
Abstract
Although proteomics is extensively used in immune research, there is currently no publicly accessible spectral assay library for the comprehensive proteome of immune cells. This study generated spectral assay libraries for five human immune cell lines and four primary immune cells: CD4 T, CD8 T, natural killer (NK) cells, and B cells. This was achieved by utilizing data-dependent acquisition (DDA) and employing fractionated samples from over 100 µg of proteins, which was applied to acquire the highest-quality MS/MS spectral data. In addition, Data-indedendent acquisition (DIA) was used to obtain sufficient data points for analyzing proteins from 10,000 primary CD4 T, CD8 T, NK, and B cells. The immune cell spectral assay library generated included 10,544 protein groups and 127,106 peptides. The proteomic profiles of 10,000 primary human immune cells obtained from 15 healthy volunteers analyzed using DIA revealed the highest heterogeneity of B cells among other immune cell types and the similarity between CD4 T and CD8 T cells. All data and spectral library are deposited in ProteomeXchange (PXD047742).
Similar content being viewed by others


Background & Summary
The immune system serves as a protective shield against various infections and tissue damage1,2. A comprehensive understanding of its intricate functions is vital for developing pharmaceutical treatments for various diseases3,4. Proteomics of immune cells is a crucial tool for gaining insights into the functioning of these cells in both health and disease, significantly advancing our understanding of immune processes4,5,6. As immune cells play a pivotal role in regulating various aspects of the immune system, tissues, organs, and overall biological homeostasis1,7,8,9, having a high-quality, publicly available spectral library of human immune cell proteins holds the potential to streamline and enhance future experiments by saving valuable resources, time, samples, and analysis efforts10,11,12,13.
A pan-human spectral library containing more than 10,000 proteins was published in 201413, but it had the limitation of not including primary immune cells. In 2020, a primary human T-cell spectral library with 4,833 proteins was established10. However, because the immune system is regulated by communication among various immune cells1,14,15,16, understanding a limited type of cells, focusing on abundant T cells in the blood14, provides only fragmentary information for understanding the regulation of immune compartments and the overall homeostasis of the immune system.
To address the various types of immune cells, we assembled a comprehensive spectral library of human immune cells using data-dependent acquisition (DDA) and data-independent acquisition (DIA) methods. Specifically, a DDA method was applied across five different types of human immune cell lines, including T lymphocyte unstimulated, and stimulated Jurkat, T lymphoblast Molt4, B cell Daudi, and Ramos, monocyte THP-1, and five types of primary cells, including peripheral blood mononuclear cells (PBMCs), CD4 T, CD8 T, natural killer (NK), and B cells. In addition, DIA was used to analyze 10,000 primary CD4 T, CD8 T, NK, and B cells (Fig. 1). The comprehensive spectral library encompassed 127,106 peptides mapped to 10,544 human proteins. Library quality was examined using the spectral library evaluation tool DIALib-QC17. All instrument raw data and spectral library were deposited in the ProteomeXchange Consortium18 (http://proteomecentral.proteomexchange.org) via the mass spectrometry interactive virtual environment (MassIVE) and an allied repository with the dataset identifier PXD04774219.

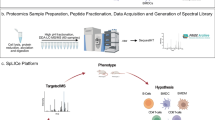
Workflow for the proteomic analysis of five immune cell lines and major primary immune cells and generation of the immune cell spectral library. In the process of generating the spectral library, proteins were extracted from both cell lines and primary cells of immune cells. Specifically, 200 μg of cell line proteins and 100 μg of primary cell proteins underwent tryptic digestion using the FASP method. To alleviate sample complexity, the peptide pre-fractionation step was implemented, involving high-pH fractionation of over 100 μg of proteins from cell lines and primary cells, resulting in 12 and 24 fractions, respectively. The fractionated tryptic peptides from both cell lines and primary cells were then subjected to analysis using the DDA method with Q-Exactive plus and Exploris 240, respectively. For trypsin digestion of proteins from a limited number of primary immune cells (10,000 cells per primary cell type), the SP3 method was employed, and the DIA method with Exploris 240 was applied to obtain the proteome profiles. The raw files acquired from DDA and DIA methods were searched and employed to create the combined immune cell library via Spectronaut (version 18). This combined library was subsequently applied to identify the proteome of the small number of primary immune cells (10,000 cells per primary cell type). The resulting proteome profiles were visualized using SIMCA-P (version 17). Statistical analysis, aiming to identify significant proteins among the four types of primary immune cells, was conducted using the Kruskal-Wallis test through SPSS (version 27). Violin plots, generated using the ggplot2 package in R, visually represented some statistically evaluated proteins.
Through the application of a combined immune cell spectral library to DIA results, we successfully identified and quantified a vast number of proteins, ranging from 2,157 to 3,444 proteins across 10,000 primary human immune cell sorted samples. Furthermore, we directly compared the performance of our purified human immune cell spectral library with public spectral libraries, including Pan-human13 and T cell10, revealing substantial overlap.
In this study, the human immune cell spectral library provided comprehensive insights into the expression and abundance of proteins in various types of immune cells. So far, it is becoming evident that protein abundance cannot be directly predicted from mRNA abundance20. Protein abundance is regulated not only by mRNA levels but also by mechanisms affecting other stages of protein metabolism, such as translation and degradation21,22. Therefore, a comprehensive understanding on cellular function requires proteomic information. Therefore, the proposed immune cell library could be a valuable data source for extensively identifying and characterizing the cellular features of low-abundance proteins and determining protein expression levels in immune cells. Information on the proteins expressed in immune cells may help screen novel drug targets for immunotherapy, which is expected to provide a valuable foundation for future investigations in the field of immune cell proteomics.
Methods
Ethics oversight
Venous blood from healthy volunteers and healthy athletes was collected under an IRB-approved protocol using an EDTA-coated BD Vacutainer (NJ, USA). Blood samples from healthy volunteers were collected at the S-medicenter hospital (Seoul, Korea) for hematology analyzer instrumental quality control purposes, in accordance with the ethical principles of the Declaration of Helsinki. Blood samples from healthy athletes were collected by a Blood Collection Officer (BCO) for anti-doping purposes, following the guidelines of the World Anti-Doping Agency. Spare samples, for which written informed consent for additional research use had been obtained, were subsequently utilized after each respective test. Ethics approval was obtained from the Institutional Review Board (IRB) of the Korea Institute of Science and Technology (KIST-2021-E-004). The IRB waived consent for this study due to its minimal risk to participants as no personally identifiable information was collected. The samples were anonymized using alpha-numeric labels upon collection, refrigerated, and then conveyed to the laboratory.
Cell culture and primary cell acquisition
Human immune cell lines were obtained from Korean cell line bank (Seoul, Korea). All cell lines were cultured in RPMI 1640 with L-glutamine (300 mg/L), 25 mM HEPES, and 25 mM NaHCO3 (Gibco, USA) with 10% inactivated fetal bovine serum (Gibco, USA) in an incubator at 37 °C with 5% CO2. Cell lines were washed and lysed in SDT buffer (4% sodium dodecyl sulfate (SDS), 100 mM Tris-HCl pH 8.5, 10 mM TCEP, and 50 mM CAA) followed by heating at 95 °C for 15 min. The lysate was centrifuged, and the supernatant was transferred to a protein lobind tube (Eppendorf, Germany).
For primary immune cell acquisition, PBMCs were isolated from venous blood using Sepmate and Histopaque-1077 (density, 1.077 g/mL; Sigma Aldrich). Diluted blood with phosphate-buffered saline (PBS) was transferred to the top layer of Hitopaque contained in a Sepmate tube (Stemcell, Canada) and centrifuged at 1,200 × g, 10 min at 25 °C. The supernatant was centrifuged to collect the PBMCs. The remaining red blood cells within PBMCs were lysed with RBC lysis buffer (BD, USA), and the residual lysis buffer was firmly washed with PBS.
Immunophenotyping and sorting of PBMCs were performed using FACS Aria Fusion (BD Biosciences) using anti-CD3-APC-H7 (BD), anti-CD4-BV480 (BD), anti-CD8-APC (BD), anti-CD19-PE-Cy7 (BD), anti-CD56-BB515 (BD), and anti-CD16-BV421 (BD Biosciences). CD4 T, CD8 T, NK, and B cells were sorted using a precision mode of 4-way purity to ensure high purity (Supplementary Figure 1). For spectral library generation, over 100 µg of proteins were acquired by pooling cell pellets of each cell originated from 50 individuals after centrifugation and washing with PBS. The immune cell pellets were lysed using SDT buffer and boiled at 95 °C for 10 min. The collection of 10,000 immune cells per each cell type from 15 individuals was performed at 96-well plates that contained 50 µL of 4% SDS, and the solvent with 10,000 immune cells per cell type was boiled and transferred into the lobind tube. The bulk immune cells and 10,000 individually sorted immune cell lysates were stored at −80 °C for further usage.
Sample preparation
The protein concentration in the lysates of bulk immune cells was measured using a tryptophan assay23. The amount of proteins used for spectral library generation is shown in Table 1. The protein lysate was mixed with cold acetone to precipitate proteins. After the storage of the mixture at −20 °C overnight, the supernatant was discarded and the pellet was re-suspended with SDT buffer. Protein digestion was conducted by filter-aided sample preparation using a 30-kDa Amicon ultra-0.5 centrifugal filter24 (Merck, USA). Next, 50 mM HEPES containing trypsin at a trypsin-to-protein ratio of 1:100 (w/w) (Promega, USA) was added to the ultrafilter. Ultra-filters were incubated at 37 °C for 16 h, and then peptide mixtures were obtained into clean tubes through centrifugation. The flow-through peptide solution was acidified to the final concentration of 0.5% trifluoroacetic acid and desalted using reverse-phase Oasis HLB. After heat-drying using a Speedvac, the sample was fractionated into 12 or 24 fractions according to StageTip-based high-pH reversed-phase fractionation25 (Table 1). Finally, the dried fractions were reconstituted with 20 μL of 0.1% formic acid (FA) in 2% acetonitrile fortified with 1 μL of the iRT Kit (Biognosys, Switzerland).
Single-pot solid-phase-enhanced sample preparation (SP3) was used to purify and digest proteins from 10,000 immune cells, according to previous studies that demonstrated the efficiency of SP3 in low-input application26,27. Sera-Mag Speed Beads A (Cytiva, USA, cat. no. 45152105050250) and B (Cytiva, USA, cat. no. 65152105050250) were mixed and washed and re-suspended with deionized water for the working concentration of 100 μg/μL. The 10,000 immune cell lysate was boiled at 95 °C for 10 min with 10 mM TCEP and 40 mM CAA in 4% SDS. Ethanol and prewashed beads were added to each sample to reach a final concentration of 80% EtOH. For binding protein to beads, the sample was shaken at 1400 rpm for 18 min at 25 °C using thermomixer and immobilized on the magnetic rack for 2 min. After the removal of the supernatant, the beads were washed three times with EtOH. 50 mM ABC containing 0.5 μg of trypsin was used to re-suspend washed beads and incubated at 37 °C for 16 h. The tryptic digests were acidified with 5% FA and fortified with 1 μL of iRT. After immobilizing the beads on the magnetic rack, the solvent was transferred to a vial.
DDA-based analysis
Immune cell tryptic peptides were analyzed using an Easy NanoLC Ultimate 3000 fitted with trapping (cat. no. 164535, Thermo Fisher, Germany) and a 50-cm analytical column (cat. no. ES903, Thermo Fisher, Germany). Solvent A was 0.1% FA in distilled water (DW), and solvent B was 0.1% FA in 80% acetonitrile (ACN). The analytical column flow was constant at 0.3 μL/min at 60 °C. For the DDA method, it took 130 minutes; the percentage of solvent B was as follows: 0–7 min (6–8% B), 7–90 min (8–30% B), 90–95 min (30–95% B), 95–105 min (95% B), 105–110 min (95–6% B), 110–130 min (6% B).
The elution of cell lines or primary human cells from the analytical column was injected into a Q-Exactive Plus or Exploris 240 mass spectrometer coupled with an NSI ion source (Thermo Fisher Scientific, Germany) (Table 1). The peptides eluted from the analytical column were analyzed using the following parameters: For the analytical method using Q-Exactive plus, the capillary temperature was maintained at 275 °C. The voltage of the ion spray was 2 kV in the positive mode. S-lens RF level was 60%, and the resolution of the full mass scan was 70,000. The maximum injection time (IT) was 25 ms, and automatic gain control (AGC) was set at 1 × 106. The scanning mass range was 350–1600 m/z. Subsequent data-dependent MS/MS was performed for the top 12 ions with an isolation window of 1.6 m/z at a resolution of 17,500 and fragmented by higher energy collisional dissociation (HCD) with a collision energy of 28. The maximum IT was 75 ms, and the AGC was 1 × 105.
For the analytical method using Exploris 240, the ion transfer tube temperature was set at 300 °C. The voltage of the ion spray was 1.8 kV in positive mode. S-lens RF level was 50%, and the resolution of the full mass scan was 60,000. The normalized AGC was set to 300%. The scanning mass range was 300–1800 m/z. Subsequent data-dependent MS/MS was performed for the top 15 ions with an isolation window of 1.2 m/z at a resolution of 15,000 and fragmented by HCD with a collision energy of 30%. The normalized AGC target was 100%.
DIA-based analysis
For the DIA of 10,000 immune cells, the analytical method took 60 min with a flow rate of 0.3 μL/min; the percentage of solvent B was as follows: 0–5 min (1–10% B), 5–35 min (10–40% B), 35–40 minutes (40–95% B), 40–45 min (95% B), 45–45.1 min (95–6% B), 45.1–60 min (6% B). For the 10,000 immune cell analysis using Exploris 240, the ion transfer tube temperature was set to 300 °C. The full MS resolution was set to 120,000 with a scan range of 495–905 m/z. The normalized AGC target was 300%, and the maximum IT for the full and DIA scans was set to auto. AGC target value for fragment spectra was set to 1000%. Twenty windows of 20 Da were designed with a window overlap of 1 Da. The resolution of the DIA scan was 120,000, and the precursor mass range was 500–900 with stepped HCD collision energies of 22, 26, and 30.
Spectral library generation and assessment
DDA and DIA data obtained from human immune cell lines and primary immune cells were used to generate a spectral library28 using Spectronaut (version 18, Biognosys, Switzerland) with default settings. The detailed information for the library generation, and DIA analysis parameter in Spectronaut is available in Supplementary Tables 1, 2, respectively. The UniProt SwissProt database (Homo sapiens) was downloaded on 17/05/2023, with 40,912 entries containing 20,407 reviewed with 96 contaminants, and iRT sequences were used to build the spectral library. Cysteine carbamidomethylation was used as the fixed modification, whereas protein N-terminal acetylation and methionine oxidation were used as variable modifications. The false discovery rate (FDR) was controlled at <0.01 at peptide and protein levels. The generated spectral library was evaluated using DIALib-QC version 1.2, which is a freely available software designed to assess different aspects of a library, such as its properties, comprehensiveness, and correctness across 62 specific compliance criteria.
Statistical analysis
The immune cell spectral library was used to identify and quantify proteins from the DIA data using Spectronaut. The proteomic profiles of 10,000 immune cells were evaluated using multivariate statistical analysis. All protein data were Pareto-scaled before principal component analysis (PCA) and partial least squares-discriminant analysis (PLS-DA) using SIMCA (v.17). After validating the PLS-DA plot by cross-validation (n = 200), proteins that showed more than a variable importance in the projection (VIP) of 1 were selected to understand the significant characteristics of each immune cell. The filtered proteins were subjected to the Kruskal-Wallis test and Dunn’s test as post-hoc tests with Bonferroni adjustment using SPSS. The quantitative data and statistical significance for 682 proteins with a VIP score above 1 and a p-value less than 0.05 between cell expression levels are presented in Supplementary Table 3. After selecting differentially expressed proteins among the four types of primary immune cells, the proteins were visualized with a violin plot using ggplot2 in R.
Functional annotation and enrichment analysis
Ontology enrichment analysis of uniquely identified proteins in immune cell library, compared to the previous pan-human29 and T cell libraries30, was administrated using ClueGo (v.2.5.10) in Cytoscape (v.3.10.2) to identify significant GO biological process terms. A total of 1,702 genes associated with the unique proteins were analyzed. The GO tree interval selection criteria ranged from 6 to 13, and a kappa score of 0.4 was used for GO term and pathway connectivity. For statistical assessment, the Bonferroni step-down method with two-sided enrichment/depletion was employed based on the hypergeometric distribution. Go terms with a p-value of less than 0.05 were considered significant.
Data Records
The MS data were used to generate a spectral library (.raw and.mzmL files) and spectral library (.kit file and.tsv), and the evaluation results using DIALib-QC (.csv file) were deposited in the ProteomeXchange Consortium via the MassIVE partner repository with the dataset identifier PXD04774219.
Technical Validation
Generation of human immune cell spectral library
To acquire as many proteins as possible, the combined spectral library of the human immune cells was obtained by both DIA and DDA analysis with delicate fractionations of large amounts of proteins (≥100 μg). The acquired spectral library contained 198,798 precursors, 127,106 peptides, and 10,544 proteins. Primary human immune cells, including CD4 T, CD8 T, NK, and B cells, contained 7,843, 7,529, 7,681, and 7,299 protein groups, respectively (Fig. 2a). Among primary human immune cells, 6,280 proteins were universal (Fig. 2b). Comparison with public spectral libraries, including the pan-human29 and T-cell spectral libraries30, showed that 3,850 protein groups were common in the three libraries, and 8,790 protein groups and 3,902 protein groups were common in the pan-human library29 and human T-cell library30, respectively (Fig. 2c). The human immune cell library generated in the present study contained 1,702 unique protein groups. In addition, 19,918 peptides were common among the spectral libraries. A total of 50,734 peptides were unique to the human immune cell library (Fig. 2d). To identify biological pathways of the 1,702 uniquely identified proteins in the immune cell library, GO enrichment analysis was investigated. As a result, this analysis highlighted several key functional terms closely related to the immune system. the most important functional terms were regulation of lymphocyte mediated immunity (GO:0002706), regulation of lymphocyte activation (GO:0051249), DNA-binding transcription factor activity (GO:0003700), DNA-binding transcription repressor activity (GO:0001217), alpha-beta T cell activation (GO:0046631), cytokine receptor activity (GO:0004896), positive regulation of interferon-gamma production (GO:0032729) (Supplementary Table 4).

Characteristics of human primary immune cell-based spectral library generated by Spectronaut. The number of proteins in individual primary immune cell libraries and the combined library is compared in a bar graph (a). The Venn diagram of individual primary immune cell libraries represents the characteristics of each library (b). Venn diagram showing the number of proteins in the three spectral libraries: ours, public pan-human library, and public t-cell library (c). Venn diagram showing the number of peptides in three spectral libraries: ours, the public pan-human library, and the public T cell library (d).
Evaluation of the human immune cell library
The quality and features of the immune cell spectral library were evaluated using the DIALib-QC. The immune cell library identified 127,106 peptides with 198,798 peptide ions and 5,197,264 fragment ions. Only 626 peptide ions, 0.31% of the total, were shared among different protein. The precursors having charge states of +2 and +3 were 38.1% and 43.6%, respectively (Supplementary table 5). A comparison of the retention time (RT) between doubly and triply charged ions of identical precursor peptides revealed nearly identical values, indicating exceptional chromatographic quality, with an R2 value of 1 (Fig. 3a). The normalized RT was determined using standard peptides from a spectral library. The average number of fragment per precursor was 26.14. The library included 58,415 mass-modified amino acids, comprising 2,705 acetylated (protein N-term), 38,360 carbamidomethylated (C), and 17,350 oxidized (M) residues (Supplementary table 5). A higher percentage of y ions (65.9%) was observed compared with b ions (34.1%), indicating higher-energy collision-induced dissociation fragmentation in the Orbitrap31,32 (Fig. 3b). Furthermore, 98.29% of peptide fragment ions in the library exhibited 1+ or 2+ charge states (Fig. 3c). In addition, 96.9% of the precursor charge states ranged from +2 to +4 (Fig. 3d). In terms of precursor m/z values, 97.3% of the precursors ranged from 400 to 1,250 m/z (Fig. 3e). More than 94.7% of all peptides in the library consisted of 30 or fewer sequences (Fig. 3f). Precursor ions with fewer than five fragment ions constituted only 4.19% of the library (Fig. 3g). Proteins containing more than five corresponding peptides accounted for more than 66.4% of the total. In addition, among proteins represented in the library, those with two or more corresponding peptides comprised 9,384 peptides and 87.55% of the total (Fig. 3h).

Quality assessment of the human immune cell library. Retention time (RT) fit of +2 and +3 charge states of the sample peptides in the spectral library (a), Number of observed b- and y-ion fragments in the spectral library (b), distribution of fragment ions by observed fragment charge states in the spectral library (c), distribution of precursors by precursor charge states (d), distribution of precursors by precursor m/z values across the acquired mass range in the assay library (e), distribution of the observed peptides by peptide length in the assay library (f), distribution of precursors by the number of fragment ions per precursor (g), and distribution of proteins by the number of peptides per protein in the spectral library (h).
Identification of 10,000 immune cells using the human immune cell library
To validate the quality of the immune cell spectral library and demonstrate its application, 10, 000 human primary immune cell proteomic results were processed using DIA. DIA has been used to obtain more MS/MS spectra and sufficient data points for quantitation with precursors and fragmented ions for low-abundance proteins33,34. The number of proteins and peptides identified from 10,000 cells using the combined spectral library ranged from 2,157 to 3,444 and 6,476 to 15,258, respectively (Fig. 4a,b).

DIA data obtained from 10,000 primary immune cells. Number of proteins of individual 10,000 primary immune cells (a) and number of peptides in individual 10,000 primary immune cells (b). Principal component analysis (PCA) score plot(c), partial least squares discriminant analysis (PLS-DA) score plot (d), and cross-validation plots for 10,000 primary immune cells (e).
The dataset composed of 4,331 proteins and 32,944 peptides was analyzed using PCA to monitor the patterns of group aggregation and differentiation and to visualize the similarities and distinctions among samples (Fig. 4c)35. The R2X and Q2 values of the PCA plots were 0.328 and 0.218, respectively. PLS-DA plots were applied to obtain a list of differential proteins among immune cells that were filtered for system noise36. As shown in Fig. 4d, CD4 T cells and CD8 T cells were closely clustered together, and NK and B cells effectively separating themselves from other groups. This observation underscores the complete distinguishability of the immune cell types. The PLS-DA model demonstrated robust performance, with R2X, R2Y, and Q2 values of 0.266, 0.793, and 0.605, respectively, indicating its excellent capability and predictability. To further validate the model, a permutation test (n = 200) was conducted, and the results indicated that the PLS-DA model was not over-fitted, as evidenced by R2 (0.0, 0.325 and Q2 (0.0, –0.283) (Fig. 4e). In the analysis of differentiated proteins, VIP was used to identify proteins that contributed significantly to the variation among the four different types of immune cells. A total of 1289 differentially expressed proteins had more than one VIP value. These proteins were subjected to the identification and validation of their significant differences using a Kruskal-Wallis test (p < 0.05) with Dunn’s post-hoc analysis with Bonferroni adjustment. As a result, 682 proteins were evaluated for their different profiles among the four types of immune cells; among them, expression levels of representative proteins were visualized with violin plots in Fig. 5. Within the violin plots, B cells showed the most unique characteristics, and CD4 T cells and CD8 T cells showed similar protein expression patterns in accordance with the PLS-DA plot results.

Violin plots of representative proteins among the 682 statistically evaluated proteins. The statistical analysis with Kruskal-Wallis test coupled with Dunn’s post-hoc analysis with Bonferroni adjustment clarified significantly differentiated proteins among the four types of primary immune cells. B cells represented the most unique characteristics.
Usage Notes
Application of the immune cell spectral library in low-protein contained samples
A comprehensive immune cell spectral library was successfully applied to analyze DIA datasets acquired using different analytical methods. To apply a spectral library, spiking iRT peptides into a sample is recommended for accurate RT alignment and calibration during subsequent data processing.
Code availability
A spectral library was generated using the commercial software Spectronaut and evaluated using DIALib-QC. No custom code was generated in this work.
References
Parkin, J. & Cohen, B. An overview of the immune system. Lancet 357, 1777–1789, https://doi.org/10.1016/s0140-6736(00)04904-7 (2001).
Sattler, S. The Role of the Immune System Beyond the Fight Against Infection. Adv Exp Med Biol 1003, 3–14, https://doi.org/10.1007/978-3-319-57613-8_1 (2017).
Nyman, T. A., Lorey, M. B., Cypryk, W. & Matikainen, S. Mass spectrometry-based proteomic exploration of the human immune system: focus on the inflammasome, global protein secretion, and T cells. Expert Review of Proteomics 14, 395–407, https://doi.org/10.1080/14789450.2017.1319768 (2017).
Rathore, D., Marino, M. J. & Nita-Lazar, A. Omics and systems view of innate immune pathways. PROTEOMICS 23, 2200407, https://doi.org/10.1002/pmic.202200407 (2023).
Berge, T. et al. Quantitative proteomic analyses of CD4+ and CD8+ T cells reveal differentially expressed proteins in multiple sclerosis patients and healthy controls. Clinical Proteomics 16, 19, https://doi.org/10.1186/s12014-019-9241-5 (2019).
Benedict, K. F. & Lauffenburger, D. A. Insights into proteomic immune cell signaling and communication via data-driven modeling. Curr Top Microbiol Immunol 363, 201–233, https://doi.org/10.1007/82_2012_249 (2013).
Sun, L., Su, Y., Jiao, A., Wang, X. & Zhang, B. T cells in health and disease. Signal Transduction and Targeted Therapy 8, 235, https://doi.org/10.1038/s41392-023-01471-y (2023).
Kansler, E. R. & Li, M. O. Innate lymphocytes—lineage, localization and timing of differentiation. Cellular & Molecular Immunology 16, 627–633, https://doi.org/10.1038/s41423-019-0211-7 (2019).
Jameson, S. C. Maintaining the norm: T-cell homeostasis. Nature Reviews Immunology 2, 547–556, https://doi.org/10.1038/nri853 (2002).
Weerakoon, H. et al. A primary human T-cell spectral library to facilitate large scale quantitative T-cell proteomics. Scientific Data 7, 412, https://doi.org/10.1038/s41597-020-00744-3 (2020).
Li, H. et al. A novel spectral library workflow to enhance protein identifications. Journal of Proteomics 81, 173–184, https://doi.org/10.1016/j.jprot.2013.01.026 (2013).
Deutsch, E. W. et al. Expanding the Use of Spectral Libraries in Proteomics. Journal of Proteome Research 17, 4051–4060, https://doi.org/10.1021/acs.jproteome.8b00485 (2018).
Rosenberger, G. et al. A repository of assays to quantify 10,000 human proteins by SWATH-MS. Scientific Data 1, 140031, https://doi.org/10.1038/sdata.2014.31 (2014).
Brodin, P. & Davis, M. M. Human immune system variation. Nature Reviews Immunology 17, 21–29, https://doi.org/10.1038/nri.2016.125 (2017).
Zhou, Y., Cheng, L., Liu, L. & Li, X. NK cells are never alone: crosstalk and communication in tumour microenvironments. Molecular Cancer 22, 34, https://doi.org/10.1186/s12943-023-01737-7 (2023).
Kumar, A., Swain, C. A. & Shevde, L. A. Informing the new developments and future of cancer immunotherapy. Cancer and Metastasis Reviews 40, 549–562, https://doi.org/10.1007/s10555-021-09967-1 (2021).
Midha, M. K. et al. DIALib-QC an assessment tool for spectral libraries in data-independent acquisition proteomics. Nature Communications 11, 5251, https://doi.org/10.1038/s41467-020-18901-y (2020).
Deutsch, E. W. et al. The ProteomeXchange consortium at 10 years: 2023 update. Nucleic Acids Research 51, D1539–D1548, https://doi.org/10.1093/nar/gkac1040 (2022).
Hyeon-Jeong Lee, H. M. Human immune cell proteomic library. MassIVE, MSV000093644. https://doi.org/10.25345/C5D50G78R (2024).
Vogel, C. & Marcotte, E. M. Insights into the regulation of protein abundance from proteomic and transcriptomic analyses. Nature Reviews Genetics 13, 227–232, https://doi.org/10.1038/nrg3185 (2012).
Wang, X., Liu, Q. & Zhang, B. Leveraging the complementary nature of RNA-Seq and shotgun proteomics data. PROTEOMICS 14, 2676–2687, https://doi.org/10.1002/pmic.201400184 (2014).
Vogel, C. et al. Sequence signatures and mRNA concentration can explain two-thirds of protein abundance variation in a human cell line. Mol Syst Biol 6, 400, https://doi.org/10.1038/msb.2010.59 (2010).
Wiśniewski, J. R. & Gaugaz, F. Z. Fast and Sensitive Total Protein and Peptide Assays for Proteomic Analysis. Analytical Chemistry 87, 4110–4116, https://doi.org/10.1021/ac504689z (2015).
Wiśniewski, J. R., Zougman, A., Nagaraj, N. & Mann, M. Universal sample preparation method for proteome analysis. Nature Methods 6, 359–362, https://doi.org/10.1038/nmeth.1322 (2009).
Kim, H. et al. An efficient method for high-pH peptide fractionation based on C18 StageTips for in-depth proteome profiling. Analytical Methods 11, 4693–4698, https://doi.org/10.1039/C9AY01269A (2019).
Müller, T. et al. Automated sample preparation with SP 3 for low‐input clinical proteomics. Molecular systems biology 16, e9111, https://doi.org/10.15252/msb.20199111 (2020).
Hughes, C. S. et al. Single-pot, solid-phase-enhanced sample preparation for proteomics experiments. Nature protocols 14, 68–85, https://doi.org/10.1038/s41596-018-0082-x (2019).
Yu, F. et al. Analysis of DIA proteomics data using MSFragger-DIA and FragPipe computational platform. Nature Communications 14, 4154, https://doi.org/10.1038/s41467-023-39869-5 (2023).
Rosenberger, G. et al. The Pan-Human Library: A repository of assays to quantify 10 000 proteins by SWATH-MS/SWATH-MS validation data. PRIDE, PXD000954. https://identifiers.org/pride.project:PXD000954 (2014).
Weerakoon, H. et al. A primary human T-cell spectral library to facilitate large scale quantitative T-cell proteomics. PRIDE, PXD019542. https://identifiers.org/pride.project:PXD019542 (2020).
Lau, K. W. et al. Observations on the detection of b- and y-type ions in the collisionally activated decomposition spectra of protonated peptides. Rapid Communications in Mass Spectrometry 23, 1508–1514, https://doi.org/10.1002/rcm.4032 (2009).
Ahn, H.-S. et al. Generating Detailed Spectral Libraries for Canine Proteomes Obtained from Serum and Urine. Scientific Data 10, 241, https://doi.org/10.1038/s41597-023-02139-6 (2023).
Chen, C.-J., Lee, D.-Y., Yu, J., Lin, Y.-N. & Lin, T.-M. Recent advances in LC-MS-based metabolomics for clinical biomarker discovery. Mass Spectrometry Reviews 42, 2349–2378, https://doi.org/10.1002/mas.21785 (2023).
Guo, J., Yu, H., Xing, S. & Huan, T. Addressing big data challenges in mass spectrometry-based metabolomics. Chemical Communications 58, 9979–9990, https://doi.org/10.1039/D2CC03598G (2022).
Guo, X.-H. et al. Identification of velvet antler and its mixed varieties by UPLC-QTOF-MS combined with principal component analysis. Journal of Pharmaceutical and Biomedical Analysis 165, 18–23, https://doi.org/10.1016/j.jpba.2018.10.009 (2019).
Yang, P. et al. Dietary effects of fish meal substitution with Clostridium autoethanogenum on flesh quality and metabolomics of largemouth bass (Micropterus salmoides). Aquaculture Reports 23, 101012, https://doi.org/10.1016/j.aqrep.2022.101012 (2022).
Acknowledgements
This research was supported by a grant (23212MFDS2023) from the Ministry of Food and Drug Safety and intramural grants (2V08320 and 2V10011) from the Korea Institute of Science and Technology (KIST).
Author information
Authors and Affiliations
Contributions
This study was conceived by H.M. (Hophil Min). The sample were prepared by H.-J.L. (Hyeon-Jeong Lee) and D.H. (Dohyun Han). The spectral library was generated by H.-J.L. and Y.S. (Yoondam Seo). The LC-MS was conducted by H.-J.L. The data was analysed by H.-J.L. and Y.P. (Yoon Park). Lastly, the manuscript was written by H.-J.L., E.Y. (Eugene C. Yi), and H.M. with contributions from all authors.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
41597_2024_3721_MOESM4_ESM.xlsx
Supplementary table 3. Quantitative data and statistical significance for 682 proteins with VIP score more than1 and p-value less than 0.05 between cell expression levels
41597_2024_3721_MOESM5_ESM.xlsx
Supplementary table 4. Result table of ClueGO analysis based on the 1702 uniquely detected proteins of immune cell spectral library
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Lee, HJ., Seo, Y., Park, Y. et al. Comprehensive immune cell spectral library for large-scale human primary T, B, and NK cell proteomics. Sci Data 11, 871 (2024). https://doi.org/10.1038/s41597-024-03721-2
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41597-024-03721-2
