
Abstract
Redox processes are at the heart of energetic metabolism that drives life on earth. By extension, complex and efficient electron transfer wires are necessary to connect the various metabolic pathways that are often located in distinct cellular compartments. Here, we uncovered a structural module that enables channeling of quinones from the membrane to various water-soluble redox catalytic units in prokaryotes. Using X-ray crystallography and cryo-electron microscopy, we determined the structure of the unusual bacterial formate dehydrogenase ForCE that contains four ForC catalytic subunits docked around a membrane-associated tetrameric ForE central scaffold. In the latter, a conserved domain that we propose to name helical membrane plugin (HMP) was identified as essential to link formate oxidation, in Bacillus subtilis, to the aerobic respiratory chain. Our bioinformatic analysis indicates that this HMP is associated with different quinone-reducing oxidoreductases, highlighting its broad importance as a functional unit to wire electrons between a given catalytic redox center and the quinone pool.
This is a preview of subscription content, access via your institution
Access options
Access Nature and 54 other Nature Portfolio journals
Get Nature+, our best-value online-access subscription
$32.99 / 30 days
cancel any time
Subscribe to this journal
Receive 12 print issues and online access
$259.00 per year
only $21.58 per issue
Buy this article
- Purchase on SpringerLink
- Instant access to the full article PDF.
USD 39.95
Prices may be subject to local taxes which are calculated during checkout




Similar content being viewed by others


Data availability
The atomic coordinates of the final models, diffraction data and corresponding cryo-EM maps were deposited to the PDB and EMDB under accession codes PDB 8RQZ, PDB 8RR0 and EMD-19452 for BsForCE1 and PDB 9GZQ for BsForCE1-ΔHMP. Source data are provided with this paper.
References
Hay Mele, B. et al. Oxidoreductases and metal cofactors in the functioning of the earth. Essays Biochem. 67, 653–670 (2023).
Garcia, A. K. et al. Nitrogenase resurrection and the evolution of a singular enzymatic mechanism. eLife 12, e85003 (2023).
Peters, J. W. et al. [FeFe]- and [NiFe]-hydrogenase diversity, mechanism, and maturation. Biochim. Biophys. Acta Mol. Cell Res. 1853, 1350–1369 (2015).
Wells, M. et al. Methane, arsenic, selenium and the origins of the DMSO reductase family. Sci. Rep. 10, 10946 (2020).
Schoepp-Cothenet, B. et al. The ineluctable requirement for the trans-iron elements molybdenum and/or tungsten in the origin of life. Sci. Rep. 2, 263 (2012).
Raanan, H., Pike, D. H., Moore, E. K., Falkowski, P. G. & Nanda, V. Modular origins of biological electron transfer chains. Proc. Natl Acad. Sci. USA 115, 1280–1285 (2018).
Magalon, A., Ceccaldi, P. & Schoepp-Cothenet, B. The prokaryotic Mo/W-bisPGD enzymes family. In Molybdenum and Tungsten Enzymes (eds Hille, R., Schulzke, C. & Kirk, M. L.) (Royal Society of Chemistry, 2016).
Grimaldi, S., Schoepp-Cothenet, B., Ceccaldi, P., Guigliarelli, B. & Magalon, A. The prokaryotic Mo/W-bisPGD enzymes family: a catalytic workhorse in bioenergetic. Biochim. Biophys. Acta Bioenerg. 1827, 1048–1085 (2013).
Arias-Cartín, R. et al. Identification and characterization of a noncanonical menaquinone-linked formate dehydrogenase. J. Biol. Chem. 298, 101384 (2022).
Radon, C. et al. Cryo-EM structures reveal intricate Fe–S cluster arrangement and charging in Rhodobacter capsulatus formate dehydrogenase. Nat. Commun. 11, 1912 (2020).
Yoshikawa, T. et al. Multiple electron transfer pathways of tungsten-containing formate dehydrogenase in direct electron transfer-type bioelectrocatalysis. Chem. Commun. 58, 6478–6481 (2022).
Zuchan, K., Baymann, F., Baffert, C., Brugna, M. & Nitschke, W. The dyad of the Y-junction- and a flavin module unites diverse redox enzymes. Biochim. Biophys. Acta Bioenerg. 1862, 148401 (2021).
Peters, J. W., Lanzilotta, W. N., Lemon, B. J. & Seefeldt, L. C. X-ray crystal structure of the Fe-only hydrogenase (CpI) from Clostridium pasteurianum to 1.8 angstrom resolution. Science 282, 1853–1858 (1998).
Sazanov, L. A. & Hinchliffe, P. Structure of the hydrophilic domain of respiratory complex I from Thermus thermophilus. Science 311, 1430–1436 (2006).
Shomura, Y. et al. Structural basis of the redox switches in the NAD+-reducing soluble [NiFe]-hydrogenase. Science 357, 928–932 (2017).
Feng, X., Schut, G. J., Haja, D. K., Adams, M. W. W. & Li, H. Structure and electron transfer pathways of an electron-bifurcating NiFe-hydrogenase. Sci. Adv. 8, eabm7546 (2022).
Harmer, J. R., Hakopian, S., Niks, D., Hille, R. & Bernhardt, P. V. Redox characterization of the complex molybdenum enzyme formate dehydrogenase from Cupriavidus necator. J. Am. Chem. Soc. 145, 25850–25863 (2023).
Duffus, B. R., Gauglitz, M., Teutloff, C. & Leimkühler, S. Redox potentials elucidate the electron transfer pathway of NAD+-dependent formate dehydrogenases. J. Inorg. Biochem. 253, 112487 (2024).
Grinter, R. et al. Structural basis for bacterial energy extraction from atmospheric hydrogen. Nature 615, 541–547 (2023).
Greening, C., Berney, M., Hards, K., Cook, G. M. & Conrad, R. A soil actinobacterium scavenges atmospheric H2 using two membrane-associated, oxygen-dependent [NiFe] hydrogenases. Proc. Natl Acad. Sci. USA 111, 4257–4261 (2014).
Marreiros, B. C., Sena, F. V., Sousa, F. M., Batista, A. P. & Pereira, M. M. Type II NADH:quinone oxidoreductase family: phylogenetic distribution, structural diversity and evolutionary divergences. Environ. Microbiol. 18, 4697–4709 (2016).
Sousa, F. M., Pereira, J. G., Marreiros, B. C. & Pereira, M. M. Taxonomic distribution, structure/function relationship and metabolic context of the two families of sulfide dehydrogenases: SQR and FCSD. Biochim. Biophys. Acta Bioenerg. 1859, 742–753 (2018).
Winstedt, L. & von Wachenfeldt, C. Terminal oxidases of Bacillus subtilis strain 168: one quinol oxidase, cytochrome aa3 or cytochrome bd, is required for aerobic growth. J. Bacteriol. 182, 6557–6564 (2000).
Ojha, S., Meng, E. C. & Babbitt, P. C. Evolution of function in the ‘two dinucleotide binding domains’ flavoproteins. PLoS Comput. Biol. 3, e121 (2007).
Vernède, X. & Fontecilla-Camps, J. C. A method to stabilize reduced and/or gas-treated protein crystals by flash-cooling under a controlled atmosphere. J. Appl. Crystallogr. 32, 505–509 (1999).
Kabsch, W. XDS. Acta Crystallogr. D Biol. Crystallogr. 66, 125–132 (2010).
Tickle, I. J. et al. STARANISO. Global Phasing http://staraniso.globalphasing.org (2018).
Agirre, J. et al. The CCP4 suite: integrative software for macromolecular crystallography. Acta Crystallogr. D Struct. Biol. 79, 449–461 (2023).
Liebschner, D. et al. Macromolecular structure determination using X-rays, neutrons and electrons: recent developments in PHENIX. Acta Crystallogr. D Struct. Biol. 75, 861–877 (2019).
Cherrier, M. V. et al. Oxygen-sensitive metalloprotein structure determination by cryo-electron microscopy. Biomolecules 12, 441 (2022).
Emsley, P., Lohkamp, B., Scott, W. G. & Cowtan, K. Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 66, 486–501 (2010).
Schorb, M., Haberbosch, I., Hagen, W. J. H., Schwab, Y. & Mastronarde, D. N. Software tools for automated transmission electron microscopy. Nat. Methods 16, 471–477 (2019).
Punjani, A., Rubinstein, J. L., Fleet, D. J. & Brubaker, M. A. cryoSPARC: algorithms for rapid unsupervised cryo-EM structure determination. Nat. Methods 14, 290–296 (2017).
Pettersen, E. F. et al. UCSF ChimeraX: structure visualization for researchers, educators, and developers. Protein Sci. 30, 70–82 (2021).
Jollie, D. R. & Lipscomb, J. D. Formate dehydrogenase from Methylosinus trichosporium OB3b. Purification and spectroscopic characterization of the cofactors. J. Biol. Chem. 266, 21853–21863 (1991).
Hartmann, T. & Leimkühler, S. The oxygen-tolerant and NAD+-dependent formate dehydrogenase from Rhodobacter capsulatus is able to catalyze the reduction of CO2 to formate. FEBS J. 280, 6083–6096 (2013).
Wang, S., Huang, H., Kahnt, J. & Thauer, R. K. Clostridium acidurici electron-bifurcating formate dehydrogenase. Appl. Environ. Microbiol. 79, 6176–6179 (2013).
Laukel, M., Chistoserdova, L., Lidstrom, M. E. & Vorholt, J. A. The tungsten-containing formate dehydrogenase from Methylobacterium extorquens AM1: purification and properties. Eur. J. Biochem. 270, 325–333 (2003).
Jumper, J. et al. Highly accurate protein structure prediction with AlphaFold. Nature 596, 583–589 (2021).
Nguyen, L.-T., Schmidt, H. A., von Haeseler, A. & Minh, B. Q. IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 32, 268–274 (2015).
Kalyaanamoorthy, S., Minh, B. Q., Wong, T. K. F., von Haeseler, A. & Jermiin, L. S. ModelFinder: fast model selection for accurate phylogenetic estimates. Nat. Methods 14, 587–589 (2017).
Hoang, D. T., Chernomor, O., von Haeseler, A., Minh, B. Q. & Vinh, L. S. UFBoot2: improving the ultrafast bootstrap approximation. Mol. Biol. Evol. 35, 518–522 (2018).
Letunic, I. & Bork, P. Interactive Tree Of Life (iTOL) v5: an online tool for phylogenetic tree display and annotation. Nucleic Acids Res. 49, W293–W296 (2021).
Wheeler, T. J., Clements, J. & Finn, R. D. Skylign: a tool for creating informative, interactive logos representing sequence alignments and profile hidden Markov models. BMC Bioinformatics 15, 7 (2014).
Potter, S. C. et al. HMMER web server: 2018 update. Nucleic Acids Res. 46, W200–W204 (2018).
Rasmussen, S., Nielsen, H. B. & Jarmer, H. The transcriptionally active regions in the genome of Bacillus subtilis. Mol. Microbiol. 73, 1043–1057 (2009).
Martin-Verstraete, I., Débarbouillé, M., Klier, A. & Rapoport, G. Levanase operon of Bacillus subtilis includes a fructose-specific phosphotransferase system regulating the expression of the operon. J. Mol. Biol. 214, 657–671 (1990).
Konkol, M. A., Blair, K. M. & Kearns, D. B. Plasmid-encoded ComI inhibits competence in the ancestral 3610 strain of Bacillus subtilis. J. Bacteriol. 195, 4085–4093 (2013).
Stoll, S. & Schweiger, A. EasySpin, a comprehensive software package for spectral simulation and analysis in EPR. J. Magn. Reson. https://doi.org/10.1016/j.jmr.2005.08.013 (2006).
Dhawan, I. K. et al. Spectroscopic studies of the tungsten-containing formaldehyde ferredoxin oxidoreductase from the hyperthermophilic archaeon Thermococcus litoralis. J. Biol. Inorg. Chem. 5, 313–327 (2000).
Lanciano, P. et al. New method for the spin quantitation of [4Fe–4S]+ clusters with S = 3/2. Application to the FS0 center of the NarGHI nitrate reductase from Escherichia coli. J. Phys. Chem. B 111, 13632–13637 (2007).
Artz, J. H. et al. Reduction potentials of [FeFe]-hydrogenase accessory iron-sulfur clusters provide insights into the energetics of proton reduction catalysis. J. Am. Chem. Soc. 139, 9544–9550 (2017).
Gladyshev, V. N. et al. Characterization of crystalline formate dehydrogenase H from Escherichia coli. Stabilization, EPR spectroscopy, and preliminary crystallographic analysis. J. Biol. Chem. 271, 8095–8100 (1996).
Brondino, C. D. et al. Incorporation of either molybdenum or tungsten into formate dehydrogenase from Desulfovibrio alaskensis NCIMB 13491; EPR assignment of the proximal iron-sulfur cluster to the pterin cofactor in formate dehydrogenases from sulfate-reducing bacteria. J. Biol. Inorg. Chem. 9, 145–151 (2004).
Oliveira, A. R. et al. Toward the mechanistic understanding of enzymatic CO2 reduction. ACS Catal. 10, 3844–3856 (2020).
Friedrich, T. The NADH:ubiquinone oxidoreductase (complex I) from Escherichia coli. Biochim. Biophys. Acta 1364, 134–146 (1998).
Ohnishi, T. Iron-sulfur clusters/semiquinones in complex I. Biochim. Biophys. Acta 1364, 186–206 (1998).
Yakovlev, G., Reda, T. & Hirst, J. Reevaluating the relationship between EPR spectra and enzyme structure for the iron–sulfur clusters in NADH:quinone oxidoreductase. Proc. Natl Acad. Sci. USA 104, 12720–12725 (2007).
Reda, T., Plugge, C. M., Abram, N. J. & Hirst, J. Reversible interconversion of carbon dioxide and formate by an electroactive enzyme. Proc. Natl Acad. Sci. USA 105, 10654–10658 (2008).
Gauquelin, C. et al. Roles of the F-domain in [FeFe] hydrogenase. Biochim. Biophys. Acta Bioenerg. 1859, 69–77 (2018).
Acknowledgements
We are grateful to E. Foulquier-Khadaroo and A. Galinier (Aix Marseille Univ, CNRS, LCB) for providing protocols and advice for B. subtilis genetics and culture, C. Aubert and B. Py (Aix Marseille Univ, CNRS, LCB) for lending us the anaerobic chamber, E. Launay (Univ. Grenoble-Alpes) for her contribution to the cryo-EM analysis, G. Brandelet (Aix Marseille Univ, CNRS, CEA, BIAM) for her contribution to the XRC analysis, B. Legeret (Aix Marseille Univ, CNRS, CEA, BIAM), H. Le Guenno (Plateforme de microscopie, IMM, Aix Marseille Univ, CNRS, Marseille, France) and A. Kosta (Plateforme de microscopie, IMM, Aix Marseille Univ, CNRS, Marseille, France), the EPR facilities available at the Aix-Marseille EPR center, the financial support from the IR INFRANALYTICS FR2054 for conducting the research, the ESRF for provision of synchrotron radiation facilities under proposal number mx2398 and the local contact A. Gautam for assistance and support in using beamline ID23-eh2 (https://doi.org/10.15151/ESRF-ES-925342287). This work was supported by the French National Research Agency in the framework of the Investissements d’Avenir program (ANR-15-IDEX-02-YN) through the funding of the ‘Origin of Life’ project of the Univ. Grenoble-Alpes. This work used the platform PBSIM de l’AFMB supported by FRISBI and the platforms of the Grenoble Instruct-ERIC center (ISBG; UAR 3518 CNRS-CEA-UGA-EMBL) within the Grenoble Partnership for Structural Biology, which is supported by FRISBI (ANR-10-INBS-05-02-YN) and GRAL and financed within the Univ. Grenoble-Alpes graduate school (Ecoles Universitaires de Recherche) CBH-EUR-GS (ANR-17-EURE-0003-YN). The EM facility is supported by the Auvergne–Rhône–Alpes region, the Fondation pour la Recherche Médicale, the fonds FEDER and the GIS Infrastructures en Biologie Santé et Agronomie. I.B.S. acknowledges integration into the Interdisciplinary Research Institute of Grenoble. This work was also supported by the French National Research Agency (ANR-22-CE44-0035-01-AW), the CNRS and the CNRS Energy Unit (Cellule Energie) through the project MICROBIOCO2. U.A. and B.M. were supported by an MESR fellowship and A.-C.R. was supported by an ANR MOLYERE (ANR 16-CE29-0010-01-BG) project postdoctoral fellowship. The funders had no role in study design, data collection and analysis, decision to publish or preparation of the manuscript.
Author information
Authors and Affiliations
Contributions
W.A., A.-C.R., A.P., M.A., C.M.V., B.M. and G.S. designed the experiments. U.A., W.A., P.F., S.F., G.B., B.G., L.P., B.M., A.-C.R., M.M.J., A.P., M.L., S.G., C.M.V., M.A. and G.S. performed the experiments. M.A., W.A., G.B., A.-C.R., C.M.V., N.Y., B.M., A.P. and G.S. wrote, reviewed and edited the manuscript. M.A., W.A., G.B., G.S., A.P. and N.Y. provided the resources and obtained funding.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Structural & Molecular Biology thanks the anonymous reviewers for their contribution to the peer review of this work. Peer reviewer reports are available. Primary Handling Editor: Dimitris Typas, in collaboration with the Nature Structural & Molecular Biology team.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data
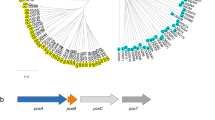
Extended Data Fig. 1 In-depth bioinformatics analysis of cytoplasmic multidomain FDHs.
a. Phylogenetic tree generated of the N-terminal domain of 442 FDH catalytic subunit sequences (Cata SU) using a maximum likelihood approach. Inter-sequence identity was reduced to a maximum of 90% between any two sequences. Sequences forming a clade have been collapsed. The presence of specific redox centers was deduced from the identification of conserved motifs in the primary amino acid sequence and the partner subunit(s) from the genomic organization. The arrow indicates the position at scale between the N-terminal and the C-terminal domain. Characterized representatives are indicated: Bacillus subtilis (1), Methylobacterium extorquens AM1 (2), Rhodobacter capsulatus (3) and Gottschalkia acidurici (4). b. Phylogenetic tree of the C-terminal domain of 442 FDH sequences using a maximum likelihood approach. Each branch tip represents a single sequence. ForC sequences are colored red. Both trees were built with IQ-TREE using the WAG + I + G4 model. Supports at nodes are ultrafast bootstrap values above 95% and are indicated by a black circle. iTOL 6.8.1 was used to generate the figures.
Extended Data Fig. 2 Biochemical and structural analysis of BsForCE1 and variants.
a. ForC and ForE form a complex with a 4:4 stoichiometry as shown by SEC-MALS analysis of BsForCE1 WT (blue curve), BsForCE1F128D-R132D (green curve) and BsForCE1ΔHMP (orange curve). Left y-axis, molar mass; right y-axis normalized UV chromatograms, measured molar mass in coloured stars. For BsForCE1 WT and BsForCE1F128D-R132D, complexes have a MW of 512 ± 1.5 and 495 ± 1.5 kDa respectively. For BsForCE1ΔHMP, the complex has a MW of 470 ± 1.9 kDa. In addition, two proteins of 117 ± 1.2 kDa and 111 ± 0.8 kDa could correspond to a monomer of ForC1. b., c. Inhibition of the formate:O2 consumption by respiratory chain inhibitors. WT strain was grown with 12.5 mM formate. O2 consumption was measured using an oxygen electrode after formate spiking, in presence of 1.29 to 27.1 μM HQNO (b) or 0.16 to 7.6 mM KCN (c). 100% corresponds to 153 nmol O2/min/mg. d., e. Michaelis Menten plot of BsForCE1F128-DR132D (d) and BsForCE1ΔHMP (e) coupling formate oxidation to menadione reduction in vitro. Data from n = 3 biological replicates are presented as mean values +/− SD. f. Conservation of hydrophobic and positively charged residues at the respective F128 and R132 positions in the DUF1641 family. Sequences from Interpro IPR012440 (DUF1641) family were clustered and restricted to 644 representatives using MMseqs2 (Many-against-Many searching, https://toolkit.tuebingen.mpg.de/tools/mmseqs2) online software (minimum sequence identity>0.5; minimum alignment coverage>0.8). Then, multiple sequence alignment (MSA) was performed using Clustal Ω (default parameters). Finally, HMM logo was generated using http://skylign.org/ with the previously described MSA (from K118 to Q155, BsForE1 numbering) to consider both helix 7 and 8. Each stack represents one residue position within the MSA. Height of a letter within a stack represents the frequency of the corresponding residue at this position. The sequence of BsForE1 is indicated below the HMM logo and helices 7 and 8 are indicated. g. X-ray structure of BsForCE1ΔHMP superimposes with BsForCE1. BsForCE1ΔHMP is colored according to the b-factor value while BsForCE1 is colored in gray. For clarity, one ForC subunit is shown on both structures.
Extended Data Fig. 3 ForCE interaction with lipids.
a. Crystallographic and b. cryo-EM lipid composition inside the cavity formed by ForE tetramer. Structures are displayed in surface for the protein residues. Lipids, menaquinone and FeS clusters are depicted as sticks. ForC and ForE proteins surfaces are colored in dark blue and orange respectively. Non-protein carbon, oxygen, sulfur and iron atoms are colored in light blue, red, yellow and brown, respectively. c. Crystallographic and d. cryo-EM ForE central cavity access. ForC and ForE proteins surface are colored in dark blue and orange respectively. Glycero-phospholipid carbon, oxygen, and phosphorus atoms are colored in light blue, red and orange respectively. e. Hydrophobic (left) and electrostatic (center and right) surface calculations of the ForCE protein. Hydrophobic and hydrophilic residues are colored in light orange and light blue respectively. The cutaway shows the internal surface of ForE lined with hydrophobic residues (left). Positively charged amino acids are colored in blue, negatively charged amino acids in red, and neutral residues in white. The membrane was positioned at the level of the C-terminal end of the ForE tube capped by fatty acids (center). The right view shows the electrostatic potential of the HMP interacting with phospholipids.
Extended Data Fig. 4 Comparison between the Q-site in ForC (a) and HucS (b).
a. α-helices 223–236 and 240–257 of the ForC V-shape motif are represented in cartoon colored in dark blue with the side chain (T100 and E237). Electron transfer distance between the menaquinone and the His-coordinated [4Fe–4S] cluster is indicated. α-helices of ForE are colored in yellow and brown and numbered. b. α-helix 298–311 from the HucS subunit is represented in cartoon colored in dark blue and light blue for the two symmetry related molecules, with the side chain of Y301 and Y229 and the backbone carbonyl of K212. Electron transfer distance between the menaquinone and the distal [3Fe–4S] cluster is indicated. α-helices of HucM are colored in yellow and brown and numbered. a. and b. Clusters are represented in balls and sticks. Oxygen, sulfur and iron atoms are colored in red, yellow and brown respectively.
Extended Data Fig. 5 Comparison between ForC and canonical formate dehydrogenase.
a. Structural superimposition of ForC1 with FdsA from the FDH of R. capsulatus (6TGA) colored according to the RMSD value. The specific ForC1 regions (that is the V-shape) are colored in grey. b. Comparison of the active site of BsForCE1 (left) to the one of R. capsulatus FDH (right). Protein atoms and bis-pyranopterin guanosine dinucleotides (PGD) atoms are represented in sticks. Molybdenum and sulfur atoms are represented in balls. Carbon, oxygen, nitrogen, sulfur and molybdenum atoms are colored in gray, red, blue, yellow and turquoise respectively.
Extended Data Fig. 6 EPR and redox analysis of [Fe-S] clusters in BsForCE1.
a. Representative EPR spectra of BsForCE1 S = 1/2 [Fe-S] clusters. Simulations (red traces) are superimposed on experimental spectra (black traces). The g values of [Fe-S] clusters are indicated with vertical dotted lines. Experimental conditions: Temperature, 15 K (1, 3), 30 K (2), 100 K (4); microwave power, 100 mW (1), 10 mW (2–4); field modulation amplitude, 1 mT; microwave frequency, 9.4812 GHz (1), 9.4791 GHz (2), 9.4801 GHz (3), 9.4809 GHz (4); redox potential (pH = 6.0), −288 mV (1), −280 mV (2), −391 mV (3), −395 mV (4). Simulation parameters are given in Extended Data Table 1. b. and c. EPR-monitored redox titration experiments of [Fe-S] clusters in BsForCE1 buffered at pH = 6 (b) and pH = 7.5 (c). EPR signal amplitudes of the Fe/S1 [4Fe-4S] (g3 = 1.825, green filled squares), the proximal [4Fe-4S] (g1 = 2.044, blue filled triangles), Fe/S2 (g2 = 1.964, red filled circles), and the [2Fe-2S] cluster (g1 = 2.018, pink filled triangles) were measured against the ambient redox potential. They were fitted to Nernst plots (solid lines) as described in the Methods, yielding estimated midpoint potential values as given in Fig. 2b. Experimental conditions: 15 K (Fe/S1 and Fe/S2) or 50 K (proximal [4Fe-4S], [2Fe-2S]), 100 mW (Fe/S1), 10 mW (proximal [4Fe-4S] at pH = 7.5, Fe/S2, [2Fe-2S]), 1 mW (proximal [4Fe-4S] at pH = 6.0), modulation amplitude, 1 mT (Fe/S1 and proximal [4Fe-4S] at pH = 6.0, Fe/S2), 0.5 mT (proximal [4Fe-4S] at pH = 7.5, [2Fe-2S]). d. Low field region of EPR spectra of dithionite-reduced BsForCE1 buffered at pH 6.0 and poised at −245 mV (top) and −390 mV (bottom). The signal with a maximum at geff2 = 5.19 and a shoulder at geff1 = 5.45 is ascribed to a [4Fe-4S]+ cluster with an S = 3/2 ground state (Supplementary Note 2 and Supplementary Figs. 6, 7). Experimental conditions: temperature, 8 K, microwave frequency, 9.480 GHz, microwave power, 100 mW, field modulation amplitude, 3 mT, 4 scans. The inset shows a potentiometric titration of the geff1 = 5.45 signal. Experimental data were fitted to a n = 1 Nernst curve using an Em = −275 mV, with an estimated accuracy of ± 20 mV. The signals from the S = 3/2 [4Fe-4S] (inset) and from the S = 1/2 proximal cluster (panel b) titrate with the same redox potential value (within the experimental error, Fig. 2b). Based on this similarity, they are assigned to the same cluster, that is, the proximal one, which consequently exists in a mixture of S = 1/2 and S = 3/2 spin states, as reported for clusters coordinated exclusively by cysteines or by a mixed 3-Cys and 1-His50,51.
Extended Data Fig. 7 Comparison of ForCE lipid composition in the crystal structure (left) and cryoEM reconstruction (right).
Main chain of the protein is represented in cartoon with the side chain in sticks. Lipids are represented in sticks. Carbon atoms are colored in dark blue for ForC and orange for ForE. Oxygen, nitrogen, phosphorus and sulfur, are colored in red, blue, orange and yellow respectively. For the crystallographic structure, Fo-Fc difference Fourier electron density map (omit map) is displayed as green mesh and contoured at 3σ. For the cryoEM reconstruction, Coulomb potential density is displayed as a beige semi-transparent surface and contoured at 4σ for ForE C-ter region, glycerophospholipid 1 and 2 and contoured at 3.4σ for ForE N-ter region.
Extended Data Fig. 8 Analysis of the HMP/DUF1641 family.
a. The nodes/sequences are colored according to cluster type, b. kingdom of the organisms and c. sequence length. A good separation between archeal and bacterial sequences suggests a divergent evolution of the family. We also noted that NDH-2 organization seems to be present only in bacteria, and that some clusters tend to contain sequences of homogeneous length. d. Sequence alignment of HMP/DUF1641-containing proteins. Two sequences from two different clusters from each type of functional arrangement, type I (clusters 1 and 3), type II (clusters 2 and 6), type III (clusters 4 and 6) and type IV (clusters 7 and 17), are aligned using Muscle and visualized in Jalview using the clustal coloring of residues and a transparency linked to conservation threshold of 30%. The position of the HMP/DUF1641 is boxed.
Extended Data Fig. 9 Cryo-EM reconstruction.
a. ForCE cryo-EM data processing workflow. b. Motion-corrected micrograph of ForCE complex. c. Fourier-shell-correlation plots for independently refined half-maps. Horizontal blue lines indicate Fourier-shell-correlation values of 0.143. d. Examples of 2D classes of the final selection of particles. Number of the particles and estimated resolution in each 2D class are indicated. e. Guinier plot of the final homogeneous refinement indicating a sharpening B-factor of 119.5 Å2. f. Angular distribution of the particles used for the final reconstruction.
Supplementary information
Supplementary Information
Supplementary Tables 1 and 4, Figs. 1–9, Notes 1–4 and References.
Supplementary Table 1
All GI numbers used for the MSA.
Supplementary Table 2
All sequences used for phylogeny analysis.
Source data
Source Data
Statistical source data.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Broc, M., Cherrier, M.V., Uzel, A. et al. A scaffold for quinone channeling between membrane and soluble bacterial oxidoreductases. Nat Struct Mol Biol 32, 2196–2202 (2025). https://doi.org/10.1038/s41594-025-01607-4
Received:
Accepted:
Published:
Version of record:
Issue date:
DOI: https://doi.org/10.1038/s41594-025-01607-4
