
Abstract
PepSeq is an in vitro platform for building and conducting highly multiplexed proteomic assays against customizable targets by using DNA-barcoded peptides. Starting with a pool of DNA oligonucleotides encoding peptides of interest, this protocol outlines a fully in vitro and massively parallel procedure for synthesizing the encoded peptides and covalently linking each to a corresponding cDNA tag. The resulting libraries of peptide/DNA conjugates can be used for highly multiplexed assays that leverage high-throughput sequencing to profile the binding or enzymatic specificities of proteins of interest. Here, we describe the implementation of PepSeq for fast and cost-effective epitope-level analysis of antibody reactivity across hundreds of thousands of peptides from <1 µl of serum or plasma input. This protocol includes the design of the DNA oligonucleotide library, synthesis of DNA-barcoded peptide constructs, binding of constructs to sample, preparation for sequencing and data analysis. Implemented in this way, PepSeq can be used for a number of applications, including fine-scale mapping of antibody epitopes and determining a subject’s pathogen exposure history. The protocol is divided into two main sections: (i) design and synthesis of DNA-barcoded peptide libraries and (ii) use of libraries for highly multiplexed serology. Once oligonucleotide templates are in hand, library synthesis takes 1–2 weeks and can provide enough material for hundreds to thousands of assays. Serological assays can be conducted in 96-well plates and generate sequencing data within a further ~4 d. A suite of software tools, including the PepSIRF package, are made available to facilitate the design of PepSeq libraries and analysis of assay data.
This is a preview of subscription content, access via your institution
Access options
Access Nature and 54 other Nature Portfolio journals
Get Nature+, our best-value online-access subscription
$32.99 / 30 days
cancel any time
Subscribe to this journal
Receive 12 print issues and online access
$259.00 per year
only $21.58 per issue
Buy this article
- Purchase on SpringerLink
- Instant access to the full article PDF.
USD 39.95
Prices may be subject to local taxes which are calculated during checkout






Similar content being viewed by others
Data availability
Data used to generate Figs. 2 and 4 and Extended Data Fig. 3 are publicly available through Open Science Framework (https://osf.io/kwxtc/).
Code availability
All custom software described in the protocol is publicly available through GitHub: Library-Design: https://github.com/LadnerLab/Library-Design; PepSIRF: https://github.com/LadnerLab/PepSIRF; and Qiime2 plug-ins: https://github.com/LadnerLab/q2-pepsirf, https://github.com/LadnerLab/q2-ps-plot and https://github.com/LadnerLab/q2-autopepsirf.
Change history
16 May 2024
A Correction to this paper has been published: https://doi.org/10.1038/s41596-024-01010-1
References
Vengesai, A. et al. A systematic and meta-analysis review on the diagnostic accuracy of antibodies in the serological diagnosis of COVID-19. Syst. Rev. 10, 155 (2021).
Pollán, M. et al. Prevalence of SARS-CoV-2 in Spain (ENE-COVID): a nationwide, population-based seroepidemiological study. Lancet 396, 535–544 (2020).
Lequin, R. M. Enzyme immunoassay (EIA)/enzyme-linked immunosorbent assay (ELISA). Clin. Chem. 51, 2415–2418 (2005).
Taylor, C. T. et al. Detection of specific ZIKV IgM in travelers using a multiplexed flavivirus microsphere immunoassay. Viruses 10, 253 (2018).
Graham, H., Chandler, D. J. & Dunbar, S. A. The genesis and evolution of bead-based multiplexing. Methods 158, 2–11 (2019).
Tokarz, R. et al. A multiplex serologic platform for diagnosis of tick-borne diseases. Sci. Rep. 8, 3158 (2018).
Buus, S. et al. High-resolution mapping of linear antibody epitopes using ultrahigh-density peptide microarrays. Mol. Cell. Proteom. 11, 1790–1800 (2012).
Xu, G. J. et al. Viral immunology. Comprehensive serological profiling of human populations using a synthetic human virome. Science 348, aaa0698 (2015).
Mohan, D. et al. PhIP-Seq characterization of serum antibodies using oligonucleotide-encoded peptidomes. Nat. Protoc. 13, 1958–1978 (2018).
Larman, H. B. et al. Autoantigen discovery with a synthetic human peptidome. Nat. Biotechnol. 29, 535–541 (2011).
Mina, M. J. et al. Measles virus infection diminishes preexisting antibodies that offer protection from other pathogens. Science 366, 599–606 (2019).
Roberts, R. W. & Szostak, J. W. RNA-peptide fusions for the in vitro selection of peptides and proteins. Proc. Natl Acad. Sci. USA 94, 12297–12302 (1997).
Kozlov, I. A. et al. A highly scalable peptide-based assay system for proteomics. PLoS One 7, e37441 (2012).
Shiryaev, S. A. et al. High-resolution analysis and functional mapping of cleavage sites and substrate proteins of furin in the human proteome. PLoS One 8, e54290 (2013).
Shiryaev, S. A. et al. Substrate cleavage profiling suggests a distinct function of Bacteroides fragilis metalloproteinases (fragilysin and metalloproteinase II) at the microbiome-inflammation-cancer interface. J. Biol. Chem. 288, 34956–34967 (2013).
Shiryaev, S. A. et al. Structural and functional diversity of metalloproteinases encoded by the Bacteroides fragilis pathogenicity island. FEBS J. 281, 2487–2502 (2014).
Kukreja, M. et al. High-throughput multiplexed peptide-centric profiling illustrates both substrate cleavage redundancy and specificity in the MMP Family. Chem. Biol. 22, 1122–1133 (2015).
Ladner, J. T. et al. Epitope-resolved profiling of the SARS-CoV-2 antibody response identifies cross-reactivity with endemic human coronaviruses. Cell Rep. Med. 2, 100189 (2021).
Elko, E. A. et al. COVID-19 vaccination elicits an evolving, cross-reactive antibody response to epitopes conserved with endemic coronavirus spike proteins. Cell Rep. 40, 11102 (2022).
Bielec, K. et al. Kinetics and equilibrium constants of oligonucleotides at low concentrations. Hybridization and melting study. Phys. Chem. Chem. Phys. 21, 10798–10807 (2019).
Björck, L. & Kronvall, G. Purification and some properties of streptococcal protein G, a novel IgG-binding reagent. J. Immunol. 133, 969–974 (1984).
Fink, Z. W., Martinez, V., Altin, J. & Ladner, J. T. PepSIRF: a flexible and comprehensive tool for the analysis of data from highly-multiplexed DNA-barcoded peptide assays. Preprint at https://arxiv.org/abs/2007.05050 (2020).
Bolyen, E. et al. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat. Biotechnol. 37, 852–857 (2019).
Brown, A. M., Bolyen, E., Raspet, I., Altin, J. A. & Ladner, J. T. PepSIRF + QIIME 2: software tools for automated, reproducible analysis of highly-multiplexed serology data. Preprint at https://arxiv.org/abs/2207.11509 (2022).
Acknowledgements
This work was supported by the NIAID (U24AI152172, U24AI152172-01S1 and U24AI152172-IOF; Principal Investigator: J.A.A.), the National Institute on Minority Health and Health Disparities of the National Institutes of Health under Award Number U54MD012388 and the State of Arizona Technology and Research Initiative Fund (TRIF, administered by the Arizona Board of Regents, through Northern Arizona University). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Author information
Authors and Affiliations
Contributions
Conceptualization, J.A.A. and J.T.L.; data curation, E.A.E., Z.F., E.J.K., H.L.M., J.A.A. and J.T.L.; formal analysis, E.A.E., Z.F., E.J.K., H.L.M., J.A.A. and J.T.L.; methodology, S.N.H., A.L.E., A.S.B., S.J.F., F.R., G.A.N., J.A.A. and J.T.L.; investigation, S.N.H., A.L.E, A.P., A.S.B., S.J.F., F.R. and G.A.N.; visualization, E.A.E., J.A.A. and J.T.L.; software, E.A.E., Z.F., V.M., A.B., E.J.K., I.R., J.A.A. and J.T.L.; validation, S.N.H., E.A.E., A.L.E., S.J.F., F.R., G.A.N. and H.L.M.; project administration, S.N.H., A.L.E., G.A.N. and J.A.A.; writing—original draft, S.N.H., E.A.E., P.M.S., A.L.E., G.A.N., J.A.A. and J.T.L.; writing—review and editing, S.N.H., E.A.E., A.L.E., A.P., F.R., G.A.N., H.L.M., J.A.A. and J.T.L.; funding acquisition, J.A.A. and J.T.L.; resources, P.M.S., Y.L. and E.J.K.; supervision, P.M.S., J.A.A. and J.T.L.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Protocols thanks the anonymous reviewers for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Related links
Key references using this protocol
Ladner, J. T. et al. Cell Rep. Med. 2, 100189 (2021): https://doi.org/10.1016/j.xcrm.2020.100189
Elko, E. A. et al. Cell Rep. 40, 111022 (2022): https://doi.org/10.1016/j.celrep.2022.111022
Schuettenberg, A. et al. Microbiol. Spect. 10, e02873-22 (2022): https://doi.org/10.1128/spectrum.02873-22
Extended data
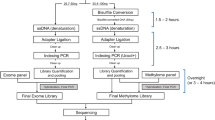
Extended Data Fig. 1 Sequence alignment for a representative PepSeq probe with amplification and sequencing primers.
Top: For DNA amplification, the forward or reverse primers bind to the PepSeq probe via the 19-nt constant regions added to either end of the DNA tag. The forward DNA amplification primer contains a T7 promoter, NEB untranslated region, start codon and TEV cleavage site sequences. The reverse DNA amplification primer contains an S6 tag and a CP1 annealing site. Bottom: For sequencing, the forward indexing primer contains a 12-nt randomer (N) and a 10-nt barcode sequence (B). The reverse indexing primer contains a separate 8-nt barcode (B). Both indexing primers bind to the DNA tags via the 19-nt constant regions. For the reverse primers, we are showing the reverse complement sequences to clearly indicate annealing regions. See Supplementary Table 1 for oligonucleotide sequences to order.
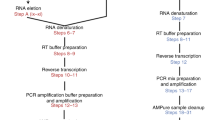
Extended Data Fig. 2 Bioinformatic pipeline for design of the PepSeq library and analysis of sequencing results.
Graphical depiction of a typical analysis workflow through peptide design and encoding (left) and bioinformatic analysis (right). Each box represents a step in the bioinformatic pipeline and includes a basic description of the step, along with a recommended piece of software (left) or PepSIRF module (right) for accomplishing the described step (contained in square brackets). Arrows indicate a direct connection, with the output file from the upstream box being used as an input file for the downstream box. The dashed box indicates a step that needs to be performed the first time running the analysis but is not required to be run for every analysis.
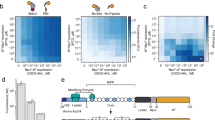
Extended Data Fig. 3 Effect of Illumina sequencing cluster density on PepSeq demultiplexed read yield.
The relationship between flowcell cluster density (x-axis) and the total number of reads successfully demultiplexed to peptides and samples (y-axis) across a series of representative sequencing runs by using mid-output (blue dots) or high-output (red dots) 150-cycle Illumina NextSeq kits. We observe a cluster density between 250 and 325 (green shaded region) to yield the greatest number of usable PepSeq reads per run, which substantially exceeds the cluster density recommended by Illumina (blue shaded region). Cluster densities for high-output kits have been normalized by a factor of 3 to allow accurate comparison with mid-output kits.
Supplementary information
Supplementary Information
Supplementary Methods and Table 1
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Henson, S.N., Elko, E.A., Swiderski, P.M. et al. PepSeq: a fully in vitro platform for highly multiplexed serology using customizable DNA-barcoded peptide libraries. Nat Protoc 18, 396–423 (2023). https://doi.org/10.1038/s41596-022-00766-8
Received:
Accepted:
Published:
Version of record:
Issue date:
DOI: https://doi.org/10.1038/s41596-022-00766-8
This article is cited by
-
Addressing gaps in the diagnoses of genetic errors of immunity
Nature Immunology (2025)
-
High-throughput multiplexed serology via the mass-spectrometric analysis of isotopically barcoded beads
Nature Biomedical Engineering (2025)
-
Molecular and physiological changes in the SpaceX Inspiration4 civilian crew
Nature (2024)
-
Virome-wide detection of natural infection events and the associated antibody dynamics using longitudinal highly-multiplexed serology
Nature Communications (2023)
