
Abstract
The Cape hare (Lepus capensis) is among the most widely distributed hare species globally, inhabiting extensive regions across Africa, the Middle East, and Central Asia. However, evolutionary and genetic research on L. capensis was seriously impeded by the absence of a reference genome. Here, we assembled and constructed a chromosome-level genome of L. capensis (with scaffolds anchored to 25 chromosomes and a total assembled length of 2.9 Gb, achieving a contig N50 length of 124.44 Mb) using PacBio HiFi sequencing and Hi-C assembly technology. Evaluation using BUSCO indicated the genome assembly to be 98.2% complete. The de novo prediction revealed that repetitive sequences constitute 46.13% of the entire genome, and long interspersed nuclear elements (LINEs) constituted the largest portion. We annotated a total of 13, 868 protein-coding genes using transcriptomes from two tissues (muscle and skin). This high-quality reference genome serves as a valuable genomic resource for advancing genetic studies in this species.
Similar content being viewed by others

Background & Summary
The genus of hares (Lepus), belonging to the Lagomorpha order, is renowned for its extensive adaptability, allowing them to thrive across diverse terrestrial habitats such as plateaus, coastal plains, polar regions, and deserts1. Lepus is well studied in almost all research areas in biology. However, it has faced long-standing controversies regarding species classification. The genus’s phylogenetic relationships are equally complex, marked by conflicting theories and intricate evolutionary histories. Classification challenges arise due to convergent evolution in morphology and anatomy, significant intraspecific variation in widely distributed species due to rapid radiation and environmental adaptability, and extensive hybridization and gene introgression among hares2.
The identification of hare species in China is still under debate, with the number of recognized species varying between seven and nine3. The Cape hare (Lepus capensis), belongs to the genus Lepus, found extensively across to Africa, the Middle East, and Central Asia, and is one of the most widely distributed species of hares in China. This hare is characterized by its well-developed legs for leaping and running, alongside large eyes and ears adapted for detecting environmental threats. Typically, it features a distinctive white ring around its eyes and sports a soft, fine coat that ranges in hues from light brown to reddish or sandy grey. However, a scarcity of a reference genome of L. capensis has hampered the study on evolutionary history and genetic differentiation in this species.
In the present study, we assembled a chromosome-level of genome of L. capensis using approximate 100 Gb of Pacbio HiFi reads and 200 Gb of Hi-C reads. The final assembled genome size is 2.9 Gb and the twenty-five chromosomes contribute 2.66 Gb, including the pseudo-chromosome X of 134.8 Mb and pseudo-chromosome Y of 27.5 Mb. The mitochondria sequence is assembled to 17.5 kb. The Benchmarking Universal Single-Copy Orthologs (BUSCO) evaluated the genome, suggesting the high quality of the completeness of the assembly, 98.2% of the gene sets of rabbit genome were covered.
Methods
Sample collection and sequencing
Blood samples were collected from a male Cape hare in Xunyi County, Shanxi Province. Genomic DNA was extracted from blood using the TIANamp Genomic DNA kit. The purity and concentration of the DNA were assessed with a Qubit 3.0 Fluorometer (Life Technologies, Carlsbad, CA, USA).
For PacBio HiFi sequencing, libraries were prepared using the SMRTbell Express Template Prep Kit 3.0, following the manufacturer’s instructions. PCR-free SMRT bell libraries were sequenced on the PacBio Sequel nova platform. The Hi-C library was constructed by the DpnII restriction enzyme, following an in situ ligation protocol. Formaldehyde-crosslinked DNA was digested with DpnII and treated with biotin. Cyclization of DNA after end-repair was used to identify intersecting DNA locations. After reversing the crosslinking, ligated DNA fragments were fragmented into 300–700 bp sizes and purified using a biotin-streptavidin approach for library construction. Additionally, a DNA sequencing library was constructed using the NEB Ultra DNA Library Prep Kit (NEB, USA) as per the manufacturer’s instructions. Both the Hi-C and DNA sequencing libraries were quantified and sequenced on the Illumina NovaSeq platform.
In total, approximately 100 Gb of PacBio HiFi reads, 200 Gb of Hi-C reads, and 88 Gb of filtered Illumina short-read sequencing data were obtained for the assembly of the L. capensis genome. Detailed statistics of the sequenced reads are provided in Supplementary Table 1–3.
Furthermore, RNA samples from the skin and muscle of the same animal were used to constructed bulk RNA-seq library. Messenger RNA was purified from total RNA using poly-T oligo-attached magnetic beads. The library preparation involved end repair, A-tailing, adapter ligation, size selection, amplification, and purification. Following quality control, the quantified libraries were pooled and sequenced on Illumina platforms based on the effective library concentration. These RNA-seq data facilitated the assembly of transcriptome and gene annotation. Detailed statistics of the sequenced reads are provided in Supplementary Table 4.
Genome survey and assembly
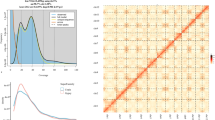
The genome size, heterozygosity, and duplication ratio were estimated based on the K-mer depth using Jellyfish (v2.3.1)4 and GenomeScope5. The estimated genome size of the Cape hare was approximately 2.1 Gb, as determined from 21-mers histogram (Fig. 1a). Primary contigs were assembled using HiFi and Hi-C reads with Hifiasm (v0.19.9-r616)6 employing default parameters. Based on the sequenced Hi-C data, we utilized Juicer7 and 3D-DNA pipeline8 to scaffold the primary contigs to a near-chromosome level. Subsequently, manual curation was performed using Juicebox9 software, based on chromosomal interaction strengths. Ultimately, a chromosome-level genome assembly was achieved (Fig. 1b). To identify sex-linked sequences, the short sequencing reads were aligned against the preliminary assembly. Contigs with half-coverage were extracted as potential sex-linked sequences. The pseudo-Y chromosome was identified through BLASTn10 analysis of the known rabbit Y-specific gene SRY (AY785433.1). The pseudo-X chromosome was identified by its high similarity to the rabbit X chromosome and Lepus europaeus.

Genome survey and assembly. (a) Genome size estimated by GenomeScope using 21-mers. (b) HiC alignments and interactions among chromosomes of the assembled Lepus capensis genome.
The final assembled genome size is 2.96 GB, with 2.66 GB of the sequence anchored to 23 autosomes, and two sex chromosomes (X and Y) (Fig. 2). The contig N50 is 93.12 Mb, and the scaffold N50 is 124.44 Mb (Table 1). Chromosome lengths and statistics are listed in Table 2.

Characteristics of the Lepus capensis genome. The tracks from the outside to the center of the circle on the chromosomes are the repetitive elements density; gene density; GC content; chromosomes karyotype; and the major inter-chromosomal relationships in the Lepus capensis genome.
Mitochondrial DNA
Animal mitochondrial (MT) DNA typically measures approximately 16 kb in size11. In the preliminary assembly, a contig of 23.2 kb was identified, covering 88.5% of the rabbit mitochondrial DNA sequence (NC_001913.1) based on BLASTn10 analysis. This contig contains a complete mitochondrial sequence of 17,542 bp, with the remainder of the sequence being a partial duplicate. Annotation of protein-coding genes, tRNAs, and rRNAs was manually adjusted according to the annotations of NC_001913.1.
Gene prediction and annotation
RNA-seq reads from liver tissue were firstly aligned to the genome using STAR (v2.7.11b)12, with the parameters “—alignIntroMax 100000 –outWigType wiggle” to generate the hints for exonic regions from the output coverage wig file. Then, transcripts were assembled de novo using Trinity13 and mapped to the reference genome to determine gene coordinates. Gene prediction was further refined with the Braker214, utilizing RNA-seq reads to assist in the process. The EVidenceModeler (EVM) software (v2.1.0)15 was then employed to integrate predicted genes and transcript alignments into weighted consensus gene models, creating a unified automated gene structure annotation system. This approach yielded a total of 16,079 transcripts from 13,868 genes in L. capensis genome (Table 3). Functional annotation of these genes was performed by eggnog-mapper (v2.1.12)16 diamond tool with the eggNOG 5.0 database17.
Repetitive elements identification
Repetitive sequences were annotated using both de novo prediction and homology-based methods. For de novo prediction, RepeatModeler (v2.0.1)18 was employed to create a repeat library and TEclass219 was used to annotate unclassified repeat sequences. RepeatMasker (v 4.1.1)20 was then utilized to annotate the repetitive elements, identifying 46.13% (1,365,670,423 bp) of the assembled genomes as repeat sequences, with long interspersed nuclear elements (LINEs) being the predominant type. Additionally, repetitive elements were detected through comparison with the RepBase database21, yielding a repeat content of 43.54% (1,289,204,280 bp), with short interspersed nuclear elements (SINEs) being the most prevalent (Table 4).
Genome synteny analysis
Synteny analysis was performed on our assembly of L. capensis with the domestic rabbit, Oryctolagus cuniculus22 (Genome assembly accession no. UM_NZW_1.0), and the European hare, Lepus europaeus23 (Genome assembly accession no. mLepTim1.pri) using MCScanX24 module implemented in the Tbtools software25. The analysis revealed that L. capensis and Lepus europaeus exhibit higher genomic homology with Oryctolagus cuniculus (Fig. 3), Chromosomes of L. capensis were numbered consistently with those of Lepus europaeus, but inconsistently with Oryctolagus cuniculus.

Synteny analysis between assembly of Lepus capensis and other two species (Oryctolagus cuniculus and Lepus europaeus). Homologous genes are represented by grey lines connecting chromosomes.
Ethics statement
All experimental procedures and animal use were approved by the Institutional Animal Care and Use Committee at Northwest A&F University (approval no. DK2024050).
Data Records
All raw sequencing data, including genomic short sequencing reads, PacBio HiFi reads, Hi-C data, and RNA-Seq reads, reported in this study have been deposited in the China National Genomics Data Center (CNGB)26,27 Genome Sequence Archive (GSA) database under accession number CRA01805728, with the BioProject accession number PRJCA02833529. The genome assembly is available in the NCBI GenBank with accession number JBHEQN00000000030. Furthermore, the genome assembly has also been deposited in Genome Warehouse (GWH) of CNGB under accession number GWHFCZH00000000.131. Additionally, the genome annotation in GFF format is accessible via the Figshare database32.
Technical Validation
The quality of the genome assembly was evaluated using BUSCO (v4.1.4)33 (glires_odb10 database, parameters: -evalue 1e-05) to assess completeness and accuracy. The BUSCO analysis indicated that, 98.2% of genes were fully represented, with 95.7% as single-copy genes and 2.5% resulting from duplication events. Additionally, 89 Gb of short sequencing reads were aligned back to the finally assembled genome using BWA-MEM34, achieving an alignment rate of 99.5%.
Code availability
All software used in this work is in the public domain, with parameters being clearly described in Methods. If no detail parameters were mentioned for a software, default parameters were used as suggested by developer.
References
Smith, A. T., Johnston, C. H., Alves, P. C. & Hackländer, K. Lagomorphs: Pikas, Rabbits, and Hares of the World. (Johns Hopkins University Press, 2008).
Shan, W., Li, Z., Dai, H., Dong, P. & Zhang, Y. Taxonomic status of hares (Lepus spp.) in Xinjiang, China (Lagomorpha: Leporidae): An integrative approach. Zoologica Scripta 53, 282–298 (2024).
Liu, J. et al. Reticulate evolution: frequent introgressive hybridization among chinese hares (genus lepus) revealed by analyses of multiple mitochondrial and nuclear DNA loci. BMC Evol Biol 11, 223 (2011).
Marçais, G. & Kingsford, C. A fast, lock-free approach for efficient parallel counting of occurrences of k -mers. Bioinformatics 27, 764–770 (2011).
Vurture, G. W. et al. GenomeScope: fast reference-free genome profiling from short reads. Bioinformatics 33, 2202–2204 (2017).
Cheng, H., Concepcion, G. T., Feng, X., Zhang, H. & Li, H. Haplotype-resolved de novo assembly using phased assembly graphs with hifiasm. Nat Methods 18, 170–175 (2021).
Durand, N. C. et al. Juicer Provides a One-Click System for Analyzing Loop-Resolution Hi-C Experiments. Cell Systems 3, 95–98 (2016).
Dudchenko, O. et al. De novo assembly of the Aedes aegypti genome using Hi-C yields chromosome-length scaffolds. Science 356, 92–95 (2017).
Robinson, J. T. et al. Juicebox.js Provides a Cloud-Based Visualization System for Hi-C Data. Cell Systems 6, 256–258.e1 (2018).
Camacho, C. et al. BLAST+: architecture and applications. BMC Bioinformatics 10, 421 (2009).
Boore, J. L. Animal mitochondrial genomes. Nucleic Acids Research 27, 1767–1780 (1999).
Dobin, A. et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21 (2013).
Grabherr, M. G. et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat Biotechnol 29, 644–652 (2011).
Brůna, T., Hoff, K. J., Lomsadze, A., Stanke, M. & Borodovsky, M. BRAKER2: automatic eukaryotic genome annotation with GeneMark-EP+ and AUGUSTUS supported by a protein database. NAR Genomics and Bioinformatics 3, lqaa108 (2021).
Haas, B. J. et al. Automated eukaryotic gene structure annotation using EVidenceModeler and the Program to Assemble Spliced Alignments. Genome Biol 9, R7 (2008).
Cantalapiedra, C. P., Hernández-Plaza, A., Letunic, I., Bork, P. & Huerta-Cepas, J. eggNOG-mapper v2: Functional Annotation, Orthology Assignments, and Domain Prediction at the Metagenomic Scale. Molecular Biology and Evolution 38, 5825–5829 (2021).
Huerta-Cepas, J. et al. eggNOG 5.0: a hierarchical, functionally and phylogenetically annotated orthology resource based on 5090 organisms and 2502 viruses. Nucleic Acids Research 47, D309–D314 (2019).
Flynn, J. M. et al. RepeatModeler2 for automated genomic discovery of transposable element families. Proc. Natl. Acad. Sci. USA 117, 9451–9457 (2020).
Abrusán, G., Grundmann, N., DeMester, L. & Makalowski, W. TEclass—a tool for automated classification of unknown eukaryotic transposable elements. Bioinformatics 25, 1329–1330 (2009).
Tarailo‐Graovac, M. & Chen, N. Using RepeatMasker to Identify Repetitive Elements in Genomic Sequences. CP in Bioinformatics 25, (2009).
Bao, W., Kojima, K. K. & Kohany, O. Repbase Update, a database of repetitive elements in eukaryotic genomes. Mobile DNA 6, 11 (2015).
Bai, Y. et al. Improving the genome assembly of rabbits with long-read sequencing. Genomics 113, 3216–3223 (2021).
Michell, C. et al. High quality genome assembly of the brown hare (Lepus europaeus) with chromosome-level scaffolding. Peer Community Journal 4, e26 (2024).
Wang, Y. et al. MCScanX: a toolkit for detection and evolutionary analysis of gene synteny and collinearity. Nucleic Acids Research 40, e49–e49 (2012).
Chen, C. et al. TBtools: An Integrative Toolkit Developed for Interactive Analyses of Big Biological Data. Molecular Plant 13, 1194–1202 (2020).
Chen, T. et al. The Genome Sequence Archive Family: Toward Explosive Data Growth and Diverse Data Types. Genomics, Proteomics & Bioinformatics 19, 578–583 (2021).
CNCB-NGDC Members and Partners. et al. Database Resources of the National Genomics Data Center, China National Center for Bioinformation in 2022. Nucleic Acids Research 50, D27–D38 (2022).
National Genomics Data Center (NGDC) Genome Sequence Archive https://ngdc.cncb.ac.cn/gsa/browse/CRA018057 (2024).
National Genomics Data Center (NGDC) BioProject https://ngdc.cncb.ac.cn/bioproject/browse/PRJCA028335 (2024).
Dong, X. Genbank https://identifiers.org/ncbi/insdc:JBHEQN000000000 (2024).
National Genomics Data Center (NGDC) Genome Warehouse https://ngdc.cncb.ac.cn/gwh/Assembly/86060/show (2024).
Dong, X. Lepus capensis genome assembly and annotation. figshare https://doi.org/10.6084/m9.figshare.26820676.v2 (2024).
Manni, M., Berkeley, M. R., Seppey, M., Simão, F. A. & Zdobnov, E. M. BUSCO Update: Novel and Streamlined Workflows along with Broader and Deeper Phylogenetic Coverage for Scoring of Eukaryotic, Prokaryotic, and Viral Genomes. Molecular Biology and Evolution 38, 4647–4654 (2021).
Li, H. & Durbin, R. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 25, 1754–1760 (2009).
Acknowledgements
This work was supported by the Chinese Universities Scientific Fund (no. 2452020187). We thank the High-Performance Computing Center of NWAFU for providing computing resources.
Author information
Authors and Affiliations
Contributions
X.G.D., Y.L. and Y.Y.Z. conceived and designed the experiments; X.G.D., Y.C. and X.X.P. conducted the experiments; Y.L. performed the genome assembly and repeat elements detection; X.G.D. conducted the gene homologues and genomic synteny analysis; Y.Y.Z. performed the gene annotation; Z.J.R. recruited animal resources; X.G.D., Y.L. and Y.Y.Z. wrote the paper; and all authors read, edited and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Dong, X., Liu, Y., Chen, Y. et al. A chromosome-level genome assembly of Cape hare (Lepus capensis). Sci Data 11, 1081 (2024). https://doi.org/10.1038/s41597-024-03953-2
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41597-024-03953-2
This article is cited by
-
Chromosome-level genome assembly of Triplophysa scleroptera
Scientific Data (2025)
