
Abstract
Magnesium (Mg2+) uptake systems are present in all domains of life, consistent with the vital role of this ion. P-type ATPase Mg2+ importers are required for bacterial growth when Mg2+ is limiting or during pathogenesis. However, insights into their mechanisms of action are missing. Here we solved the cryo-EM structure of the Mg2+ transporter MgtA from Escherichia coli. We obtained high-resolution structures of both homodimeric (2.9 Å) and monomeric (3.6 Å) forms. The dimer structure is formed by multiple contacts between residues in adjacent soluble N and P subdomains. Our structures revealed an ion, assigned as Mg2+, in the transmembrane segment. Moreover, we detected two cytoplasmic ion-binding sites and determined the structure of the N-terminal tail. Sequence conservation, mutagenesis and ATPase assays indicate dimerization, the ion-binding sites and the N-terminal tail facilitate cation transport or serve regulatory roles.
This is a preview of subscription content, access via your institution
Access options
Access Nature and 54 other Nature Portfolio journals
Get Nature+, our best-value online-access subscription
$32.99 / 30 days
cancel any time
Subscribe to this journal
Receive 12 print issues and online access
$259.00 per year
only $21.58 per issue
Buy this article
- Purchase on SpringerLink
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout






Similar content being viewed by others

Data availability
Cryo-EM maps have been deposited in the Electron Microscopy Data Bank (EMDB) under accession codes EMD-42794 (apo-dimer, C2), EMD-42795 (apo-dimer, C1), EMD-42796 (apo-monomer, C1), EMD-42797 (ATP-dimer, C2), EMD-42798 (ATPγS-dimer, C2) and EMD-42799 (ADP-dimer, C2). The atomic coordinates have been deposited in the PDB under accession codes 8UY7 (apo-dimer, C2), 8UY8 (apo-dimer, C1), 8UY9 (apo-monomer, C1), 8UYA (ATP-dimer, C2), 8UYB (ATPγS-dimer, C2) and 8UYC (ADP-dimer, C2). Trajectories and AMBER input files for simulations with and without ATP are publicly available via Zenodo at https://doi.org/10.5281/zenodo.10017395 (ref. 77). The MS proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository84 with the dataset identifier PXD047999. The MgtA (POABB8) and MgtS (A5A616) sequences can be readily downloaded from UniProtKB. Additional source data for Figs. 2–4 and Extended Data Fig. 2 are available via figshare at https://doi.org/10.6084/m9.figshare.26820568.v1 (ref. 58). All other data supporting the findings of this study are available from the corresponding authors on reasonable request. Source data are provided with this paper.
Code availability
Calculation of water pathway code is available via GitHub at https://github.com/alexsodt/WaterPathMGTA.
References
Moomaw, A. S. & Maguire, M. E. The unique nature of Mg2+ channels. Physiology 23, 275–285 (2008).
Franken, G. A. C., Huynen, M. A., Martínez-Cruz, L. A., Bindels, R. J. M. & de Baaij, J. H. F. Structural and functional comparison of magnesium transporters throughout evolution. Cell. Mol. Life Sci. 79, 418 (2022).
Jin, F., Huang, Y. & Hattori, M. Recent advances in the structural biology of Mg2+ channels and transporters. J. Mol. Biol. 434, 167729 (2022).
Groisman, E. A., Duprey, A. & Choi, J. How the PhoP/PhoQ system controls virulence and Mg2+ homeostasis: lessons in signal transduction, pathogenesis, physiology, and evolution. Microbiol. Mol. Biol. Rev. 85, e00176–00120 (2021).
Cunrath, O. & Bumann, D. Host resistance factor SLC11A1 restricts Salmonella growth through magnesium deprivation. Science 366, 995–999 (2019).
Neef, J., Andisi, V. F., Kim, K. S., Kuipers, O. P. & Bijlsma, J. J. E. Deletion of a cation transporter promotes lysis in Streptococcus pneumoniae. Infect. Immun. 79, 2314–2323 (2011).
Park, S.-Y. & Groisman, E. A. Signal-specific temporal response by the Salmonella PhoP/PhoQ regulatory system. Mol. Microbiol. 91, 135–144 (2014).
Yeom, J., Shao, Y. & Groisman, E. A. Small proteins regulate Salmonella survival inside macrophages by controlling degradation of a magnesium transporter. Proc. Natl Acad. Sci. USA 117, 20235–20243 (2020).
Hattori, M. et al. Mg2+-dependent gating of bacterial MgtE channel underlies Mg2+ homeostasis. EMBO J. 28, 3602–3612 (2009).
Dyla, M., Kjærgaard, M., Poulsen, H. & Nissen, P. Structure and mechanism of P-type ATPase ion pumps. Annu. Rev. Biochem. 89, 583–603 (2020).
Jumper, J. et al. Highly accurate protein structure prediction with AlphaFold. Nature 596, 583–589 (2021).
Varadi, M. et al. AlphaFold Protein Structure Database: massively expanding the structural coverage of protein-sequence space with high-accuracy models. Nucleic Acids Res. 50, D439–D444 (2022).
Albers, R. W. Biochemical aspects of active transport. Annu. Rev. Biochem. 36, 727–756 (1967).
Post, R. L. Seeds of sodium, potassium ATPase. Annu. Rev. Physiol. 51, 1–15 (1989).
Lutsenko, S. & Kaplan, J. H. Organization of P-type ATPases: significance of structural diversity. Biochemistry 34, 15607–15613 (1995).
Subramani, S., Perdreau-Dahl, H. & Morth, J. P. The magnesium transporter A is activated by cardiolipin and is highly sensitive to free magnesium in vitro. eLife 5, e11407 (2016).
Møller, J. V., Olesen, C., Winther, A.-M. L. & Nissen, P. The sarcoplasmic Ca2+-ATPase: design of a perfect chemi-osmotic pump. Q. Rev. Biophys. 43, 501–566 (2010).
Aguayo-Ortiz, R. & Espinoza-Fonseca, L. M. Linking biochemical and structural states of SERCA: achievements, challenges, and new opportunities. Int. J. Mol. Sci. 21, 4146 (2020).
Håkansson, K. O. The structure of Mg-ATPase nucleotide-binding domain at 1.6 Å resolution reveals a unique ATP-binding motif. Acta Crystallogr. D 65, 1181–1186 (2009).
Burroughs, A. M., Allen, K. N., Dunaway-Mariano, D. & Aravind, L. Evolutionary genomics of the HAD superfamily: understanding the structural adaptations and catalytic diversity in a superfamily of phosphoesterases and allied enzymes. J. Mol. Biol. 361, 1003–1034 (2006).
Wang, H. et al. Increasing intracellular magnesium levels with the 31-amino acid MgtS protein. Proc. Natl Acad. Sci. USA 114, 5689–5694 (2017).
Bublitz, M. et al. Ion pathways in the sarcoplasmic reticulum Ca2+-ATPase. J. Biol. Chem. 288, 10759–10765 (2013).
Takahashi, M., Kondou, Y. & Toyoshima, C. Interdomain communication in calcium pump as revealed in the crystal structures with transmembrane inhibitors. Proc. Natl Acad. Sci. USA 104, 5800–5805 (2007).
Winther, A. M. L. et al. Critical roles of hydrophobicity and orientation of side chains for inactivation of sarcoplasmic reticulum Ca2+-ATPase with thapsigargin and thapsigargin analogs. J. Biol. Chem. 285, 28883–28892 (2010).
Heit, S. et al. Structure of the hexameric fungal plasma membrane proton pump in its autoinhibited state. Sci. Adv. 7, eabj5255 (2021).
Focht, D., Croll, T.I., Pedersen, B.P. & Nissen, P. Improved model of proton pump crystal structure obtained by interactive molecular dynamics flexible fitting expands the mechanistic model for proton translocation in P-type ATPases. Front. Physiol. 8, 202 (2017).
Chen, Z. et al. Cryo-EM structures of human SPCA1a reveal the mechanism of Ca2+/Mn2+ transport into the Golgi apparatus. Sci. Adv. 9, eadd9742 (2023).
Diaz, D. & Clarke, R. J. Evolutionary analysis of the lysine-rich N-terminal cytoplasmic domains of the gastric H+,K+-ATPase and the Na+,K+-ATPase. J. Membr. Biol. 251, 653–666 (2018).
Jephthah, S., Månsson, L. K., Belić, D., Morth, J. P. & Skepö, M. Physicochemical characterisation of KEIF—the intrinsically disordered N-terminal region of magnesium transporter A. Biomolecules 10, 623 (2020).
Kanai, R., Ogawa, H., Vilsen, B., Cornelius, F. & Toyoshima, C. Crystal structure of a Na+-bound Na+,K+-ATPase preceding the E1P state. Nature 502, 201–206 (2013).
Zhang, Z. et al. Detailed characterization of the cooperative mechanism of Ca2+ binding and catalytic activation in the Ca2+ transport (SERCA) ATPase. Biochemistry 39, 8758–8767 (2000).
Nielsen, J. M., Pedersen, P. A., Karlish, S. J. & Jorgensen, P. L. Importance of intramembrane carboxylic acids for occlusion of K+ ions at equilibrium in renal Na,K-ATPase. Biochemistry 37, 1961–1968 (1998).
Bovo, E. et al. Dimerization of SERCA2a enhances transport rate and improves energetic efficiency in living cells. Biophys. J. 119, 1456–1465 (2020).
Clarke, R. J. & Kane, D. J. Two gears of pumping by the sodium pump. Biophys. J. 93, 4187–4196 (2007).
Dean, W. L. & Tanford, C. Reactivation of lipid-depleted Ca2+-ATPase by a nonionic detergent. J. Biol. Chem. 252, 3551–3553 (1977).
Hidalgo, C., Thomas, D. D. & Ikemoto, N. Effect of the lipid environment on protein motion and enzymatic activity of sarcoplasmic reticulum calcium ATPase. J. Biol. Chem. 253, 6879–6887 (1978).
Jilka, R. L., Martonosi, A. N. & Tillack, T. W. Effect of the purified (Mg2+ + Ca2+)-activated ATPase of sarcoplasmic reticulum upon the passive Ca2+ permeability and ultrastructure of phospholipid vesicles. J. Biol. Chem. 250, 7511–7524 (1975).
Maire, M. L., Lind, K. E., Jørgensen, K. E., Røigaard, H. & Møller, J. V. Enzymatically active Ca2+ ATPase from sarcoplasmic reticulum membranes, solubilized by nonionic detergents. Role of lipid for aggregation of the protein. J. Biol. Chem. 253, 7051–7060 (1978).
Møller, J. V., Lind, K. E. & Andersen, J. P. Enzyme kinetics and substrate stabilization of detergent-solubilized and membraneous (Ca2+ + Mg2+)-activated ATPase from sarcoplasmic reticulum. Effect of protein–protein interactions. J. Biol. Chem. 255, 1912–1920 (1980).
Taylor, K., Dux, L. & Martonosi, A. Structure of the vanadate-induced crystals of sarcoplasmic reticulum Ca2+-ATPase. J. Mol. Biol. 174, 193–204 (1984).
Vanderkooi, J. M., Ierokomas, A., Nakamura, H. & Martonosi, A. Fluorescence energy transfer between Ca2+ transport ATPase molecules in artificial membranes. Biochemistry 16, 1262–1267 (1977).
Maire, M. L., Moller, J. V. & Tanford, C. Retention of enzyme activity by detergent-solubilized sarcoplasmic Ca2+-ATPase. Biochemistry 15, 2336–2342 (1976).
Toyoshima, C., Sasabe, H. & Stokes, D. L. Three-dimensional cryo-electron microscopy of the calcium ion pump in the sarcoplasmic reticulum membrane. Nature 362, 467–471 (1993).
Blackwell, D. J., Zak, T. J. & Robia, S. L. Cardiac calcium ATPase dimerization measured by cross-linking and fluorescence energy transfer. Biophys. J. 111, 1192–1202 (2016).
Ushimaru, M. & Fukushima, Y. The dimeric form of Ca2+-ATPase is involved in Ca2+ transport in the sarcoplasmic reticulum. Biophys. J. 414, 357–361 (2008).
Calì, T., Brini, M. & Carafoli, E. Regulation of cell calcium and role of plasma membrane calcium ATPases. Int. Rev. Cell. Mol. Biol. 332, 259–296 (2017).
Hari, S. B., Morehouse, J. P., Baker, T. A. & Sauer, R. T. FtsH degrades kinetically stable dimers of cyclopropane fatty acid synthase via an internal degron. Mol. Microbiol. 119, 101–111 (2022).
Akin, B. L., Hurley, T. D., Chen, Z. & Jones, L. R. The structural basis for phospholamban inhibition of the calcium pump in sarcoplasmic reticulum. J. Biol. Chem. 288, 30181–30191 (2013).
Winther, A.-M. L. et al. The sarcolipin-bound calcium pump stabilizes calcium sites exposed to the cytoplasm. Nature 495, 265–269 (2013).
Anderson, D. M. et al. A micropeptide encoded by a putative long noncoding RNA regulates muscle performance. Cell 160, 595–606 (2015).
Nelson, B. R. et al. A peptide encoded by a transcript annotated as long noncoding RNA enhances SERCA activity in muscle. Science 351, 271–275 (2016).
Silberberg, J. M. et al. Inhibited KdpFABC transitions into an E1 off-cycle state. eLife 11, e80988 (2022).
Huang, C.-S., Pedersen, B. P. & Stokes, D. L. Crystal structure of the potassium-importing KdpFABC membrane complex. Nature 546, 681–685 (2017).
Stock, C. et al. Cryo-EM structures of KdpFABC suggest a K+ transport mechanism via two inter-subunit half-channels. Nat. Commun. 9, 4971 (2018).
Morth, J. P. et al. Crystal structure of the sodium–potassium pump. Nature 450, 1043–1049 (2007).
Shinoda, T., Ogawa, H., Cornelius, F. & Toyoshima, C. Crystal structure of the sodium–potassium pump at 2.4 Å resolution. Nature 459, 446–450 (2009).
Laskowski, R. A. & Swindells, M. B. LigPlot+: multiple ligand-protein interaction diagrams for drug discovery. J. Chem. Inf. Model. 51, 2778–2786 (2011).
Storz, G. Zeinert, R. Sodt, A. & Matthies, D. P-type ATPase magnesium transporter MgtA acts as a dimer. figshare https://doi.org/10.6084/m9.figshare.26820568.v1 (2025).
Toyoshima, C., Nomura, H. & Tsuda, T. Lumenal gating mechanism revealed in calcium pump crystal structures with phosphate analogues. Nature 432, 361–368 (2004).
Nyblom, M. et al. Crystal structure of Na+, K+-ATPase in the Na+-bound state. Science 342, 123–127 (2013).
Pettersen, E. F. et al. UCSF Chimera—a visualization system for exploratory research and analysis. J. Comput. Chem. 25, 1605–1612 (2004).
Zöller, J. et al. Ligand binding and conformational dynamics of the E. coli nicotinamide nucleotide transhydrogenase revealed by hydrogen/deuterium exchange mass spectrometry. Comput. Struct. Biotechnol. J. 20, 5430–5439 (2022).
Mastronarde, D. N. Automated electron microscope tomography using robust prediction of specimen movements. J. Struct. Biol. 152, 36–51 (2005).
Heymann, J. B. Guidelines for using Bsoft for high resolution reconstruction and validation of biomolecular structures from electron micrographs. Protein Sci. 27, 159–171 (2018).
Grant, T., Rohou, A. & Grigorieff, N. cisTEM, user-friendly software for single-particle image processing. eLife 7, e35383 (2018).
Punjani, A., Rubinstein, J. L., Fleet, D. J. & Brubaker, M. M. cryoSPARC: algorithms for rapid unsupervised cryo-EM structure determination. Nat. Methods 14, 290–296 (2017).
Håkansson, K. O. The structure of Mg-ATPase nucleotide-binding domain at 1.6 Å resolution reveals a unique ATP-binding motif. Acta Crystallogr. D 65, 1181–1186 (2009).
Emsley, P., Lohkamp, B., Scott, W. G. & Cowtan, K. Features and development of Coot. Acta Crystallogr. D 66, 486–501 (2010).
Liebschner, D. et al. Macromolecular structure determination using X-rays, neutrons and electrons: recent developments in Phenix. Acta Crystallogr. D 75, 861–877 (2019).
Williams, C. J. et al. MolProbity: more and better reference data for improved all-atom structure validation. Protein Sci. 27, 293–315 (2018).
Goddard, T. D. et al. UCSF ChimeraX: meeting modern challenges in visualization and analysis. Protein Sci. 27, 14–25 (2018).
Jo, S., Kim, T., Iyer, V. G. & Im, W. CHARMM-GUI: a web-based graphical user interface for CHARMM. J. Comput. Chem. 29, 1859–1865 (2008).
Bogdanov, M. et al. Phospholipid distribution in the cytoplasmic membrane of Gram-negative bacteria is highly asymmetric, dynamic, and cell shape-dependent. Sci. Adv. 6, eaaz6333 (2020).
Huang, J. et al. CHARMM36m: an improved force field for folded and intrinsically disordered proteins. Nat. Methods 14, 71–73 (2017).
Klauda, J. B. et al. Update of the CHARMM all-atom additive force field for lipids: validation on six lipid types. J. Phys. Chem. B 114, 7830–7843 (2010).
Phillips, J. C., Zheng, G., Kumar, S. & Kale, L. V. NAMD: biomolecular simulation on thousands of processors. In Proc. 2002 ACM/IEEE Conference on Supercomputing 36 (IEEE, 2002).
Sodt, A. & Lessen, H. Simulations of the bacterial magnesium transporter MgtA with and without bound ATP. Zenodo https://doi.org/10.5281/zenodo.10017395 (2025).
Beaven, A. H. et al. Gramicidin A channel formation induces local lipid redistribution I: experiment and simulation. Biophys. J. 112, 1185–1197 (2017).
Kaufman, L. & Rousseeuw, P. J. in Wiley Series in Probability and Statistics Ch. 2, 68–125 (Wiley, 1990).
Sodt, A. Shells. GitHub http://github.com/alexsodt/shells (2025).
Sodt, A. Clustering. GitHub https://github.com/alexsodt/clustering (2025).
Baek, M. et al. Accurate prediction of protein structures and interactions using a three-track neural network. Science 373, 871–876 (2021).
FigTree. Molecular Evolution, Phylogenetics and Epidemiology http://tree.bio.ed.ac.uk/software/figtree/ (2025).
Perez-Riverol, Y. et al. The PRIDE database resources in 2022: a hub for mass spectrometry-based proteomics evidences. Nucleic Acids Res. 50, D543–D552 (2022).
Acknowledgements
We thank A. Banerjee for the use of his lab’s AKTA and both A. Banerjee and M. Britt for comments on the paper; J. Petersen and J. Zimmerberg for access to the T20 electron microscope; the NICE facility with A. Zeher, Z. Lang and R.K. Huang for support on the G1 Krios electron microscope; E. Viverette, J. Bouvette and M. Borgnia for support on the G4 Krios electron microscope; and S. Mahé and J. Cometa for technical support on the T20 and G1 Krios. We also thank K. Ito for providing us the Mg2+-auxotrophic E. coli strain (BW25113 ΔmgtA, ΔcorA, ΔyhiD DE3). This research was supported by the Intramural Research Programs of the Eunice Kennedy Shriver National Institute of Child Health and Human Development (NICHD) (grant nos. ZIA HD008955 (to A.J.S.), ZIA HD008855 (to G.S.) and ZIA HD008998 (to D.M.)) and the National Library of Medicine (grant no. LM594244 (to L.A.)) as well as a NIGMS PRAT fellowship (grant no. 1FI2GM146628-01 (to R.Z.)) and NICHD Early Career Awards to R.Z. and D.M. and funding from the German Research Foundation (grant no. DFG 3542/1-1 (to J.D.L.)). This work utilized the computational resources of the NIH HPC Biowulf cluster (http://hpc.nih.gov).
Author information
Authors and Affiliations
Contributions
R.Z. expressed and purified protein, performed SDS–PAGE and western blots, mutagenesis, in vivo assays and ATP hydrolysis; made negative-staining and cryo-EM grids; analyzed data, generated figures and wrote the paper. F.Z. performed native PAGE, made negative-staining and cryo-EM grids, screened negative-staining and cryo-EM grids, and helped collect negative-staining and cryo-EM data. P.F. performed MS analysis of crosslinked samples, data evaluation and generated figures. J.Z. performed HDX–MS experiments and HDX–MS data evaluation and generated figures. Z.K.M. expressed and purified protein, performed SDS–PAGE and western blots and ATP hydrolysis. H.L. performed MD simulations. L.A. carried out computation sequence/structural and evolutionary analysis, identified sequence motifs and generated the large sequence alignment. J.D.L. analyzed crosslinking and HDX–MS data. A.J.S. performed MD simulations, analyzed data, generated figures, wrote the paper and designed and supervised the project. G.S. analyzed data, generated figures, wrote the paper, initiated and designed and supervised the project. D.M. collected and processed negative-staining data, processed cryo-EM data, produced structural models, analyzed data, generated figures, wrote the paper and designed and supervised the project. All authors contributed to the paper.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Structural & Molecular Biology thanks Kazuhiro Abe and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Peer reviewer reports are available. Primary Handling Editor: Katarzyna Ciazynska, in collaboration with the Nature Structural & Molecular Biology team.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data
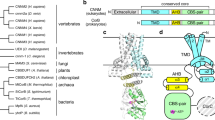
Extended Data Fig. 1 Multisequence alignment of MgtA and MgtB illustrating conserved structural and functional features.
Sequence alignment of E. coli MgtA (EcMgtA), S. enterica serovar Typhimurium MgtA (SeMgtA), and S. enterica serovar Typhimurium MgtB (SeMgtB) generated using Clustal Omega. Domains are colored and named according to Fig. 1. Both the canonical P-type ATPase nomenclature and more detailed description of the fold of the domain with boundaries are given. The soluble A domain is split into two regions: a and b. The b segment of the A domain is comprised of a Double Stranded beta-Helix fold (DSbH). The soluble P subdomain is a noncontiguous segment comprised of two regions a and b that house the key catalytic residues required for phosphorylation. D373 is phosphorylated in EcMgtA. The N or CAP subdomain is a contiguous sequence that binds the nucleotide and aids in catalysis. The P and N subdomains comprise the haloacid dehalogenase (HAD) domain20. The TM-spanning alpha-helical regions, as determined by a residue’s alpha-carbon position residing, on average, within the hydrocarbon bilayer interior of the molecular dynamics (MD) simulations (+/- 15 Angstroms from the bilayer midplane) are denoted by TM1-10. Gray circles denote residues present at the dimer interface, red circles denote residues near ATP, green circles denote residues near Mg2+ in the transmembrane domain, and black circles denote residues near Mg2+ in the cytoplasmic domain in our dimeric cryo-EM structures, as annotated in Figs. 2–4. Black dashed boxes indicate sequences conserved across all P-type ATPases as shown in the logos in Extended Data Fig. 2b. Red dashed boxes indicate sequences specific to Mg2+ importers as shown in the logos in Extended Data Fig. 7b.
Extended Data Fig. 2 Tree of P-type ATPases and amino acids related to catalysis and structural architecture that are highly conserved across the P-type ATPase family.
a, In the tree, the clades are labeled and colored as per their known or predicted transport substrates. The P-type ATPase subclass is provided in brackets next to the aforestated label. All the major clades with IQtree bootstrap sport of 90% or higher are marked with a filled circle. Branches of special representatives, namely MgtA, MgtB, SERCA and SPCA1 are separately labeled and indicated with bold branches. The tree is based on a multiple sequence alignment of representatives of the major clades of P-type ATPase, which is in figshare. b, Sequence logos showing conservation of amino acid residues involved in ATP hydrolysis and structural architecture conserved among entire family of P-type ATPases (indicated by black dashed boxes in Extended Data Fig. 1). Letters represent amino acid abbreviations and height represents the probability of conservation in the P-type ATPase family. As in Extended Data Fig. 1, gray circles denote residues present at the dimer interface, red circles denote residues involved in ATP hydrolysis, green circles denote residues near Mg2+ in the transmembrane domain, and black circles denote residues near Mg2+ in the cytoplasmic domain in our dimeric structures, as annotated in Figs. 2–4. Gray, red, green and black arrows, respectively, indicate residues located at the dimer interface, involved in ATP hydrolysis, surrounding the transmembrane or cytoplasmic Mg2+, which were mutated in subsequent experiments.
Extended Data Fig. 3 Purification of MgtA and ATPase activity of purified protein.
a, Solubilization of membranes expressing MgtA or MgtAS with detergents of varying strengths show differences in native protein interactions when analyzed by Native-PAGE and Western blot analysis. Membranes from cells overexpressing MgtA or MgtAS were solubilized with the detergents LMNG (L), DDM (D), or GDN (G) before Native-PAGE and Western blot analysis using α-FLAG antibodies against tagged MgtS (left two panels; middle panel is a longer exposure of the left panel) or α-His6 antibodies against tagged MgtA (right panel). b, Purification of MgtA results in two distinct MW protein complexes. SEC profile (left) of MgtA used to solve the dimer and monomer structures of MgtA in Fig. 1. Fractions were analyzed by SDS-PAGE (middle) and Native-PAGE gels (right). V indicates the void volume of the SEC column. Fractions that were used to solve the structure are referred to as pooled. c, Dimeric MgtA can be separated from monomeric MgtA. SEC profile (left) of MgtA used to solve the nucleotide-bound dimer structures of MgtA in Fig. 3 and Supplementary Fig. 3 and 5. Fractions were analyzed by SDS-PAGE (middle) and Native-PAGE gels (right). Fraction 18 which possessed predominantly dimer species was used for structural analysis. d, SDS-PAGE gel showing purity of purified WT and D373N mutant proteins assayed in f. e, ATPase activity of WT MgtA in GDN in the presence of 0.5 or 5 mM MgCl2 and with and without the addition of cardiolipin. f, ATPase activity of WT and D373N mutant MgtA in GDN in different concentrations of MgCl2 between 0.5 to 20 mM. For e and f, protein was purified in 0.5 mM MgCl2 and assays were supplemented with the indicated concentrations of MgCl2. Assays were performed with 6.25 µg MgtA and 2 mM ATP at 37˚C. Free phosphate was quantified using molybdate/malachite green-based assays (see methods). Data are shown for duplicate experiments with mean. Separate preparations of WT MgtA were assays for e and f.
Extended Data Fig. 4 Quality of cryo-EM dimer map for key structural features.
Cryo-EM map and atomic model of the TM segments (1-10), soluble domains (A, P, N), N-terminus, and Mg2+ ions colored as in Fig. 1. Atomic model of each structural element is shown in stick representation, the atoms are colored by heteroatom within the cryo-EM map and the corresponding map features are represented in gray mesh.
Extended Data Fig. 5 Experimental support of transmembrane borders and lipid distribution.
a, Side views of cryo-EM maps at different thresholds showing extra densities in orange corresponding to the detergent micelle, detergent molecules, or potential copurified lipids near the transmembrane region. b, Side view of MgtA simulated in a native lipid environment displayed in surface representation colored by electrostatic potential (UCSF Chimera coloring varies from red [-10 kcal/mol/e] to blue [+10 kcal/mol/e] with distance-dependent dielectric constant 4, distance from surface 1.4). Phospholipids corresponding to phosphatidylethanolamine (PE), phosphatidylglycerol (PG) and cardiolipin are colored tan, yellow, and green, respectively. c, Arginine and lysine residues that interact with anionic lipids during at least 20% of the simulation, with cutoff enclosing the first peak of the radial distribution function. d, Side view of MgtA with arginine (R), lysine (K), and tryptophan (W) residues near the lipid membrane borders highlighted in blue spheres. e, Voronoi decomposition of lipid centers-of-geometry in the cytoplasmic leaflet initially (initial) and at the end of the simulation (final). Yellow and green are anionic PG and cardiolipin lipids, respectively. At right, average enrichment or depletion of lipids based on solvation shell as assigned by Voronoi decomposition averaged over the trajectory. Data are shown for independent replicates (n = 10) from the same simulation with mean and standard deviation (error bars) for each solvation shell.
Extended Data Fig. 6 More splayed conformation of monomeric MgtA structure relative to the dimer with major changes in TM1-4, N and A domains.
a, Dimeric structure with two subunits in two different gray tones and Mg2+ ions as green spheres. b, Monomeric structure colored as in Fig. 1. c-d, To visualize structural changes the colored monomer and a single dimer subunit in gray were superimposed (c) and RMSD was calculated (d). The monomer structure is shown colored by RMSD indicating the largest differences when comparing the two structures by high RMSD values in red. e-f, Structural differences between the dimer (gray) and monomer (purple) for TM1-2 (e) and TM3-4 (f). The monomer colored according to RMSD is on the right with densities in mesh indicating the fit. g, Monomer density features and fitted model of TM5 and TM7 near ion-binding site A in the middle of the membrane which is similar to the dimer structures. h-j, Differences in position in the overall conformation between the soluble domains of the dimer (gray) and monomer (colored).
Extended Data Fig. 7 Stability of Mg2+ binding in simulation and conservation of key Mg2+-binding residues.
a, Adaptive-biasing MD simulations perturbing Mg2+ from the three binding sites and K+ from site A. Simulated barriers are similar for Mg2+ at each site at 13-15 kcal/mol, with K+ much weaker. Data are for independent simulations (n = 3) with mean (colored solid line) and standard deviation (shaded band) indicated. b, Sequence logos displaying conservation of amino acids in different members of the P-type ATPase transporters. Letters represent amino acid abbreviations; the height of each letter represents the relative probability of conservation among members of the P-type ATPase family. Logos correspond to the Mg2+ TM binding site A to illustrate residues conserved in the MgtA clade (top) compared to the Ca2+ clade (bottom). The sequence logos are also highlighted in the reduced multisequence alignment in Extended Data Fig. 1. Green circles denote residues near Mg2+ in the transmembrane domain.
Extended Data Fig. 8 Water accessibility in the TM domain of MgtA.
Figures are arranged such that rows correspond with overlapping 1.5 nanometer thick cuts spaced every 1 nanometer. The left column is the side view from the simulation, while the middle column is from the top-down starting on the periplasmic side of the transporter. For the simulation, waters (including hydrogens) are shown in sphere representation. At right is the corresponding top-down view of the MgtA dimer from cryo-EM, with resolved water molecules shown as red spheres. The protein ribbon model is shown opaque through the cut of the simulation, while outside of the cut waters are not shown, and the protein is transparent. The transmembrane Mg2+ is a green sphere and residues E331 and D780 are shown in stick representation.
Extended Data Fig. 9 Extended N-terminus cannot be predicted by AlphaFold, conceals negative patches but does not affect ATPase activity.
a, Alignment showing extended N-terminus present in MgtA, H+-pumps, H+/K+-ATPases and Na+/K+-ATPases but not Ca2+-pump SERCA. The sequence alignment of E. coli MgtA (EcMgtA), Plant Pma2, Fungal Pma1, ATP4A (H+/K+ transporter) from pig and human, ATP1A1 (Na+/K+ transporter) from pig and human, and SERCA (ATP2A1) from rabbit and human was generated using Clustal Omega. Domains are colored and named according to Fig. 1. A more complete alignment can be found in figshare. b, AlphaFold models of Fungal Pma1, SeMgtB and EcMgtA. AlphaFold’s low confidence score for the N-terminal tails predicts them to be disordered. c, RMSD calculated between the AlphaFold model and a single subunit of the dimeric MgtA structure to visualize differences. d, Front and side view of the surface representation of the electrostatic potential displayed for WT and the deletion of the N-terminus (∆1-36). Side view is rotated 90˚. e, Native-PAGE gel of SEC fractions 1-6 following Superose 6 fractionation of ∆1-31 in 5 mM MgCl2. Bolded fractions 2 and 5 were used in the ATPase assay. f, ATPase activity of dimer and monomer fractions with either 5 mM or 20 mM MgCl2. Assays were performed with 6.25 µg MgtA and 2 mM ATP at 37˚C and free phosphate was quantified using molybdate/malachite green-based assays (see methods). Data are shown for duplicate experiments with mean. g, Levels of MgtA are reduced by mutations of the N-terminus, to either eliminate the N-terminus or mutate residues conserved in the Mgt family and suggested to play a role in membrane sensing29. Cells were grown uninduced (- IPTG) overnight at 37˚C in LB supplemented with 100 mM MgSO4 and normalized in lysis buffer before western blot analysis with polyclonal anti-MgtA antibodies. Non-specific bands, also observed with just the vector control, are detected with the native antibody for the low levels of MgtA assayed here. Ponceau S-stained membrane (right) serves as a loading control.
Extended Data Fig. 10 Predicted and documented interactions of small proteins with P-type ATPase proteins.
a, Dimer cryo-EM structure of E. coli MgtA from this study with E. coli MgtS binding predicted by AlphaFold Multimer beta. b, Models of E. coli MgtA monomer and E. coli MgtS and S. enterica MgtA and S. enterica MgtS, MgtU and MgtR predicted by AlphaFold Multimer beta. c, Selected structures of indicated P-type ATPases solved with small α-helical proteins: SERCA1a + phospholamban (PLN)48, SERCA1a + sarcolipin (SLN)22, ATP1A1 + FXYD2 subunit60, and KdpABC + KdpF53. P-type ATPases are in gray with small protein in red.
Supplementary information
Supplementary Information
Supplementary Figs. 1–7.
Supplementary Video 1
A 360° view of the dimeric cryo-EM map of E. coli Mg2+ transporter MgtA.
Supplementary Video 2
A 360° view of the monomeric cryo-EM map of E. coli Mg2+ transporter MgtA.
Supplementary Video 3
Morph between the structural model of a single subunit of the dimeric and monomeric E. coli Mg2+ transporter MgtA. The morph starts in the open monomeric state, morphs into the more compact state observed in all dimer structures and then morphs back into the monomeric state to indicate the large conformational changes observed.
Supplementary Video 4
MD simulation movie of the E. coli Mg2+ transporter MgtA showing the full dimer (in color) aligned to the cryo-EM structure (gray).
Supplementary Video 5
Zoomed in MD simulation movie of the E. coli Mg2+ transporter MgtA dimer interface.
Supplementary Video 6
The E. coli Mg2+ transporter MgtA dimer with ATP bound.
Supplementary Video 7
MD simulation movie of the Mg2+ ion in the middle of the transmembrane domains of the E. coli Mg2+ transporter MgtA. Analysis of the radial distribution of atoms around the transmembrane Mg2+ ion indicates that it is coordinated by six oxygens for the last half of the simulation; it is fully coordinated.
Supplementary Video 8
MD simulation movie of the N-terminal tail of the E. coli Mg2+ transporter MgtA.
Supplementary Tables 1–8
Table 1 Residues involved in dimer interface; Table 2 Summary of all mutants generated and their outcome; Table 3 Residues involved in ATP binding; Table 4 Primers; Table 5 Plasmids; Table 6 Strains; Table 7 Cryo-EM data collection parameters and analysis; Table 8 Cryo-EM map and model analysis.
Source data
Source data Fig. 2
Unprocessed western blots and/or gels.
Source data Fig. 3
Unprocessed growth assays.
Source data Fig. 4
Unprocessed gels and growth assays.
Source data Extended Data Fig. 3
Unprocessed western blots and/or gels.
Source data Extended Data Fig. 9
Unprocessed western blots and/or gels.
Source data Figs. 2 and 4 and Extended Data Figs. 3, 5, 7 and 9f
Statistical source data for Figs. 2c and 4f and Extended Data Figs. 3e,f, 5e, 7a and 9f.
Rights and permissions
About this article

Cite this article
Zeinert, R., Zhou, F., Franco, P. et al. P-type ATPase magnesium transporter MgtA acts as a dimer. Nat Struct Mol Biol 32, 1633–1643 (2025). https://doi.org/10.1038/s41594-025-01593-7
Received:
Accepted:
Published:
Issue date:
DOI: https://doi.org/10.1038/s41594-025-01593-7
