
Abstract
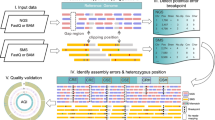
Long-read sequencing technologies yield extended DNA sequences capable of spanning intricate, repetitive genome regions, thereby facilitating the generation of more precise and comprehensive genome assemblies. However, assembly errors are inevitable owing to inherent genomic complexity and limitations of sequencing technology and assembly algorithms, making assembly evaluation crucial. The genome assembly evaluation tool Inspector presents several advantages over existing long-read de novo assembly evaluation tools, including (1) both reference-free and reference-guided assembly evaluation; (2) the ability to detect both small- and large-scale structural errors; (3) the option of assembly error correction, which can improve the quality value of the original assembly; and (4) the ability to perform haplotype-resolved assembly evaluation. Inspector can provide not only basic contig and alignment statistics, but also the precise locations and types of the different structural errors. These advantages provide a robust framework for long-read assembly evaluation. In this Protocol, we showcase four procedures to demonstrate the different applications of Inspector for long-read assembly evaluation. Inspector software and additional guides can be found at https://github.com/ChongLab/Inspector_protocol.
Key points
-
Long-read sequencing has been instrumental in improving de novo assembly of genomes. However, genome complexity and limitations of sequencing technology and assembly algorithms necessitate comprehensive evaluation of the accuracy of the assembled genomes. Inspector is a flexible tool for reference-free or reference-guided assembly evaluation, showcased in four use-case scenarios described in this protocol.
-
Inspector identifies the types and precise locations of small- and large-scale structural errors and provides an error-correction module that can improve the quality of the original assembly.
This is a preview of subscription content, access via your institution
Access options
Access Nature and 54 other Nature Portfolio journals
Get Nature+, our best-value online-access subscription
$32.99 / 30 days
cancel any time
Subscribe to this journal
Receive 12 print issues and online access
$259.00 per year
only $21.58 per issue
Buy this article
- Purchase on SpringerLink
- Instant access to the full article PDF.
USD 39.95
Prices may be subject to local taxes which are calculated during checkout



Similar content being viewed by others


Data availability
The dataset used in this protocol can be downloaded with the provided links.
Code availability
All code and commands used in this protocol are available as Supplementary Data. Additionally, the code and commands used for this protocol can be found at https://github.com/ChongLab/Inspector_protocol. The original code for Inspector is hosted at https://github.com/ChongLab/Inspector.
References
Sohn, J. I. & Nam, J. W. The present and future of de novo whole-genome assembly. Brief. Bioinform. 19, 23–40 (2018).
Siva, N. 1000 Genomes project. Nat. Biotechnol. 26, 256 (2008).
Ashley, E. A. Towards precision medicine. Nat. Rev. Genet. 17, 507–522 (2016).
The 1000 Genomes Project Consortium. An integrated map of genetic variation from 1,092 human genomes. Nature 491, 56–65 (2012).
Butler, J. et al. ALLPATHS: de novo assembly of whole-genome shotgun microreads. Genome Res. 18, 810–820 (2008).
Zerbino, D. R. & Birney, E. Velvet: algorithms for de novo short read assembly using de Bruijn graphs. Genome Res. 18, 821–829 (2008).
Luo, R. et al. SOAPdenovo2: an empirically improved memory-efficient short-read de novo assembler. Gigascience 1, 18 (2012).
Bradnam, K. R. et al. Assemblathon 2: evaluating de novo methods of genome assembly in three vertebrate species. Gigascience 2, 10 (2013).
Wang, Y., Zhao, Y., Bollas, A., Wang, Y. & Au, K. F. Nanopore sequencing technology, bioinformatics and applications. Nat. Biotechnol. 39, 1348–1365 (2021).
Wenger, A. M. et al. Accurate circular consensus long-read sequencing improves variant detection and assembly of a human genome. Nat. Biotechnol. 37, 1155–1162 (2019).
Logsdon, G. A., Vollger, M. R. & Eichler, E. E. Long-read human genome sequencing and its applications. Nat. Rev. Genet. 21, 597–614 (2020).
Gao, Y. et al. A pangenome reference of 36 Chinese populations. Nature 619, 112–121 (2023).
Sherman, R. M. & Salzberg, S. L. Pan-genomics in the human genome era. Nat. Rev. Genet. 21, 243–254 (2020).
Marx, V. Method of the year: long-read sequencing. Nat. Methods 20, 6–11 (2023).
Wang, Y., Yang, Q. & Wang, Z. The evolution of nanopore sequencing. Front. Genet. 5, 449 (2015).
Cheng, H., Concepcion, G. T., Feng, X., Zhang, H. & Li, H. Haplotype-resolved de novo assembly using phased assembly graphs with hifiasm. Nat. Methods 18, 170–175 (2021).
Nurk, S. et al. HiCanu: accurate assembly of segmental duplications, satellites, and allelic variants from high-fidelity long reads. Genome Res. 30, 1291–1305 (2020).
Kolmogorov, M., Yuan, J., Lin, Y. & Pevzner, P. A. Assembly of long, error-prone reads using repeat graphs. Nat. Biotechnol. 37, 540–546 (2019).
Ruan, J. & Li, H. Fast and accurate long-read assembly with wtdbg2. Nat. Methods 17, 155–158 (2020).
Lieberman-Aiden, E. et al. Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science 326, 289–293 (2009).
Cheng, H., Asri, M., Lucas, J., Koren, S. & Li, H. Scalable telomere-to-telomere assembly for diploid and polyploid genomes with double graph. Nat. Methods 1–4 (2024).
Cheng, H. et al. Haplotype-resolved assembly of diploid genomes without parental data. Nat. Biotechnol. 40, 1332–1335 (2022).
Rautiainen, M. et al. Telomere-to-telomere assembly of diploid chromosomes with Verkko. Nat. Biotechnol. 41, 1474–1482 (2023).
Chen, Y., Zhang, Y., Wang, A. Y., Gao, M. & Chong, Z. Accurate long-read de novo assembly evaluation with Inspector. Genome Biol. 22, 312 (2021).
Tanudisastro, H. A., Deveson, I. W., Dashnow, H. & MacArthur, D. G. Sequencing and characterizing short tandem repeats in the human genome. Nat. Rev. Genet. 25, 460–475 (2024).
Agustinho, D. P. et al. Unveiling microbial diversity: harnessing long-read sequencing technology. Nat. Methods 21, 954–966 (2024).
Logsdon, G. A. et al. Complex genetic variation in nearly complete human genomes. Preprint at bioRxiv https://doi.org/10.1101/2024.09.24.614721 (2024).
Simão, F. A., Waterhouse, R. M., Ioannidis, P., Kriventseva, E. V. & Zdobnov, E. M. BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 31, 3210–3212 (2015).
Mikheenko, A., Prjibelski, A., Saveliev, V., Antipov, D. & Gurevich, A. Versatile genome assembly evaluation with QUAST-LG. Bioinformatics 34, i142–i150 (2018).
Mikheenko, A., Saveliev, V. & Gurevich, A. MetaQUAST: evaluation of metagenome assemblies. Bioinformatics 32, 1088–1090 (2016).
Manchanda, N. et al. GenomeQC: a quality assessment tool for genome assemblies and gene structure annotations. BMC Genomics 21, 193 (2020).
Rhie, A., Walenz, B. P., Koren, S. & Phillippy, A. M. Merqury: reference-free quality, completeness, and phasing assessment for genome assemblies. Genome Biol. 21, 245 (2020).
Chen, Y. et al. Deciphering the exact breakpoints of structural variations using long sequencing reads with DeBreak. Nat. Commun. 14, 283 (2023).
Chen, Y. et al. Gene fusion detection and characterization in long-read cancer transcriptome sequencing data with fusionseeker. Cancer Res. 83, 28–33 (2023).
Kolmogorov, M. et al. metaFlye: scalable long-read metagenome assembly using repeat graphs. Nat. Methods 17, 1103–1110 (2020).
Li, H. Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics 34, 3094–3100 (2018).
Li, H. et al. The sequence alignment/map format and SAMtools. Bioinformatics 25, 2078–2079 (2009).
Pearson, W. R. & Lipman, D. J. Improved tools for biological sequence comparison. Proc. Natl Acad. Sci. USA 85, 2444–2448 (1988).
Cock, P. J., Fields, C. J., Goto, N., Heuer, M. L. & Rice, P. M. The Sanger FASTQ file format for sequences with quality scores, and the Solexa/Illumina FASTQ variants. Nucleic Acids Res. 38, 1767–1771 (2010).
Zook, J. M. et al. Extensive sequencing of seven human genomes to characterize benchmark reference materials. Sci. Data 3, 1–26 (2016).
Fairley, S., Lowy-Gallego, E., Perry, E. & Flicek, P. The International Genome Sample Resource (IGSR) collection of open human genomic variation resources. Nucleic Acids Res. 48, D941–D947 (2020).
Shafin, K. et al. Nanopore sequencing and the Shasta toolkit enable efficient de novo assembly of eleven human genomes. Nat. Biotechnol. 38, 1044–1053 (2020).
Nurk, S. et al. The complete sequence of a human genome. Science 376, 44–53 (2022).
Kent, W. J. et al. The human genome browser at UCSC. Genome Res. 12, 996–1006 (2002).
Robinson, J. T. et al. Integrative genomics viewer. Nat. Biotechnol. 29, 24–26 (2011).
Acknowledgements
This study was supported by the Department of Biomedical Informatics and Data Science, Marnix E. Heersink School of Medicine, University of Alabama at Birmingham, and the Biostatistics and Bioinformatics Shared Resource, Sylvester Comprehensive Cancer Center, University of Miami. Y.G. was supported by P30CA240139, R01ES030993 and R01ES035421 from NIH, USA Z.C. was supported by the MIRA award (1R35GM138212) from NIH/NIGMS.
Author information
Authors and Affiliations
Contributions
Y.G. and Z.C. conceived and managed the project. Y.S. collected all the datasets and performed all the analyses. L.J., Y.C., M.C. and M.G. were involved in testing and evaluating the tool. Y.G., Y.S., L.J. and Z.C. prepared the figures and tables, wrote the manuscript draft and revised the manuscript. All authors have read and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Protocols thanks Guangyi Fan and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Key reference
Chen, Y. et al. Genome Biol. 22, 312 (2021): https://doi.org/10.1186/s13059-021-02527-4
Supplementary information
Supplementary Tables 1–3
Supplementary Tables 1–3.
Supplementary Code 1
Source codes for the entire protocol.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Guo, Y., Song, Y., Jiang, L. et al. A detailed guide to assessing genome assembly based on long-read sequencing data using Inspector. Nat Protoc 20, 2845–2864 (2025). https://doi.org/10.1038/s41596-025-01149-5
Received:
Accepted:
Published:
Version of record:
Issue date:
DOI: https://doi.org/10.1038/s41596-025-01149-5
