three-dimensional representation15,16. Hence, by integrating these multiscale and multimodal data, a comprehensive systems biology perspective of biological tissue can be attained, offering a two-dimensional or three-dimensional view of biology in vivo. These integrative approaches promise to considerably advance understanding of the complex spatial and functional relationships among cells and tissues, leading to a more precise and nuanced understanding of biological processes in health and disease. However, most software engineering approaches are not purposefully designed to fully capture the increasing complexity associated with multiscale and multimodal datasets in a technology-agnostic manner. Furthermore, tools and methods to access and work with the full breadth of information that is available within these emerging spatial datasets are often either performed in isolated environments or completely lacking. Hence, there is a dire need for further method and data engineering development along with the spatial technologies and datasets.
The programming language R is widely used for analyzing biological data. It has seen a strong increase in users across the fields of biomedical sciences because it offers an array of tools for statistical and genomics analysis. For instance, the Bioconductor project has a central role in more advanced omics analysis and has created a collaborative community for method development and interoperability17. In parallel, the geospatial field has a long history of working with various spatial data, including raster-based and vector-based data types18. However, implementations of the many associated and established spatial data analysis or simulation methods are less frequent within the field of biomedical sciences. In the present study, we leveraged our previous expertise from Giotto19,20 to create a rich and inclusive ecosystem for spatial data analysis and engineering. This ecosystem includes an innovative and immersive spatial multimodal data framework, called Giotto Suite, and modules to represent, analyze and visualize virtually any type of spatial omics data. In addition, it maximizes and connects with other existing exploratory data ecosystems in genomics and spatial data science to provide users with easy workflows for a plethora of spatial downstream analyses. Finally, it facilitates the building of novel methods and applications by external developers that are accessible to large user groups.
Results
Giotto Suite core framework
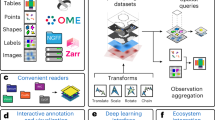
Giotto Suite is a new modular suite of R packages that together create a holistic spatial data analysis ecosystem (Fig. 1a). It is technology agnostic and designed to work with the ever-increasing size and complexity of spatial datasets, including innovative implementations for multimodal and multiresolution dataset representation and integration. It couples easy-to-use workflows on a large variety of spatial omics technologies with extended documentation, including examples for numerous downstream spatial analysis methods and visualizations (Supplementary Fig. 1a). Notably, Giotto Suite has been specifically designed to underscore the Findable, Accessible, Interoperable and Reusable (FAIR) Principles and promote community building in an open-source software environment21.

a, Giotto Suite consists of a suite of integrated R packages supported by a technology-agnostic core data framework. b, Illustration depicting the conceptual and flexible integration of spatial datasets across multiple length scales and data modalities in Giotto Suite. c, Schematic depicting how Giotto Suite’s core framework represents diverse spatial datasets by features (molecular analytes) and spatial units (for example, cell or grid), thereby facilitating various multiscale and multiomics data integration. d, Pictograms summarizing key implementations for scalability, interactivity and interoperability. Panel b was adapted from Servier Medical Art assets. mass spec, mass spectrometry; nn, nearest neighbor.
At the core of Giotto Suite is an innovative data framework that is designed to be agnostic to spatial omics technologies and to maximally capture and represent the multiscale and multimodal biological data (Fig. 1b). This framework is designed based on two core principles. First, any type of spatial omics data can be efficiently used and represented by dedicated data structures, which are built on top of classes within the geospatial ‘terra’ package and emphasize retaining raw information to a maximal degree. These structures include giottoPoints, giottoPolygon and giottoLargeImage that capture, respectively, point, vector-based and image or raster-like information (Fig. 1b and Supplementary Fig. 1b). giottoPoints represents points information (for example, individual transcripts) and their associated spatial coordinates generated by multiplexed in situ hybridization techniques, thus ensuring that subcellular resolution is retained. Similarly, it can also be used to work with sequencing-based methods that generate spatial data from individual arrays with dimensions at the subcellular scale8,22. giottoPolygon is a versatile class used to represent the spatial organization and structures of biological shapes or annotated regions, including biological segmentation results (for example, cell or organelle boundaries), flexible spatial arrays or uniform grid and tessellation structures. Hence, it can be used to represent several layers of information, ranging from (sub)cellular or tissue structures to external biological information (for example, pathology annotations). Finally, almost all spatial technologies include an associated tissue image (for example, hematoxylin and eosin (H&E) image) or build a dataset through sequential imaging (for example, protein intensities for cyclic antibody multiplexing technologies11,23), and this type of data is represented with giottoLargeImages. As such, it provides a dual role by allowing both efficient visualization and extraction of the data.
The second principle focuses on data organization. Giotto Suite implements an approach that can be easily modified to facilitate handling multiple samples and, furthermore, allow their integration across different modalities and/or resolutions (Fig. 1c). A key step herein is to organize data types based on their feature type and spatial unit (Fig. 1c and Supplementary Fig. 1c). The feature type refers to the corresponding data modalities, whereas the spatial unit refers to the underlying spatial structure (for example, nucleus, cell or abstract grid/spot) and is represented by the giottoPolygon class. In this manner, an unlimited number of feature type–spatial unit aggregations can be performed and efficiently stored for further downstream specialized integration methods (Supplementary Fig. 2a,b). In addition, provenance and hierarchy can be encoded within the aggregation or integration steps to build a custom hierarchical tissue structural model. Hence, the core framework in Giotto Suite facilitates the aggregation or integration of multiple different feature types at multiple spatial units at both technical and biological levels.
To support the core framework, the original and convoluted S4 Giotto class was redesigned and supplanted by a lightweight design that underscores the generation of independent S4 subclasses representing different data types and structures. These subclasses are both extensible and easy to maintain. This new design underscores a commitment to good practice object-oriented programming (OOP) principles, providing a platform with increased flexibility for future tooling, visualization and framework development. Additionally, this design is oriented toward ensuring backward compatibility, thereby creating a seamless experience for long-term users and adopters. Moreover, improved and unified accessor and show functions were created as well as ‘terra’ spatial generics for each subobject. Continuous integration and unit testing were added to facilitate contributions from external developers. Finally, considerable efforts to improve scalability, interactivity and interoperability (Fig. 1d) were implemented to augment the new core framework from Giotto Suite, as demonstrated through the following vignettes.
Vignettes for multiscale and multiomic analyses
To demonstrate the ability of Giotto Suite and highlight its spatial technology-agnostic (Supplementary Fig. 3), flexible and scalable implementations, we showcase various applications on specific datasets generated by some of the latest spatial technologies as follows. We also provide a summary table highlighting the Giotto Suite features compared to the previous version of Giotto and other spatial-technology-focused tools19,24,25,26,27,28,29,30 (Supplementary Table 1).
Multiscale and expansive framework
Biological processes occur at multiple scales and resolutions and will lead to different, but related, scientific questions throughout the anatomical hierarchy of the tissue (Fig. 2a). Giotto Suite’s core framework (Fig. 1c) facilitates joint data representation and analysis at any level (Fig. 2a–d). To demonstrate this utility, we use a subset of the MERFISH FFPE Human breast cancer dataset. First, tissue structures are annotated with increasing granularity, such as tissue domains, niches, individual cell types or nuclei (Fig. 2b,d). Next, the data organization facilitates efficient queries between different spatial scales and can also be used to assess how independent clustering results at different scales (for example, nuclei versus cells) are related (Fig. 2e–g).

a, Pictogram depicting the hierarchy of multiple biological scales. b, Sankey plot showing the hierarchical relationships between different spatial units in Giotto Suite. c, Spatial plots showing raw transcript locations (top) and corresponding DAPI image (bottom). d, Spatial plots illustrating annotations at different scales, including nucleus (top left), cell (top right), neighborhood (bottom left) and domain (bottom right). Highlighted annotations correspond with selection in b. e,f, UMAP and spatial plot showing Leiden clusters for cellular (e) and nuclear (f) segmentations. g, Sankey plot showing the Leiden cluster relationships between count aggregation results from cellular (e) and nuclear (f) segmentations. h, Overlay of multiscale annotations of a MERFISH dataset, depicting nuclear lumen and cytoplasmic enriched transcripts for all genes in GSEA terms related to ‘nuclear lumen’ (magenta) and ‘COPII-coated ER to Golgi transport vesicle’ (cyan), respectively. The nuclear boundary is defined with a yellow border, and the cell body is filled in with dark green. i, Overlay of multiscale annotations of a MERFISH dataset, depicting chromosome centromeric region (magenta) and collagen-containing extracellular matrix (cyan) transcripts. j,k, Waterfall plot depicting the log fold enrichment changes for all transcripts, nuclear versus cytoplasm (j) and within versus outside of the cell (k). l,m, GSEA plots showing results for nuclear versus cytoplasm (l) and inside versus outside cells (m). ER, endoplasmic reticulum; FC, fold change; MHC, major histocompatibility complex.
In addition to common analysis pipelines that typically treat each cell as the basic unit, Giotto Suite’s framework allows users to carry out subcellular analysis as well. Individual transcript locations can be queried against any predefined spatial units and used to identify genes or gene sets that are spatially enriched at subcellular organelles, such as nucleus versus cytoplasm (Fig. 2h), or detect transcripts that are found preferentially inside or outside cell boundaries (Fig. 2i). For example, gene set enrichment analysis (GSEA) identified enrichment of nuclear lumen-related genes within the nucleus, whereas genes linked to cytoplasmic membrane structures (that is, Golgi transport and endoplasmic reticulum) were found mostly within the cytoplasm (Fig. 2j,l). When comparing transcript location preferences inside or outside the cell boundaries, genes associated with chromosomal and nuclear structures were enriched within the cells, whereas genes linked to the extracellular matrix are often located outside cells (Fig. 2k,m and Methods). Finally, the Giotto Suite framework facilitates subcellular three-dimensional data analysis. To demonstrate this workflow, we use a subset of the MERFISH mouse brain dataset (version 1.0, May 2021) as an example. Within this dataset, individual transcripts are detected in seven adjacent z stacks, each separated by approximately 1.5 µm (Supplementary Fig. 4a). In addition, each z stack also contains its own cell polygon information, such that transcripts can be accurately assigned to the correct cell polygon (Supplementary Fig. 4a,b). Users can efficiently aggregate the multiple stacks to create a unified single-cell representation (Supplementary Fig. 4c) or use layer-specific information to assess technological or biological differences between layers (Methods and Supplementary Fig. 4d–h).
Registration and segmentation
Spatial omics assays can profile different molecular analytes in thin tissue sections. The use of multiple adjacent tissue sections has become a popular strategy to create three-dimensional spatial datasets15,16 or to integrate multiple technologies31,32,33 that are not directly compatible on the same section slice (Fig. 3a). This approach provides individual laboratories, or larger consortia with multiple groups, the opportunity to create more complex datasets that can unravel the intricacies of cellular decision-making in the context of tissue architecture. Tools for aligning or co-registering one or more tissue slices were developed previously34 or are currently being adapted for spatial biology purposes35,36. However, capturing and working with different spatially aligned datasets, each with its own resolution and data output, is still challenging. The Giotto Suite framework is uniquely suited to work with multiple co-registered spatial datasets due to its flexible handling of different scales and data types. To demonstrate this capacity, we use a spatial multimodal breast cancer dataset from 10x Genomics, consisting of Visium, Xenium, H&E and immunofluorescence, as a concrete example32. First, dataset registration was performed by calculating an affine transform from manually selected landmarks (Supplementary Fig. 5a,b). The transformed spatial datasets, including Xenium, Visium, immunofluorescence and H&E images, were subsequently used to create a multimodal and multiscale Giotto object for downstream analysis, including RNA, protein and morphology information (Fig. 3b). Next, correlations between corresponding spatial protein and RNA levels, such as for the B cell (CD20 encoded by MS4A1) and breast cancer (HER2 encoded by ERBB2) markers, demonstrated moderate to high spatial associations (global bivariate Moran’s I) between the protein and transcript analytes across multiple bin sizes and reached the maximum at 0.48 corresponding to 3,547.2 μm2 binning for MS4A1 gene versus CD20 protein and 0.71 at 221.7 μm2 binning for ERBB2 gene versus HER2 protein (Fig. 3c–e). Similarly, systematic comparisons were performed for transcript counts from genes present in both the Visium (sequencing) and Xenium (in situ sequencing) datasets (Fig. 3f). This analysis indicated that, on average, concordance between both technologies is good (median Pearsonʼs r = 0.414) and that lower correlations are associated with low detection levels (Fig. 3g). For example, the highly expressed FASN gene showed a similar spatial expression pattern for both Visium and Xenium (Fig. 3h); however, the opposite was seen for the low-expressed HDC gene (Fig. 3i).

a, Pictogram showing multimodal co-registration from serial tissue sections. b, Aligned Visium H&E, post-Xenium HER2 immunofluorescence (cyan), Xenium (blue) and Visium (orange) spatial datasets from a single breast cancer sample (10x Genomics). c, Overlay of HER2 protein (grayscale), 10x cell segmentations (cyan) and ERBB2 transcripts (points, red); inset shows zoomed-in region. d, CD20 intensity data (far left), MS4A1 transcript counts (middle left), HER2 intensity data (middle right) and ERBB2 transcript counts (far right) hexbinned at the 8-μm center-to-center level. e, Bivariate Moran’s I of MS4A1 versus CD20 (red) and ERBB2 versus HER2 (blue) calculated for hexbinned values at the 8-μm, 16-μm, 32-μm, 64-μm, 128-μm, 256-μm and 512-μm center-to-center levels. f, Rank of correlation scores between overlapping genes expressed in Xenium and registered Visium data. g, LOESS regression (solid curves) of mean log expression against ranked correlation scores from f. h,i, Spatial feature expression plots for registered Xenium and Visium data for genes FASN (h) and HDC (i). IF, immunofluorescence.
Another key task for many new spatial technologies is cell segmentation and subsequent transcript aggregation. Giotto Suite can read and simultaneously store various segmentation output formats, including mask files or geojson files, and this capacity was illustrated for several common methods37,38,39,40 (Fig. 4a). To assess how segmentation choices might lead to variable outcomes in cell type composition, two complementary analyses were performed. First, a Giotto object was created containing both original segmented cells (that is, provided by 10x Genomics) and cells segmented by Baysor38, which also uses transcript density. Next, transcripts were aggregated within cells from both segmentation results, filtered and co-clustered to obtain joint annotations for both methods (Fig. 4b,c). The results did not only show a difference in the number (Baysor: 223,696 versus original: 164,781) and median micron area size (Baysor: 67.45 versus original: 136.82) (Fig. 4d) of detected cells but also notable differences in cellular composition, such as for stromal cells (Fig. 4b,e,f), which are key players in breast cancer progression41,42. Next, a more controlled analysis was performed by resizing (that is, 25% downscaled) cell segmentation results from Imaging Mass Cytometry (IMC) data43 (Methods and Supplementary Fig. 6a). Unbiased clustering with k-means (k = 9) of both the original and downscaled cells showed a substantial number (11.14%) of cell annotation class switches that are dispersed across the region of interest, with an additional 43.06% of cells being Delaunay neighbors and possibly affected during niche analyses (Supplementary Fig. 6b–f). Together, these observations highlight how Giotto Suite provides a standardized ecosystem to study how segmentation choices or strategies can affect downstream spatial analysis.

a, Spatial plots depicting the segmentation results from a set of established segmentation methods on a subset of the 10x Genomics Xenium Breast Cancer data. b, Spatial plots of the 10x Genomics Xenium Breast Cancer data depicting the segmentation results and cellular annotations for the original (left) and Baysor (right) segmentation methods. c, UMAP plots showing the joint clustering results for both the manufacturer-provided and Baysor cellular segmentations. d, Violin plots showing the surface area of polygons identified by the original and Baysor segmentation methods. e, Bar plot illustrating the percentage of cellular annotations for both the original and Baysor segmentation results. f, Stacked bar plot showing the percentage of cellular annotations for both the original and Baysor segmentation results. NK, natural killer.
Multimodal data analysis
Emerging technologies provide a great opportunity to investigate the relationship between different data modalities. A major addition to Giotto Suite is the support of multimodal data analyses and integration to obtain a more comprehensive characterization of cell states. This is made possible by the addition of specific features facilitated by the core framework. First, multiple data modalities (for example, RNA and protein or RNA and epigenetics) can be stored at the same spatial locations (Fig. 5a). Here, the multiomic data may be either obtained from the same cells or assembled from different cells through image alignment, as described previously (Fig. 3a). Next, efficient structures for combining and re-weighting graph structures from different modalities have been implemented that could support various statistical integration approaches44 (Fig. 5a). To demonstrate how Giotto Suite handles multiomics data, we analyzed the Visium CytAssist Human Tonsil dataset (10x Genomics), containing multiomics information for RNA and surface proteins (Fig. 5b–e), and the spatial RNA assay for transposase-accessible chromatin using sequencing (ATAC–seq) mouse embryo ME13 dataset45 (Fig. 5f–h). By analyzing each data modality separately, we identified 12 cell clusters from RNA and protein profiles, respectively (Fig. 5b,d), whereas we observed 11 and seven clusters when analyzing separately the RNA and ATAC data, respectively (Fig. 5f,g). The difference between the clustering results suggests that these data modalities contain complementary information. By using a weighted nearest neighbor (WNN) method44, both sets of omics information were integrated, thereby obtaining 13 integrated cell clusters for the Visium CytAssist dataset (Fig. 5c) and 17 integrated clusters for the mouse embryo ME13 dataset (Fig. 5h). The multiomic clustering result offers a comprehensive clustering that reflects the information of both modalities, and its spatial pattern resembles the histological structures in the tissue image (Fig. 5c,h). This is further corroborated by the spatial distribution of different cell types, which was estimated from a cell type deconvolution algorithm (Methods). The resulting pattern follows the known architecture of the tonsil (Fig. 5e).

a, Schematic representation and workflow for multiomics integration of spatial transcriptomic datasets. b–e, 10x Genomics Human Tonsil dataset. Analysis at the RNA (b) and protein (d) levels, multiomics integration (c) and DWLS deconvolution (e) using a reference scRNA-seq were performed. f–h, Multiomics analysis of the spatial RNA ATAC–seq mouse embryo ME13 dataset. Spatial distribution of RNA (f), ATAC (g) and integrated modalities-based Leiden clusters (h) is presented. Panel a was created in BioRender.com. CD4_T, CD4 T cell; DWLS, dampened weighted least squares; FDC, follicular dendritic cell; GCBC, germinal center B cell; PDC, plasmacytoid dendritic cell; PC, plasma cell; preBC, pre-B cell; preTC, pre-T cell; NBC_MBC, naïve B cell and memory B cell.
Scalability and tiling
Advancements in spatial technologies have led to ever-growing datasets that can be stored in large databases46,47,48 and can easily contain spatial and expression information from millions of cells. Even high-performance computing infrastructure often struggles to process these datasets with conventional methods. To alleviate the challenge of scalable analysis, several complementary tools are implemented in Giotto Suite, including optimized parallel coding, delayed on-disk calculations and data projection strategies (Fig. 6a). The Giotto Suite package is also built, tested and available on terra.bio as a cloud-based solution to accommodate users who have no access to high-performance computing infrastructure or would like additional scaling (Supplementary Fig. 7a).

a, Pictograms depicting on-disk backends, parallelization and projection to improve scalability within Giotto Suite, including schematic pictograms for different tiling options and tunable parameters. b, Spatial plots showing aggregated Stereo-seq transcript counts for hexagon tiles with width = 400 (left) and width = 100 (right). Inset shows zoomed-in tail region. c, Spatial plots showing Leiden clustering results at low (hexagon, width = 100) and high (hexagon, width = 50) tile resolution for a selected Stereo-seq region. d, Heatmap (top) depicting spatial gene co-expression modules for the selected region in c. Spatial metagene plots (bottom) showing the expression of select spatial co-expression modules. e, Left, visualization illustrating the creation of a pseudo-Visium dataset from a Stereo-seq dataset. Spots with diameter of 55 µm and inter-centroid distance of 100 µm were used to aggregate bin1-level expression. This was followed by downstream spatial processing steps and the generation of Leiden clusters at the spot level. e, Right, spatial deconvolution plot showing the percentage of individual brain cell types for the pseudo-Visium dataset generated from the Stereo-seq subset. DWLS, dampened weighted least squares.
As an illustrating example, we analyzed a Stereo-seq dataset obtained from a mouse embryo at embryonic day 16.5 (‘E16.5_E2S6’) from the Mouse Organogenesis Spatiotemporal Transcriptomic Atlas (MOSTA)22. We focused on a whole sagittal section at its highest resolution (bin1). The dataset contains 378 million transcripts from 292 million bins covering the whole transcriptome. Storing the raw cell gene matrix alone takes about 40 GB of memory. To facilitate working with large spatial data, a database backend was implemented to provide on-disk S4 representations for points and polygons information as dbPointsProxy and dbPolygonProxy, respectively (Fig. 6a). These representations respond to spatial manipulation generics and can also be directly converted into corresponding ‘terra’ objects that are native to the Giotto Suite framework. To increase responsiveness and allow in-memory operations, lazy evaluation is used when possible, and spatial chunking is implemented on these S4 classes. To support interactive usage of these objects, they are also implemented with high-performing plotting methods. Finally, any resulting large aggregated expression matrices are handled by using a delayed HDF5matrix (Methods) on-disk backend (Supplementary Fig. 7b). Users can simultaneously achieve additional computing speed gains by using data projection strategies for initial data exploration. Similar to a standard exploratory data analysis pipeline, it facilitates the optimization of parameters in a computation-efficient and more responsive manner.
In parallel, Giotto Suite also offers flexible tiling and tessellations that can be interpreted by the spatial framework layer (Fig. 1c and Supplementary Fig. 2). Tiling is a popular strategy to analyze large-scale data at multiple scales or resolutions (Fig. 6b–d) but can also be used to create pseudo-datasets with a custom grid configuration to support benchmark analyses. For example, a more granular ‘pseudo-Visium’ dataset was created from a brain region in the Stereo-seq mouse embryo dataset. Next, spatialDWLS was used to deconvolve each pseudo-Visium spot and to assess deconvolution accuracy relative to the original and ground truth Stereo-seq data (Fig. 6e). Together, tiling and scalability implementations provide users with the tools needed to analyze large-scale data at custom resolutions or patterns.
Interactivity and interoperability
To aid users in their exploration of the relationship among molecular, histomorphological and pathological changes within a tissue, we created an integrated and interactive Shiny app for refined annotation and region selection in Giotto Suite (Supplementary Fig. 8a). This tool allows users to manually annotate multiple spatial regions using an HTML widget. Notably, information within the selected regions (for example, cell identities) is immediately available in the Giotto object and can, thus, be directly used in any other downstream analyses (for example, differential expression) (Supplementary Fig. 8b). Additionally, we developed an interactive tool to plot and subset three-dimensional datasets (for example, mouse brain with MERFISH16) that facilitate subsetting slices in the z axis and select cell types or clusters. The subsetted cells can be used for downstream analysis such as the comparison of gene expression patterns across diverse slices (Supplementary Fig. 8c). The integrated Shiny app offers an easy-to-use tool, although its responsiveness decreases when working with large datasets. To overcome this issue, we developed a function that facilitates the exporting of a processed Giotto object to a format compatible with the ‘vitessceR’ package (Methods), which allows not only the selection of regions of interest compatible with downstream analysis in Giotto but also an interactive visualization of the gene expression level, dimension reduction and clustering plots (Supplementary Fig. 8d).
During the past years, many groups developed innovative tools and methods for spatial transcriptomics data analysis49,50. Giotto Suite provides several utilities to facilitate interoperability with these external tools, including rich data structures, accessor functions and plugins. Giotto Suite also provides built-in converter functionality for the R/Bioconductor SpatialExperiment29, Python AnnData51, SpatialData26 and Seurat30 classes and classes used in the open-source spatial sciences field within R52. For example, Giotto Suite users can use the bidirectional converters (Supplementary Fig. 9a–d) to effortlessly use tools developed with Seurat or in Bioconductor and subsequently combine and visualize the final results within the Giotto framework (Supplementary Fig. 9a,b). Similarly, the Giotto to AnnData class converter functions can be used to download and access all datasets within the Spatial Omics DataBase46 (Supplementary Fig. 9c). In addition, a modified AnnData version for the Bento53 pipeline is also available and allows users to perform various RNA localization pattern analyses (Supplementary Fig. 9e–g). Finally, various classes (for example, ‘terra’, ‘sf’ and ‘stars’) and associated methods and statistics used in the R spatial sciences field are easily accessible (Supplementary Fig. 10a) and can be directly created from Giotto (sub)classes through ‘as’ converter functions (Supplementary Fig. 10b). In this manner, other methods and packages can utilize the accessor functions in Giotto Suite to extract information from individual Giotto Suite slots. For example, interpolation methods, such as ‘kriging’ in the ‘gstat’ package, can be easily combined with the Giotto Suite framework to develop unique ways to enhance low-resolution spatial datasets (Supplementary Fig. 10c,d). Altogether, close integration with these other large and established ecosystems represents a substantial extension of Giotto Suite’s capabilities for spatial downstream analysis and visualization.
Discussion
We present a new generation spatial analysis framework, Giotto Suite, which offers a fully integrated and comprehensive suite of tools that were built to provide end-to-end workflows encompassing every critical stage of working with data generated by the latest spatial technologies. In this manner, Giotto Suite differs considerably from the original Giotto package19 and provides an all-in-one solution that is otherwise offered only through a combination of multiple recently developed tools26,27,28,54,55. To summarize Giotto Suite’s features and allow a comprehensive comparison between the previous version of Giotto and other existing tools, we created a table that focuses on key elements for building a spatial data analysis ecosystem, including data representations and support, core and auxiliary functionalities, augmented tools and documentation and interoperability (Supplementary Table 1).
First, Giotto Suite adheres to its technology-agnostic approach by providing data ingestion pipelines that are compatible with any type of raw data structure. At the core of the Giotto Suite framework, we developed innovative data classes to represent biological datasets across multiple spatial and data modalities. In addition, these data classes form their own fully independent subobjects and can, thus, be the starting point for independent spatial workflows, such as providing solutions for imaging-based segmentation-free clustering approaches56. Notably, these core classes make it possible to generate a multimodal hierarchical representation that faithfully reflects the organizational principles of tissue architecture and provides a unified approach for data representation. Similarly, it underlies seamless integration with co-registration methods such that multiple spatial technologies can be jointly queried or analyzed together. In addition, the Giotto Suite framework facilitates handling and joining multiple samples, from either the same or different resolutions. This includes new tools for spatial data registration, mapping and transformations. All the features from the Giotto framework are extended to all the samples in the object. Although Harmony57 is currently available in Giotto to correct heterogeneity when working with multiple samples, the flexibility in the design of the Giotto object facilitates the future integration of additional methods. Notably, too, to accommodate the increasing size of spatial multimodal datasets, we developed GiottoDB, which provides the groundwork that developers and users can use to represent their data through different backends that can scale according to their needs.
Next, Giotto Suite provides users with a modular set of tools for spatial analysis and visualization, including a responsive coding environment and seamless interoperability to both spatial and other genomics software communities. In this manner, Giotto Suite provides direct access to other spatial omics ecosystems, such as those from R/Bioconductor28,29,54 and geospatial52 communities. Although similar effort has been taken in Voyager28, the underlying SpatialFeatureExperiment class currently limits working with multiple spatial layers at different scales or with different data modalities. Hence, Giotto Suite provides a more integrated and inclusive solution that combines the strengths of these methods.
Altogether, Giotto Suite offers a flexible technology-agnostic framework to systematically implement and compare a large number of spatially related methods. The results of such analyses are needed for establishing best practices and data standards. It is also well suited to adopt and adhere to any future minimum information guidelines58 or metadata standards59 that will be necessary for spatial dataset and method harmonization. These activities are especially important for coordinating large-scale collaborative efforts that involve many technology and data analysis experts, such as in various cell atlas projects6,48,60.
Although Giotto Suite aims to provide an easy-to-use and comprehensive workflow, there are limitations, such as the need for using specialized tools for initial raw data preprocessing (for example, spot calling for spatial imaging-based technologies) or the limited support for analyzing some of the new emerging spatial technologies, including spatial metabolomics. Similarly, in future improvements, Giotto Suite would benefit from integration with tools from the rapidly developing field of artificial intelligence, including to help users with support or, alternatively, to augment data analysis by creating integrated data representations.
Finally, we have demonstrated the utility of Giotto Suite through applications to multiple spatial datasets from a diverse set of technologies that span across various spatial resolutions and modalities. Taken together, the core framework and new tools implemented in Giotto Suite provide a powerful solution for a seamless harmonization and integration of diverse datasets.
Methods
Spatial data co-expression processing pipeline
A standard Giotto spatial data processing and analysis workflow was used to visualize spatial clusters starting from a balanced set of genes from spatial co-expression modules. A similar pipeline was used for data generated by NanoString CosMx, Spatial Genomics, DBiT-seq, Spatial CITE-seq, Seq-Scope, Slide-Seq, Open-ST and VisiumHD. More specifically, raw data were loaded, followed by filtering, normalization and detection of spatial genes using binSpect(). Top spatial genes (maximum = 500) were subsequently used to create spatial gene co-expression modules, followed by hierarchical clustering and the selection of an equal representation of spatial gene groups from each co-expression module with getBalancedSpatCoexpressionFeats(). These genes were then used in a typical Giotto Suite pipeline, including dimension reduction (principal component analysis (PCA)), creation of a shared nearest neighbor (SNN) network and Leiden clustering to create spatial expression-informed clusters for each tissue sample.
MERSCOPE data processing and analysis
Two Vizgen MERSCOPE datasets were used. The FFPE Human Immuno-oncology Breast Cancer (patient 1) dataset and the Mouse Brain Receptor Map (version 1.0, May 2021; slice 1, replicate 1). A key difference between both datasets is that the Breast Cancer dataset contains cellular segmentations, whereas the Brain dataset contains nuclear segmentations. In addition, the Brain dataset also provides different segmentation results associated with each of the seven provided z stacks.
For each dataset, associated images were first loaded in as giottoLargeImages and mapped to microns using the provided ‘transforms’ information via createMerscopeLargeImage(), and then cell segmentations and transcript detections were aligned to the same coordinate reference frame. These early MERSCOPE datasets provide cell segmentation information as a directory of thousands of HDF5 files, separated by fields of view. The files were first scanned through to produce an H5TileProxy object that contains a spatially indexed manifest of the files, their contents and a parse function for converting chunks of data read in from the HDF5 files into the format expected downstream. This provided a framework for spatially chunked access to the polygon information. For visualization purposes and faster processing of the analysis examples, the datasets were spatially subset to 500 × 500-µm regions. The Breast Cancer dataset was subset to 5,500, 6,000, 3,500 and 4,000, and the Mouse Brain dataset was subset to 6,700, 7,200, 3,100 and 3,600 (x_min, x_max, y_min and y_max). Because the vector data (polygons and points) have inverted y values relative to the image, the spatial selection was first flipped across the image y midline and then used to select the data. The resulting data were then flipped back across the image’s midline and then ingested as giottoPolygon and giottoPoints objects, respectively. Next, Giotto objects were then constructed from the giottoPoints, giottoPolygon and giottoLargeImage objects using createGiottoObject(). For the Breast Cancer dataset, only one of the polygon layers was loaded in because they are all identical. For the Mouse Brain dataset, all seven of the polygon z layers were loaded. Next, addSpatialCentroidLocations() was used to calculate a set of spatial location coordinates for each of the polygons. Counts matrices were then created for each set of polygons in each dataset by first running calculateOverlapRaster() to determine the points data overlapped by the polygons and then overlapToMatrix() to convert the overlaps information into matrices. At this step, aggregateStacks() was run on the Mouse Brain dataset to combine the expression and spatial information content of its seven z layers into a single spatial unit called ‘aggregate’. This step also creates a new set of matching ‘aggregate’ polygon information, with vertices defined as the combined outer boundary across layers. From here, both datasets were processed using the standard steps, including filtering, normalization, dimension reduction and clustering.
Multiscale analysis and visualization
To demonstrate data representation and analysis at multiple spatial scales, we started with a subset of the Vizgen FFPE Breast Cancer dataset, which already had a set of machine-based cell boundary segmentations. We added a set of nuclear segmentations by hand using QuPath61. Transcripts aggregation, filtering, normalization, PCA, uniform manifold approximation and projection (UMAP), nearest neighbors network creation and Leiden clustering (res = 0.7) were performed independently for both nucleus and cell spatial units. Additional spatial scales or spatial units for cell neighborhoods and domains were calculated based on cell-level expression information. Cell neighborhoods were defined by finding local niches composed of cells with similar Leiden clustering results using calculateSpatCellMetadataProportions() and k-means (k = 6). Spatial domains were detected using hidden Markov random field (HMRF)62 on genes selected from spatial co-expression modules. spatialSplitCluster() was then run to further split the resulting annotations when spatial regions were non-contiguous. Spatial intersections were used to establish relationships between spatial units (for example, which nucleus is owned by which cell). A Sankey plot was used to visualize the hierarchical relationships (cells to nuclei) and Leiden annotation concordance across the previously described spatial units.
Transcript location GSEA
Comparative analysis of transcript abundance differences within the cell-versus-extracellular and nuclear-versus-cytoplasmic segmented compartments was performed to demonstrate spatial querying. Intracellular was defined as all transcript detections that overlap within the Vizgen-provided cell segmentation annotations and extracellular as those that are outside. Nuclear was the detections that were within the manual nuclear annotations, and cytoplasmic was those that were within the cell but outside the nuclear. GSEA62 analysis was performed using clusterProfiler via ‘fgsea’63, ‘biomaRt’ and ‘org.Hs.eg.db’ packages. The results were then plotted using the ‘enrichplot’ package’s dotplot() function.
z-stack variance analysis
To illustrate variance in transcript abundance between different z-stack layers, a subset of the Vizgen Mouse Brain Receptor Map dataset was used. The polygons from each of the seven provided z layers were rasterized at 1,000 × 1,000 with the rast() function from ‘terra’. To assess gene expression variance within and between z-stack layers, transcript locations for each gene were first rasterized and summed within pixels at a predetermined pixel size (20 × 20). Hence, each gene is represented by seven aligned images, and these images were then used to calculate the coefficient of variation (COV) across the z-stack layers for each gene. To identify genes that display higher than expected variation, the COV was plotted against the summed counts.
10x Genomics data processing and analysis
To demonstrate the different steps of co-registering spatial multimodal datasets, the data associated with the 10x Genomics Breast Cancer dataset were used. These Xenium instrument data include subcellular transcript locations, polygon information, immunofluorescence and post-Xenium H&E images from the same tissue section. From an adjacent tissue section, the Visium CytAssist instrument was used to generate gene expression data with an H&E image.
Co-registering multimodal dataset
The Janesick adjacent Visium CytAssist dataset was registered to the Xenium replicate 1 dataset. First, the Visium data were read in with createGiottoVisiumObject(), and the Xenium data were read in with createGiottoXeniumObject(). Next, the additional post-Xenium immunofluorescence images were first converted from .ome.tif to .tif using ometif_to_tif() and then loaded using read10xAffineImage() with the provided alignment information in imagealignment.csv. These images were represented as giottoAffineImages, which link to the full-size image file on disk, without fully loading it, in the same way as giottoLargeImages, but additionally carry affine transformation information that is applied only when needed. Then, the images were attached to the Xenium object with setGiotto(), and the paired H&E and DAPI image objects were subsequently extracted from the Visium and Xenium Giotto objects, respectively. Using these images, interactiveLandmarkSelection() was used to manually select 14 pairs of landmarks with the Visium H&E image as the source and the Xenium DAPI image as the target image. Finally, calculateAffineMatrixFromLandmarks() was used to convert the source and target landmark pairs to a 3 × 3 affine transformation matrix. This affine transform was then applied to the Visium Giotto object using affine() to match the spatial coordinate space of the Xenium Giotto object.
Cell segmentation
The 10x Genomics Xenium Pre-Release Breast Cancer dataset was segmented with different methods to simulate different scenarios or allow method comparison within the Giotto Suite framework. The original segmentation data from the Xenium output were directly read into a giottoPolygon object using the piecewise loading importXenium() utility function. For image segmentation methods, the post-Xenium aligned immunofluorescence image was loaded as giottoAffineImage through importXenium() and cropped to the area of interest. The delayed affine transformation encoded in the giottoAffineImage was then forced with doDeferred(), converting it to giottoLargeImage, after which the image was exported as .tif with writeRaster() for downstream segmentation tasks.
For Baysor38 (version 0.5.2 with Julia version 1.7.3), the Xenium transcripts information was first loaded in and filtered to Phred-scaled quality value (QV) ≥ 20 via importXenium() as a data.table. The control probe detections were then filtered out by checking against known keywords, and any cell ID values equal to −1 were changed to 0. The resulting table was then written to .csv with data.table’s fwrite(). The generated transcripts .csv file was used for Baysor segmentation in combination with a reference segmentation—that is, the original segmentation results provided by 10x Genomics for initialization. The minimum transcripts per cell parameter was set to 30, and the confidence for the prior segmentation was set to 0.5. The resulting Baysor polygons were saved as a GeometryCollection.json file and used as input for the createGiottoPolygonsFromGeoJSON() function. Cellpose37 (version 3.1.0) was used to segment the Xenium immunofluorescence data. The immunofluorescence image was segmented using the cyto3 model from Cellpose out-of-the-box using the doCellposeSegmentation() wrapper function. The HER2 channel was defined as a cell boundary stain for the model, whereas the DAPI channel was used as the nuclei stain. No further parameters were modified. The mask output image was provided as input to the createGiottoPolygonsFromMask() function. For StarDist40, the immunofluorescence image’s DAPI channel was normalized with csbdeep64 (version 0.8.1) and then segmented using the 2D_demo model from StarDist (version 0.9.1) via the doStardistSegmentation() wrapper function. The results were written out as a mask image and used as input to createGiottoPolygonsFromMask(). The resulting polygons were nuclei segmentations, so they were expanded by 5 µm using buffer() to better represent the whole cell.
For Mesmer39, the immunofluorescence image was segmented using deepcell (version 0.12.10) via the doMesmerSegmentation() wrapper function. The HER2 channel was used as the cell boundary channel, and the DAPI channel was used as the nuclei stain. The micron scaling (multiplicative factor to convert pixels to microns) value was also provided. The output was a mask image that was then provided to createGiottoPolygonsFromMask().
Spatial correlation analysis of molecular modalities from adjacent tissue slices
Transcript and protein co-localization analysis
Xenium data were read in as a Giotto object, and post-Xenium DAPI, CD20 and HER2 immunofluorescence images were converted into single-channel .tif files, loaded as giottoAffineImage and then attached as previously described. Hexagonal giottoPolygons were then made across the whole spatial extent of the Xenium transcript coordinates using the function tessellate() with center-to-center distances of 8 μm, 16 μm, 32 μm, 64 μm, 128 μm, 256 μm and 512 μm. The functions calculateOverlap() and overlapToMatrix() were used to generate spatial bin-by-feature matrices of image intensities from the CD20 and HER2 images and transcript counts (including MS4A1 and ERBB2) for each of the tessellations. More specifically, and to improve performance and avoid alteration of raw data, the polygons are transformed using the inverse affine transform and then used to extract intensity values from the untransformed image files. The resulting hexbin giottoPolygons and aggregated matrices were then appended to the Giotto object with setGiotto(). Spatial relationships between adjacent bins were set up using createSpatialNetwork() to create k-nearest neighbor (KNN) networks (k = 6). A maximum connection distance of 1.1 times the center-to-center distance of that hexbin level was enforced, preventing incorrect connections at the edges of the tessellation. This network was then converted into a weight matrix using createSpatialWeightMatrix(). The raw numerical values of the RNA–protein feature pairs (MS4A1 versus CD20 and ERBB2 versus HER2) were then extracted with spatValues(), scaled between 0 and 1 and passed along with the weight matrix to callSpdep() to find the global bivariate Moran’s I statistic, which is implemented in spdep as moran_bv(). The above was repeated for each of the different bin resolutions, providing an understanding of spatial association between the two modalities at multiple length scales.
Xenium-aggregated transcripts and Visium
The Visium dataset was registered to the Xenium dataset as described above. The Visium spot polygons were then extracted from the Visium dataset and appended to the Xenium dataset using setGiotto(). The newly appended Visium polygons were used to aggregate the Xenium transcript detections via calculateOverlap() and overlapToMatrix() to generate a pseudo-Visium count matrix. Visium and Xenium Giotto objects were filtered to remove empty spots and undetected features. The two Giotto objects were then further subsetted for only the cell IDs that survived the filtering step on both sides, ensuring that downstream steps can be performed on spots shared by both datasets. The Pearson correlation statistic (r) was calculated using the log2-normalized values for all genes shared in the Visium and Xenium datasets. To assess the association between correlation scores and gene expression levels, we computed the locally estimated scatterplot smoothing (LOESS) trend for the log2 expression value of both Xenium and Visium assays against the rank of the correlation of intersecting genes.
Joint clustering and comparing of multiple segmentation results
Creation and processing of a multisegmentation Giotto object
Two Giotto objects were created using the 10x Genomics Xenium Pre-Release Breast Cancer dataset. The first Giotto object was created using the original cell segmentation data, and the second was created using the Baysor segmentation data read in with createGiottoPolygonsFromGeoJSON(). The gene-by-cell expression matrix for each Giotto object was calculated using the functions calculateOverlap() and overlapToMatrix(). Next, the two Giotto objects were combined using the function joinGiottoObjects(), which is typically used for analyzing multiple samples by appending sample-specific names (for example, Baysor) to cell IDs from both objects. The combined Giotto object was then processed following standard processing steps, including filtering and normalization.
Clustering and annotation
PCA and low-dimensional UMAP representations were first calculated using runPCAprojection() and runUMAPprojection() (with the top 15 principal components), respectively. To speed up exploratory data analysis and cell clustering, these were run with a randomized subset of 25% of all cells. A nearest neighbor network (createNearestNetwork()) was then created using the top 15 principal components and a k of 30. Leiden clustering was then performed with the doLeidenClusterIgraph() function, with 100 iterations and a resolution parameter of 0.15. Finally, individual Leiden clusters from the whole Giotto object were annotated based on differential gene expression information (that is, known markers) and visual overlap with the original annotations provided by 10x Genomics.
Pairwise cluster results comparison
The polygon area of each segmentation method was calculated by running addStatistics() on the joint Giotto object. Cell type and list ID metadata (which distinguishes which cells belong to which segmentation method) were extracted in tabular format from the joint object using spatValues(). This table was then used to determine cell type occurrence frequency by segmentation method. Percentages of each cell type were calculated by dividing the number of occurrences per cell type by the number of polygons in the segmentation method.
Imaging mass cytometry data processing and analysis
The human lymph node FFPE IMC dataset contains 40 images, 36 of which are for protein targets. First, the 193Ir intensity image, depicting nucleic acids, was used for segmentation in QuPath61, using the positive cell detection functionality (non-default parameters: Background Radius, 5; Minimum Area, 5; Maximum Area, 18; Threshold, 5; Smooth Boundaries, No). The polygonal data were exported from QuPath as a .geojson file. Intensity images for the 36 proteins were used to create a subcellular Giotto object using createGiottoObjectSubcellular(). The subcellular locations in the Giotto object were subsetted, and centroids were calculated for all polygons within the default spatial unit ‘cell’.
Polygon rescaling analysis
The function rescalePolygons() was used to create a new set of polygons stored in a separate spatial unit, ‘smallcell’ (arguments used: poly_info = ‘cell’, name = ‘smallcell’, fx = 0.75, fy = 0.75, calculate_centroids = TRUE). For both spatial units, overlaps between polygon and feature information (that is, intensities for each antibody representing the genes previously listed) were calculated using calculateOverlap(). The features were aggregated by summation per polygon to create an expression matrix using overlapToMatrix(). Using the function filterGiotto(), both spatial units in the Giotto object were filtered using the same parameters (expression_threshold = 0, feat_det_in_min_cells = 10, min_det_feats_per_cell = 2). The expression threshold was left at 0 in order to preserve the maximum amount of expression information. Expression matrices for each spatial unit were normalized with normalizeGiotto() via the pearson_resid method, and PCA dimension reductions were performed using runPCA() on the sets of normalized values (scale_unit = FALSE, center = FALSE, ncp = 20). UMAPs were calculated with runUMAP() for each spatial unit (dimensions_to_use = 1:10), and a shared nearest network was created with the createNearestNetwork() function (dimensions_to_use = 1:10). k-means clustering was performed on the normalized values of each spatial unit with doKmeans() and k = 9 for each spatial unit. Between the spatial units, clusters were determined to be corresponding based on the percentage overlap in cell IDs in each cluster. Clusters with maximal overlapping cell IDs were determined to be matching clusters between spatial units ‘cell’ and ‘smallcell’. The results were visualized with spatInSituPlotPoints().
Multiomics data processing and analysis
Multiomics integration
To perform multiomics data integration, individual modality dimension reduction (PCAs or latent semantic indexing (LSI)) and KNNs were used for calculating the weighted matrix and cell-specific modality weights using the method of Hao et al.44 integrated into the function runWNN(). Both results were stored within the multiomics slot of the Giotto object. The resulting weighted matrix was used for calculating an integrated KNN graph by using the function runIntegratedUMAP(). Both weighted matrix and integrated KNN graphs were used for calculating the integrated UMAP that was stored within the dimension reduction slot of the Giotto Suite object, and using both RNA and protein feature type names. Finally, the integrated KNN graph was used to calculate integrated Leiden clusters using the standard Giotto function doLeidenCluster(). The resulting cluster IDs were stored in the cell metadata slot.
Analysis of Visium CytAssist Human Tonsil dataset
For the RNA modality, a minimum of 1,000 features per cell, 50 cells with a feature and an expression threshold of 1 were used for filtering, resulting in the removal of three out of 4,194 cells and 230 out of 18,977 genes. For the protein modality, a minimum of 1 feature per cell, 50 cells with a feature and an expression threshold of 1 were used; no cells or proteins were removed. Both RNA and protein expression matrices were normalized and scaled using logbase = 2, log_offset = 1 and scalefactor = 6,000. The calculation of highly variable features (HVFs) was performed for the RNA modality using z-score threshold = 1.5. The HVFs were used for the calculation of the RNA modality PCA, and all 35 proteins were used for protein PCA; 100 principal components were calculated for both individual modalities. The first 10 principal components were used for the calculation of individual UMAP, t-distributed stochastic neighbor embedding (t-SNE) and SNNs. The resulting SNN graphs were used for calculating Leiden clusters with a resolution of 1. The individual modality PCAs were used for integrating RNA and protein modalities, and then the integrated UMAP and clusters were calculated by running Giotto functions runWNN(), runIntegratedUMAP() and doLeidenCluster(), respectively. The annotated single-cell dataset from the Atlas of Cells in the Human Tonsil at annotation level 1 and the integrated Leiden clusters were used for the deconvolution of the spatial transcriptomics dataset using the spatialDWLS65 method.
Analysis of spatial RNA/ATAC–seq mouse embryo ME13 dataset
For the RNA modality, a minimum of 50 features per cell, 50 cells with a feature and an expression threshold of 1 were used for filtering, resulting in the removal of zero out of 2,187 cells and 10,774 out of 20,900 genes. The expression matrix was normalized and scaled using logbase = 2, log_offset = 1 and scalefactor = 6,000. All genes were used to calculate 100 principal components without calculation of HVFs. The first 10 principal components were used for the calculation of individual UMAP and SNNs. The resulting SNN graph was used for calculating Leiden clusters with a resolution of 0.8. For the ATAC modality, the ArchR66 package was used to preprocess and align the fragments with the mouse genome mm10, using a minimum transcription start site (TSS) enrichment score = 0, minimum number of fragments = 0, maximum number of fragments = 1 × 107, tileSize = 5,000, offsetPlus = 0 and offsetMinus = 0. The resulting ATAC TileMatrix and RNA count matrix were used to create a multiomics Giotto object. The TileMatrix was used to reduce dimensions using the LSI method integrated in the Giotto function runIterativeLSI() using the following parameters: lsi_method = 2, resolution = 0.2, sample_cells_pre = 20,000, var_features = 30,000 and dims = 30. The first 10 LSI values were used for the calculation of individual UMAP and SNNs. The resulting SNN graph was used for calculating Leiden clusters with a resolution of 0.6. The first 10 principal components and LSI values were used to calculate an individual modality KNN network using k = 10. The individual modality PCA/LSI and KNN were used for integrating RNA and ATAC modalities, and then the integrated UMAP and clusters with a resolution of 1.2 were calculated by running Giotto functions runWNN(), runIntegratedUMAP() and doLeidenCluster(), respectively.
Stereo-seq data processing and analysis
The bin1 spatial matrix .tsv.gz file was downloaded from the China National GeneBank (CNGB) website (see ‘Data availability’ and ‘Code availability’ for details). A simple reader function for chunkwise ingestion was declared with GiottoDB’s provided stream_reader_fread(), with a callback function to format chunks to have the correct colnames. The matrix was then read into the DuckDB67 backend as a dbPointsProxy using dbvect(), with the stop condition being to end when a chunk with zero rows is encountered. tesselate() was then used to generate two sets of tiled hexagon bin polygons, with widths of 400 and 100 units (200 µm and 50 µm, respectively), which were also read into the backend as dbPolygonProxy using dbvect(). Finally, calculateOverlap() was run on the dbPolygonProxy and dbPointsProxy objects, and the overlap results were written to disk using overlapToMatrix() as HDF5Matrix count matrices. The matrices and the tesselated polygons were added to a Giotto object. The expression information was filtered and normalized with filterGiotto() and normalizeGiotto(), and then calculateHVF() was used to find highly variable genes using a randomly sampled 10% subset of the dataset. Downstream, further projection strategies were used to speed up analysis. runPCAprojection(), runUMAPprojection() and doClusterProjection() were performed with 25% subsets of the dataset, after which the results were projected onto the rest of the dataset. See the script for further details and parameters.
Resolution increase, pseudo-aggregation and deconvolution
A representative region of interest was selected in the mouse brain with subsetGiottoLocs() and coordinates x_min = 3,826, x_max = 5,826, y_min = 11,975 and y_max = 14,975. Next, a similar workflow as for the whole embryo dataset was followed, except with hexagon bin polygons with width of 50 units (25 µm). Spatially variable genes (SVGs) were then detected to improve clustering results. First, to identify spatial co-expression modules, a spatial KNN network was created with createSpatialNetwork() and parameters k = 6 and maximum_distance_knn = 60. This network was used together with binSpect() to identify the top SVGs. These SVGs were used as input for detectSpatialCorFeats() to compute spatial co-expression modules and followed by hierarchical clustering with clusterSpatialCorFeats() and k = 15 to classify the SVGs into 15 spatial co-expression modules. Individual spatial co-expression modules were converted to metafeatures (that is, metagenes) with createMetafeats() and visualized with spatInSituPlotPoints(). Up to the top 20 SVGs within each module were then selected via getBalancedSpatCoexpressionFeats() to prevent highly represented spatial modules from dominating. The PCA space was then regenerated using only the selected SVGs. A nearest neighbors network was generated with createNearestNetwork(), using the first 25 principal components and k = 20. Subsequent UMAP and Leiden clustering (resolution = 1.2) were performed with this network to produce spatially informed clustering. This clustering was further refined using niche clustering via calculateSpatCellMetadataProportions() and simple kmeans() (k = 15). Next, makePseudoVisium() was used to generate a scale-accurate Visium spot array within the region of interest. For deconvolution, spatialDWLS65 was run using runDWLSDeconv() on the pseudo-Visium spots using a previously published developmental mouse brain single-cell atlas as a reference dataset for cell typing with the blood cell type removed (see ‘Data availability’ and ‘Code availability’ for sources). Top single-cell markers in this dataset were identified using the findMarkers_one_vs_all() function in Giotto Suite using the scran method.
Scalability implementations
Integration of Giotto in the cloud
We developed a Docker image compatible with terra.bio available at giottopackage/terra_jupyter_suite_modular:latest. The image contains the latest version of Giotto 4.2.1, which allows running interactive Jupyter notebooks within a customized cloud environment. We also developed a startup script for running the RStudio app on terra.bio with an automatic Giotto installation, available at https://github.com/drieslab/Giotto_Suite_manuscript.
DelayedArray and future.apply implementation
We used the HDF5Array package to create a DelayedArray68 backend. For integrating the HDF5 backend, the function createGiottoObject() was adapted to write expression matrices within an on-disk .h5 file instead of the Giotto object, and a string with the internal path in the h5 file leading to the matrix was stored in the expression slot. Giotto ‘getter’ and ‘setter’ functions were adapted to automatically identify the HDF5 backend and manage expression information to/from the on-disk file using the chihaya package. Additionally, the ScaledMatrix package was used for storing and reading scaled matrices. Analysis functions were adapted using the DelayedMatrixStats package to handle DelayedArray calculations. To allow parallel operations, the future.apply package has been implemented, and users can follow the plan() guidelines to use the processing (for example, sequential or multisession) backend of choice.
Subsampling and projection strategies
To facilitate large-scale PCA, runPCAprojection() and runPCAprojectionBatch() were implemented. First, the expression matrix is subsetted by taking a user-defined percentage of all spatial units (for example, cells) in a random sampling manner. Next, the downscaled expression matrix is used for PCA using the standard implementations in Giotto Suite, and results are converted to an S3 prcomp class. This is then followed by the projection of the remaining expression matrix with predict.prcomp() to the same PCA space. A similar approach is followed by the batch approach, except that multiple batches will be performed and aggregated for a final PCA result. To compute UMAP coordinates from a large-scale spatial dataset, runUMAPprojection() was implemented. First, the expression matrix is subsetted by taking a user-defined percentage of all spatial units (for example, cells) in a random sampling manner. Next, the downscaled expression matrix is used with runUMAP(), and the UMAP model is stored. This UMAP model is subsequently used to transform the remaining expression matrix spatial units (for example, cells) into the same UMAP coordinate space. Finally, doClusterProjection() is implemented to transfer annotation labels from spatial units (for example, cells) to other unseen spatial units that share an identical dimension reduction space. For this approach, users can create a smaller Giotto object with one of the convenience functions (for example, subsetGiotto()), cluster the data with their preferred method (for example, k-means, hierarchical, Leiden, etc.) and subsequently provide both the original Giotto object (target) and the smaller cluster Giotto object (source) to transfer the obtained labels using a fast KNN approach as implemented by the ‘FNN’ package.
Database and spatial chunking approach
dbPointsProxy and dbPolygonProxy are S4 structures that contain dbplyr/dplyr tibbles connected to a database via Database Interface (DBI). They can be created using dbvect() from specifically formatted data.frames, ‘terra’ SpatVectors and filepath inputs. In these analyses, the database used was DuckDB. On backend creation, connection details are stored in a package-level environment, from which objects can independently retrieve connections, allowing them to function in a standalone manner and be encapsulated within larger objects similarly to normal in-memory objects. Connection handling for these objects is then abstracted away through ‘pool’. These representations respond to spatial manipulation generics and can be pulled into memory as their corresponding ‘terra’ objects using as.spatvector(), making them convenient proxies for the data that they contain. Spatially chunked processing is implemented through chunkSpatApply(), which plans and pulls out chunks of data for up to two inputs and then applies a supplied function, writing the results back to the database. Individual geometries are selected during the chunking process using a \(\min {\mathrm{value}}\le x < \max {\mathrm{value}}\) filter, ensuring that entities are not double selected. For dbPolygonProxy specifically, geometries are selected based on the x and y means of the vertices of each polygon. Utilities for table generation with constraints and chunkwise data ingestion into the database are also provided and allow flexible use of different reader, writer and callback functions.
Interactive tools
Interactive polygon selection
We developed a Shiny gadget that launches a local application to interactively draw multiple regions of interest over a Giotto spatial plot, by running the function plotInteractivePolygons(). The spatial plot may or not contain a tissue image in the background. The application provides the flexibility to assign custom names for each region of interest as well as multiple or individual colors for the polygons. The tool also provides slide bars across the x and y axes to zoom in and out over the image. The reactivity feature of this interactive plot allows users to draw polygons on the images as well as simultaneously retrieve the corresponding x and y coordinates to a user-defined variable within the R console. The resulting table with coordinates can subsequently be used or integrated within the Giotto Suite object by running the functions addGiottoPolygons() and addPolygonCells(). Polygon information can be used for downstream analysis such as the comparison of cell type abundance or gene expression patterns within the drawn areas by running the functions compareCellAbundance() and comparePolygonExpression(), respectively.
Interactive landmark selection
We implemented a Shiny gadget that accepts a pair of source and target objects and allows manual placement of landmarks by running interactiveLandmarkSelection(). These objects can be a giottoLargeImage-inheriting object or a ggplot2 gg object. Inputs are plotted side by side, with the source being on the left and the target being on the right. Slide bars are provided that allow subsetting and panning across the x and y dimensions displayed in each plot viewer. Clicking on each of the two plots allows manual placement of landmarks on the source and target plots. Clicking ‘Undo Click on Target Image’ or ‘Undo Click on Source Image’ allows removal of selected landmarks. Clicking ‘Done’ in the top right closes the Shiny gadget and returns the selected landmarks as a list of two data.frames of x and y coordinates, with the first data.frame containing landmarks from the source and the second from the target. Feeding these data.frames to calculateAffineMatrixFromLandmarks() calculates and returns a 3 × 3 affine transformation matrix that best maps the source landmarks to the target landmarks.
Interactive three-dimensional spatial plotting
To create an interactive three-dimensional visualization of three-dimensional spatial datasets, the plotly package was used within an interactive Shiny application. The implementation runs locally by calling the function plotInteractive3D(). The application is reactive to slide bars that modify the lower and upper limits of the x, y and z axes, creating custom slices across the dataset. Additionally, the plot is reactive to an optional selection of cluster IDs listed in the cell metadata table, facilitating the visualization and subsetting of cell types of interest. When closing the application, a table containing cell IDs, spatial coordinates and cluster or cell type IDs will be retrieved.
Interactive selection integration with the vitessceR package
To integrate the Giotto object framework with the interactive tools provided by the vitessceR69 package, we created the function ‘giottoToAnndataZarr’, which exports the processed Giotto object to a local Zarr folder that can be read by the vitessceR package to interactively visualize spatial and dimension reduction plots as well as select regions of interest. The selected areas can be exported to a .csv file compatible with downstream analysis in Giotto.
Interoperability
Converters between Giotto Suite and other spatial omics packages
Currently, Giotto objects created within Giotto Suite are interoperable with other spatial omics packages, including Bioconductor/SpatialExperiment29, Seurat and AnnData/Squidpy27 or SpatialData26. This promotes a bidirectional compatibility of Giotto objects with other ecosystems and simultaneously extends its applications.
For the Bioconductor group of packages, the SpatialExperiment data container is used for storing data from spatial omics experiments. It is designed to handle data from spot-based and molecule-based platforms that include spatial coordinates, images and image metadata, apart from the data already common to experiment classes. Giotto Suite provides two functions, giottoToSpatialExperiment() and spatialExperimentToGiotto(), developed by mapping the slots of the Giotto object to the corresponding slots. In brief, Giotto’s feat_metadata maps to SpatialExperiment’s rowData, expression corresponds to assays, cell_metadata corresponds to colData, dim_reductions corresponds to reducedDims and spatial_locs corresponds to spatialCoords, and images are reflected as imgData. The images in Giotto are technically stored as raster objects, and SpatialExperiment also supports the same. Giotto handles expression matrices within separate spatial units and feature types. The SpatialExperiment object can store only one spatial unit at a time; therefore, a list of SpatialExperiment objects is returned from the giottoToSpatialExperiment() function, where each element of the list corresponds to a distinct SpatialExperiment object for a specific spatial unit.
Giotto Suite also provides interoperability between Seurat and Giotto. Because Seurat has multiple versions in use with differences in object structure, we currently provide interoperability between Giotto and both the older and the newer versions of Seurat objects. Therefore, four functions are tailored for these different Seurat versions: giottoToSeuratV4() and seuratToGiottoV4() for the older versions and giottoToSeuratV5() and seuratToGiottoV5() for Seurat version 5, which now includes subcellular and image information. The version 4 functions map Giotto’s cell_metadata to Seurat’s meta.data and dimension_reduction to reductions; feat_metadata from Giotto is mapped to meta.data for each assay in Seurat and expression to assays. With version 5, additional slots such as spatial_loc and images from Giotto are mapped to the most relevant slots in Seurat. During the conversion from Giotto to Seurat, Giotto’s spatial information is stored in Seurat’s dimension reduction slot as it does not provide a separate slot for overall tissue-level coordinates. Images and subcellular information in Giotto are both passed to the images slot of the Seurat objects.
Finally, to support conversions to the AnnData class in Python, the functions anndataToGiotto() and giottoToAnnData() were created by mapping the slots of the Giotto object to the corresponding locations in a Squidpy-flavored AnnData object. In summary, Giotto’s expression slot maps to adata.X, spatial_locs to adata.obsm, cell_metadata to adata.obs, feat_metadata to adata.var, dimension_reduction to adata.obsm and nn_network and spat_network to adata.obsp. Images are currently not mapped between both classes. Of note, the Giotto object stores expression matrices within separate spatial units and feature types, whereas AnnData objects do not support this hierarchical data storage method. Thus, multiple AnnData objects will be created from a Giotto object when multiple spatial units and feature type pairs exist. The generated .h5ad files are prefixed by the spatial unit and feature type that they represent.
As an extension to the AnnData conversion methods, spatialDataToGiotto() and giottoToSpatialData() allow for further flexibility in processing and analyzing spatial omics data. SpatialData object consists of separate elements such as Images, Labels, Points, Shapes and Tables. The format of data stored in Tables is essentially analogous to an AnnData object. Therefore, the conversion functions utilize anndataToGiotto() and giottoToAnnData() to attach the AnnData information to SpatialData’s Tables element and parse AnnData within SpatialData’s Tables to convert back to the Giotto object. When .h5ad files are ingested as a Table, the spatial unit and feature type represented by the file are parsed from the filename and encoded as the name of the Table. The names are parsed for spatial unit and feature type information when converting back to the Giotto object. For the SpatialData elements Images, Points and Shapes, we provide bidirectional mappings to the GiottoImages, GiottoPoints and GiottoPolygons classes, respectively. There is currently no separate mapping for the element Labels, although this information could be stored within the GiottoImages class. When converting a Giotto object to a SpatialData object, the converted object will be stored on disk as .zarr stores, thereby creating separate folders for each element that has been converted in a specified location. This format of on-disk SpatialData object will be required when converting back to the Giotto object.
Bento integration and analysis
To integrate Bento53 analysis with Giotto, Bento (version 2.0.1) scripts were adapted and updated to ensure compatibility with Python 3.10, the current default version utilized in Giotto Suite. These modified Bento scripts are also accessible on GitHub at https://github.com/wwang-chcn/bento-tools. For this example, the Xenium Breast Cancer dataset was used and subsetted with subsetGiottoLocs() with parameters x_min = 0, x_max = 2,000, y_min = 0 and y_max = 2,000. First, transcripts, cell and nuclei coordinates information was extracted from the Giotto object. Next, cell and nucleus polygons were recreated in Python, utilizing the shapely package (version 1.8.5.post1), and subsequently stored in a geopandas (version 0.10.2) dataframe. A modified AnnData object was created using the bt.io.prepare() function from Bento. Furthermore, various application programming interfaces (APIs) were established to invoke Bento’s tools and plotting functions for shape features, point features, RNAflus, RNAforest and co-localization analyses, amounting to a total of 12 APIs.
R-Spatial open science integration and analysis
To facilitate integration between Giotto Suite and (geo)spatial open science ecosystems, we implemented several convenience and interoperability functions. First, functions and methods from the ‘terra’ package are also available for the derived giottoPoints and giottoPolygon objects. Next, to scale up and generalize accessibility for other (geo)spatial classes and dependent packages, we implemented converters between spatial data and ‘sp’, ‘sf’, ‘terra’ and ‘stars’ objects as as.sp(), as.sf(), as.terra() and as.stars(), respectively. These converters can also be directly applied on giottoPoints and giottoPolygon objects to change the underlying data representation. Finally, to make all spatial autocorrelation statistics and metrics available, the functionality from the spdep package is also available through the spdepAutoCorr() function.
Spatial resolution enhancement through interpolation and single-cell segmentation
Spatial interpolation for SVGs
The 10x Genomics Mouse Brain Coronal Section dataset was downloaded and used in this analysis (see ‘Data availability’). createGiottoVisiumObject() was used to create a Giotto object, considering only in-tissue spots. Raw data were filtered and normalized. createSpatialnetwork() was used to generate a spatial network with the ‘KNN’ method, specifying k = 5 and maximum_distance_knn = 400. This spatial network was used to identify spatially variable features using binSpect() or spdepAutoCorr(), which makes all metrics and spatial statistics from the spdep package available in Giotto Suite. The top 500 spatially variable features were selected. Next, a model using gstat gstat(), specifying formula = count~1 (where the tilde indicates R model formula syntax) and locations = ~x + y, was set up and provided to the ‘terra’ function interpolate() alongside paired spatial coordinates and raw expression values in order to compute spatial interpolated and higher resolved information for each gene as 500 rows by 379 columns (189,500 pixels) images. These images were then saved to disk as .tif files.
Building and representing a super-resolved Visium dataset
The gene images were first loaded as a list of giottoLargeImage objects using createGiottoLargeImage(), and then they were attached to the Giotto object. The high-resolution H&E image from the dataset was segmented using StarDist for QuPath. Polygons with a nucleus area less than 0.0001 px2 or more than 600 px2 were removed. The polygons were exported from QuPath in a geoJSON format and read into R as giottoPolygons using createGiottoPolygon(). These polygons were then also attached into the Giotto object as a second set of polygons, alongside the Visium spot polygons, which were generated during Giotto object creation. Expression matrices were computed for each spatial unit (that is, Visium Spots and StarDist Cells) starting from the two sets of polygons and interpolation gene images using the functions calculateOverlap() and overlapToMatrix(). Expression data for StarDist were filtered to remove all cells that did not have any expression data from the 500 genes considered in this analysis. Then, each of the expression matrices was normalized. PCA was performed on both expression matrices, and the first 10 principal components were used with 25 neighbors to calculate a UMAP and SNN network. Finally, the data were clustered using doKmeans() (k = 12).
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The following spatial datasets were used in this paper:
Spatial Genomics dataset. The mouse kidney fresh-frozen dataset was downloaded from the Spatial Genomics website at https://db.cngb.org/stomics/mosta/download/.
DBiT-seq dataset. The mouse embryo E10.5 dataset was downloaded from https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE137986.
NanoString CosMx dataset. The CosMx FFPE Non-Small Cell Lung Cancer dataset for lung sample 12 was downloaded from the NanoString website at https://nanostring.com/products/cosmx-spatial-molecular-imager/ffpe-dataset/nsclc-ffpe-dataset/.
Seq-Scope dataset. The Seq-Scope liver dataset was downloaded directly from the following link:
https://deepblue.lib.umich.edu/data/concern/data_sets/9c67wn05f.
Vizgen dataset. The MERSCOPE/MERFISH FF mouse brain (data release version 1.0, May 2021) and FFPE breast cancer (May 2022) datasets were downloaded directly from the Vizgen website at https://info.vizgen.com/mouse-brain-data and https://info.vizgen.com/ffpe-showcase, respectively.
10x Genomics Xenium dataset. Xenium, corresponding images and Visium datasets for human breast cancer were downloaded directly from the 10x Genomics website at https://www.10xgenomics.com/products/xenium-in-situ/preview-dataset-human-breast.
10x Genomics multimodal Visium CytAssist Human Tonsil dataset. The multimodal Visium CytAssist Human Tonsil dataset was downloaded from the 10x Genomics website at https://www.10xgenomics.com/resources/datasets/gene-protein-expression-library-of-human-tonsil-cytassist-ffpe-2-standard.
10x Genomics Visium Mouse Brain Section (Coronal) dataset. The Visium Mouse Brain Section (Coronal) dataset was downloaded from the 10x Genomics website at https://support.10xgenomics.com/spatial-gene-expression/datasets/1.1.0/V1_Adult_Mouse_Brain.
10x Genomics Visium HD Human Colorectal Cancer. The Visium HD Human Colorectal Cancer (FFPE) dataset was downloaded from the 10x Genomics website at https://www.10xgenomics.com/datasets/visium-hd-cytassist-gene-expression-libraries-of-human-crc.
OpenST adult mouse hippocampus dataset. The OpenST adult mouse hippocampus dataset was downloaded from https://rajewsky-lab.github.io/openst/latest/examples/datasets.
Slide-seq mouse brain. The processed counts of the Slide-seq mouse brain were downloaded from the NeMO database using the following link: https://data.nemoarchive.org/biccn/grant/rf1_macosko/macosko/spatial_transcriptome/cellgroup/Slide-seq/mouse/.
Stereo-seq dataset. The bin1 matrix files (that is, *_GEM_bin1.tsv.gz) were downloaded from the CNGB portal at the following link: https://db.cngb.org/stomics/mosta/download/.
IMC dataset. Intensity images of human lymph node FFPE tissue were downloaded from a repository created by Bost et al. 43 https://data.mendeley.com/datasets/ncfgz5xxyb/1.
Single-cell Mouse Brain Dataset. A single-cell reference dataset published by Manno et al.70 was used to identify developmental mouse brain cell types for spatialDWLS deconvolution with Stereo-seq data in this study. These data can be downloaded in the form of a .loom file from http://mousebrain.org/development/downloads.html.
Single-cell Human Tonsil Dataset. The Atlas of Cells in the Human Tonsil published by Massoni-Badosa et al.71 containing the annotation of more than 357,000 cells was used for spatialDWLS deconvolution of Visum CytAssist data in this study. The annotated SpatialExperiment object was downloaded from https://github.com/massonix/HCATonsilData.
Spatial RNA/ATAC–seq mouse embryo ME13. The mouse embryo ME13 spatial RNA/ATAC–seq dataset was originally published by Zhang et al. 45 and was downloaded from https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE205055.
Code availability
The packages that are part of Giotto Suite (version 4.2.1) can be downloaded from our GitHub page (http://github.com/drieslab/Giotto) or Zenodo (https://doi.org/10.5281/zenodo.15361655)72. Additional information, including a large set of examples, vignettes and frequently asked questions, can be found at http://giottosuite.com. The reproducible scripts used to analyze and generate the figures in this paper can be found at https://github.com/drieslab/Giotto_Suite_manuscript and on Zenodo at https://doi.org/10.5281/zenodo.15361422.
References
Moor, A. E. et al. Spatial reconstruction of single enterocytes uncovers broad zonation along the intestinal villus axis. Cell 175, 1156–1167 (2018).
Hildebrandt, F. et al. Spatial transcriptomics to define transcriptional patterns of zonation and structural components in the mouse liver. Nat. Commun. 12, 7046 (2021).
Harney, A. S. et al. Real-time imaging reveals local, transient vascular permeability, and tumor cell intravasation stimulated by TIE2hi macrophage-derived VEGFA. Cancer Discov. 5, 932–943 (2015).
Sunkin, S. M. et al. Allen Brain Atlas: an integrated spatio-temporal portal for exploring the central nervous system. Nucleic Acids Res. 41, D996–D1008 (2013).
HuBMAP Consortium. The human body at cellular resolution: the NIH Human Biomolecular Atlas Program. Nature 574, 187–192 (2019).
Rozenblatt-Rosen, O. et al. The Human Tumor Atlas Network: charting tumor transitions across space and time at single-cell resolution. Cell 181, 236–249 (2020).
Eng, C.-H. L. et al. Transcriptome-scale super-resolved imaging in tissues by RNA seqFISH+. Nature 568, 235–239 (2019).
Cho, C.-S. et al. Microscopic examination of spatial transcriptome using Seq-Scope. Cell 184, 3559–3572 (2021).
Rodriques, S. G. et al. Slide-seq: a scalable technology for measuring genome-wide expression at high spatial resolution. Science 363, 1463–1467 (2019).
Goltsev, Y. et al. Deep profiling of mouse splenic architecture with CODEX multiplexed imaging. Cell 174, 968–981 (2018).
Lin, J.-R., Fallahi-Sichani, M. & Sorger, P. K. Highly multiplexed imaging of single cells using a high-throughput cyclic immunofluorescence method. Nat. Commun. 6, 8390 (2015).
Angelo, M. et al. Multiplexed ion beam imaging of human breast tumors. Nat. Med. 20, 436–442 (2014).
Giesen, C. et al. Highly multiplexed imaging of tumor tissues with subcellular resolution by mass cytometry. Nat. Methods 11, 417–422 (2014).
Baysoy, A., Bai, Z., Satija, R. & Fan, R. The technological landscape and applications of single-cell multi-omics. Nat. Rev. Mol. Cell Biol. 24, 695–713 (2023).
Kiemen, A. L. et al. CODA: quantitative 3D reconstruction of large tissues at cellular resolution. Nat. Methods 19, 1490–1499 (2022).
Fang, R. et al. Three-dimensional single-cell transcriptome imaging of thick tissues. eLife 12, RP90029 (2024).
Gentleman, R. C. et al. Bioconductor: open software development for computational biology and bioinformatics. Genome Biol. 5, R80 (2004).
Bivand, R. S. Progress in the R ecosystem for representing and handling spatial data. J. Geogr. Syst. 23, 515–546 (2021).
Dries, R. et al. Giotto: a toolbox for integrative analysis and visualization of spatial expression data. Genome Biol. 22, 78 (2021).
Del Rossi, N., Chen, J. G., Yuan, G.-C. & Dries, R. Analyzing spatial transcriptomics data using Giotto. Curr. Protoc. 2, e405 (2022).
Barker, M. et al. Introducing the FAIR Principles for research software. Sci. Data 9, 622 (2022).
Chen, A. et al. Spatiotemporal transcriptomic atlas of mouse organogenesis using DNA nanoball-patterned arrays. Cell 185, 1777–1792 (2022).
Saka, S. K. et al. Immuno-SABER enables highly multiplexed and amplified protein imaging in tissues. Nat. Biotechnol. 37, 1080–1090 (2019).
Manukyan, A. et al. VoltRon: a spatial omics analysis platform for multi-resolution and multi-omics integration using image registration. Preprint at bioRxiv https://doi.org/10.1101/2023.12.15.571667 (2023).
Larsson, L., Franzén, L., Ståhl, P. L. & Lundeberg, J. Semla: a versatile toolkit for spatially resolved transcriptomics analysis and visualization. Bioinformatics 39, btad626 (2023).
Marconato, L. et al. SpatialData: an open and universal data framework for spatial omics. Nat. Methods 22, 58–62 (2025).
Palla, G. et al. Squidpy: a scalable framework for spatial omics analysis. Nat. Methods 19, 171–178 (2022).
Moses, L. et al. Voyager: exploratory single-cell genomics data analysis with geospatial statistics. Preprint at bioRxiv https://doi.org/10.1101/2023.07.20.549945 (2023).
Righelli, D. et al. SpatialExperiment: infrastructure for spatially resolved transcriptomics data in R using Bioconductor. Bioinformatics 38, 3128–3131 (2022).
Stuart, T. et al. Comprehensive integration of single-cell data. Cell 177, 1888–1902 (2019).
Zhang, W. et al. Integration of multiple spatial omics modalities reveals unique insights into molecular heterogeneity of prostate cancer. Preprint at bioRxiv https://doi.org/10.1101/2023.08.28.555056 (2023).
Janesick, A. et al. High resolution mapping of the tumor microenvironment using integrated single-cell, spatial and in situ analysis. Nat. Commun. 14, 8353 (2023).
Androvic, P. et al. Spatial transcriptomics-correlated electron microscopy maps transcriptional and ultrastructural responses to brain injury. Nat. Commun. 14, 4115 (2023).
Arganda-Carreras, I., Sorzano, C., Kybic, J. & Ortíz-de-Solórzano, C. Consistent and elastic registration of histological sections using vector-spline regularization. In Computer Vision Approaches to Medical Image Analysis (eds Beichel, R.R. & Sonka, M.) 85–95 (2008).
Gatenbee, C. D. et al. Virtual alignment of pathology image series for multi-gigapixel whole slide images. Nat. Commun. 14, 4502 (2023).
Clifton, K. et al. STalign: alignment of spatial transcriptomics data using diffeomorphic metric mapping. Nat. Commun. 14, 8123 (2023).
Stringer, C., Wang, T., Michaelos, M. & Pachitariu, M. Cellpose: a generalist algorithm for cellular segmentation. Nat. Methods 18, 100–106 (2021).
Petukhov, V. et al. Cell segmentation in imaging-based spatial transcriptomics. Nat. Biotechnol. 40, 345–354 (2022).
Greenwald, N. F. et al. Whole-cell segmentation of tissue images with human-level performance using large-scale data annotation and deep learning. Nat. Biotechnol. 40, 555–565 (2022).
Weigert, M., Schmidt, U., Haase, R., Sugawara, K. & Myers, G. Star-convex polyhedra for 3D object detection and segmentation in microscopy. In 2020 IEEE Winter Conference on Applications of Computer Vision (WACV) 3655–3662 (IEEE, 2020).
Houthuijzen, J. M. & Jonkers, J. Cancer-associated fibroblasts as key regulators of the breast cancer tumor microenvironment. Cancer Metastasis Rev. 37, 577–597 (2018).
Risom, T. et al. Transition to invasive breast cancer is associated with progressive changes in the structure and composition of tumor stroma. Cell 185, 299–310 (2022).
Bost, P., Schulz, D., Engler, S., Wasserfall, C. & Bodenmiller, B. Optimizing multiplexed imaging experimental design through tissue spatial segregation estimation. Nat. Methods 20, 418–423 (2023).
Hao, Y. et al. Integrated analysis of multimodal single-cell data. Cell 184, 3573–3587 (2021).
Zhang, D. et al. Spatial epigenome-transcriptome co-profiling of mammalian tissues. Nature 616, 113–122 (2023).
Yuan, Z. et al. SODB facilitates comprehensive exploration of spatial omics data. Nat. Methods 20, 387–399 (2023).
Xu, Z. et al. STOmicsDB: a comprehensive database for spatial transcriptomics data sharing, analysis and visualization. Nucleic Acids Res. 52, D1053–D1061 (2024).
Jain, S. et al. Advances and prospects for the Human BioMolecular Atlas Program (HuBMAP). Nat. Cell Biol. 25, 1089–1100 (2023).
Lewis, S. M. et al. Spatial omics and multiplexed imaging to explore cancer biology. Nat. Methods 18, 997–1012 (2021).
Moses, L. & Pachter, L. Museum of spatial transcriptomics. Nat. Methods 19, 534–546 (2022).
Virshup, I., Rybakov, S., Theis, F. J., Angerer, P. & Wolf, F. A. anndata: access and store annotated data matrices. J. Open Source Softw. 9, 4371 (2024).
Páez, A. Open spatial sciences: an introduction. J. Geogr. Syst. 23, 467–476 (2021).
Mah, C. K. et al. Bento: a toolkit for subcellular analysis of spatial transcriptomics data. Genome Biol. 25, 82 (2024).
Peters Couto, B. Z., Robertson, N., Patrick, E. & Ghazanfar, S. MoleculeExperiment enables consistent infrastructure for molecule-resolved spatial omics data in bioconductor. Bioinformatics 39, btad550 (2023).
Qiu, X. et al. Spatiotemporal modeling of molecular holograms. Cell 187, 7351–7373 (2024).
Park, J. et al. Cell segmentation-free inference of cell types from in situ transcriptomics data. Nat. Commun. 12, 3545 (2021).
Korsunsky, I. et al. Fast, sensitive and accurate integration of single-cell data with Harmony. Nat. Methods 16, 1289–1296 (2019).
Schapiro, D. et al. MITI minimum information guidelines for highly multiplexed tissue images. Nat. Methods 19, 262–267 (2022).
Sarfraz, I. et al. MAMS: matrix and analysis metadata standards to facilitate harmonization and reproducibility of single-cell data. Genome Biol. 25, 205 (2024).
Maroso, M. A quest into the human brain. Science 382, 166–167 (2023).
Bankhead, P. et al. QuPath: open source software for digital pathology image analysis. Sci. Rep. 7, 16878 (2017).
Zhu, Q., Shah, S., Dries, R., Cai, L. & Yuan, G.-C. Identification of spatially associated subpopulations by combining scRNAseq and sequential fluorescence in situ hybridization data. Nat. Biotechnol. https://doi.org/10.1038/nbt.4260 (2018).
Subramanian, A. et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl Acad. Sci. USA 102, 15545–15550 (2005).
Weigert, M. et al. Content-aware image restoration: pushing the limits of fluorescence microscopy. Nat. Methods 15, 1090–1097 (2018).
Dong, R. & Yuan, G.-C. SpatialDWLS: accurate deconvolution of spatial transcriptomic data. Genome Biol. 22, 145 (2021).
Granja, J. M. et al. ArchR is a scalable software package for integrative single-cell chromatin accessibility analysis. Nat. Genet. 53, 403–411 (2021).
Raasveldt, M. & Mühleisen, H. DuckDB: an embeddable analytical database. In Proc. of the 2019 International Conference on Management of Data 1981–1984 (Association for Computing Machinery, 2019).
Pagès, H., Lun, A. & Hickey, P. DelayedArray: a unified framework for working transparently with on-disk and in-memory array-like datasets. Bioconductor version: Release (3.18). https://doi.org/10.18129/B9.bioc.DelayedArray (2023).
Keller, M. S. et al. Vitessce: integrative visualization of multimodal and spatially resolved single-cell data. Nat. Methods 22, 63–67 (2025).
La Manno, G. et al. Molecular architecture of the developing mouse brain. Nature 596, 92–96 (2021).
Massoni-Badosa, R. et al. An atlas of cells in the human tonsil. Immunity 57, 379–399.e18 (2024).
Chen, J. G. et al. Giotto Suite v4.2.1. Zenodo https://doi.org/10.5281/zenodo.15361655 (2025).
Acknowledgements
We would like to thank former and current members of the Dries and Yuan laboratories for their support, contributions and invaluable feedback on the Giotto Suite project and development over time. More specifically, we would like to thank the following individuals: E. Kelley, C. Ennis, J. Weis, Y. M. Wei, R. dos Santos Peixoto, M. M. Khan, J. Yeung, A. Zug, L. Norford, C. McCormick, N. Del Rossi, A. Krek, W. Ma, X. Cao and S. Zheng. This work was supported by the Chan Zuckerberg Initiative’s Essential Open Source Software for Science Program (2022-252544) (R.D. and G.-C.Y.), the Crazy 8 Initiative from Alex’s Lemonade Stand Foundation for Childhood Cancer (R.D.) and National Institutes of Health RF1MH128970, RF1MH133703 and R01AG066028 (G.-C.Y.). R.D. is also supported by Boston University Chobanian & Avedisian School of Medicine and Boston Medical Center.
Author information
Authors and Affiliations
Contributions
J.G.C., J.C.C.-F., G.-C.Y. and R.D. conceived the project. J.G.C., J.C.C.-F., M.O.B., J.X., E.C.R., W.W., I.A., S.V.H., I.S., P.G., A.S., V.J., S.C.S. and R.D. developed Giotto Suite and implemented and revised code. J.G.C., J.C.C.-F., M.O.B., J.X., E.C.R., W.W. and J.P.S. performed analyses and generated figures. M.O.B. and J.X. contributed equally as co-second authors. G.-C.Y. and R.D. wrote the manuscript, with input and feedback from all other authors.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Methods thanks Nikos Karaiskos, Jin Liu and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Primary Handling Editor: Lin Tang, in collaboration with the Nature Methods team. Peer reviewer reports are available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Supplementary Information
Supplementary Figs. 1–10 and Supplementary Table 1.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Chen, J.G., Chávez-Fuentes, J.C., O’Brien, M. et al. Giotto Suite: a multiscale and technology-agnostic spatial multiomics analysis ecosystem. Nat Methods 22, 2052–2064 (2025). https://doi.org/10.1038/s41592-025-02817-w
Received:
Accepted:
Published:
Version of record:
Issue date:
DOI: https://doi.org/10.1038/s41592-025-02817-w
This article is cited by
-
Spatially resolved integrative analysis of transcriptomic and metabolomic changes in tissue injury studies
Nature Communications (2026)
