
Abstract
Persicaria tinctoria (2n = 40) is an important traditional medicinal plant and natural dye source within the genus Persicaria. P. tinctoria has been utilized for its antibacterial, antiviral, anti-inflammatory, and tumor treatment properties. Additionally, it has served as a natural blue dye for thousands of years worldwide, and continues to be used in countries such as China and Japan. Here, we assembled a tetraploid chromosome-scale genome of P. tinctoria, organized into two subgenomes: subgenome A, which contains 10 pseudochromosomes with a genome size of 888.67 Mb and a scaffold N50 of 90.56 Mb, and subgenome B, which also comprises 10 pseudochromosomes with a genome size of 771.58 Mb and a scaffold N50 of 76.84 Mb. Repeat sequences constitute 77.9% of the genome. A total of 76,742 high-confidence protein-coding genes were annotated, with 94.28% of these genes assigned functional annotations. This high-quality genome assembly of P. tinctoria will provide valuable genomic resources for studying the biosynthesis and evolution of indigoids in indigo plants, as well as for further research on the Polygonaceae family.
Similar content being viewed by others
Background & Summary
Persicaria tinctoria (Aiton) Spach (Polygonum tinctorium Aiton), a species in the Persicaria genus and the Polygonaceae family, is a valuable natural blue dye and medicinal herb. It has been used and cultivated worldwide as a dye plant for thousands of years. In China, the earliest literature record of P. tinctoria is the ancient Chinese agricultural Book of Da Dai Liji volume of Xia XiaoZheng in the Xia Dynasty 4000 years ago1. In Japan, P. tinctoria became the dominant source of indigo when it was introduced in the 4th century, and its leaf fermentation product, called sukumo, was used extensively in the textile industry2,3. At the same time, P. tinctoria also has a very long history as a medicinal plant. In the Chinese Pharmacopoeia, its dried leaves are called folium isatidis, the extract of its stems and leaves is called indigo naturalis, both of them have functions including clearing heat and detoxifying, cooling blood and eliminating spots4. In addition, previous pharmacological studies have shown that P. tinctoria has antibacterial5,6, anti-HIV7, anti-inflammatory8,9,10, antioxidant11,12 and anticancer functions13,14,15, and indigo naturalis is used for treating ulcerative colitis16,17 and as an antipsoriatic18,19, antileukemic20,21,22, and anti-inflammatory23 agent.
P. tinctoria is a crop with important value as a dye and as a medicine, and many previous studies have shown that indigo and indirubin are the main dyeing and medicinal components24,25, but the lack of genomic information and insufficient research on the molecular biosynthesis mechanisms of indigo and indirubin severely restricts the application of natural indigo plants. In addition, plants in the Polygonaceae are widely distributed and well adapted, which makes this family a good target for studying plant adaptability to extreme climates. Many gene resources from Polygonaceae that are resistant to adverse environments can be used to improve the germplasm of rice, wheat and other crops. Moreover, polyploidy following hybridization (allopolyploidy) has long been recognized as a significant factor in plant evolution26. The ploidy diversity within the Polygonaceae makes it an excellent subject for study. Kim predicted the formation of allopolyploid species in several Polygonaceae species by analyzing a low-copy nuclear gene region, suggesting that approximately fifteen species may be allopolyploids, including P. tinctoria27. Our genomic data validated the reliability of his methodology and conclusions at the genomic level. And acquiring additional genomic data on Polygonaceae will enhance our understanding of the origins and evolution of plant polyploidy. However, genome resources in the family are scarce, and reference genomes are available for only a few plants, such as Polygonum cuspidatum28, Rumex hastatulu29, rhubarb30 and buckwheat31. The analysis of genomic information from P. tinctoria offers a potential foundational resource for further research on the biosynthesis mechanisms of indigo and indirubin, the breeding of new cultivars with highly effective components, and the in-depth study of Polygonaceae.
In a previous ethnobotanical study, researcher Pei Shengji conducted a preliminary investigation of the P. tinctoria used by the Yi people in Butuo County, Liangshan Yi Autonomous Prefecture, Sichuan Province, China, and collected seeds for further research. In this study, we used these previously collected P. tinctoria as samples and estimated the genome size through flow cytometry and K-mer analysis to determine the whole-genome de novo sequencing strategy. The high-quality chromosome-level genome of P. tinctoria generated using HiFi sequencing data obtained from the PacBio single-molecule real-time technique and Hi-C sequencing data produced from the high-throughput chromosome conformation capture technique. The final P. tinctoria genome of 1.66 Gb was assembled by 322 contigs with a contig N50 of 34.06 Mb, of which 84 contigs were assigned to 20 chromosomes with a scaffold N50 of 82.35 Mb. The final assembly genome of P. tinctoria contained subgenome A (888.67 Mb) and subgenome B (771.58 Mb), with scaffold N50 length of 90.56 Mb and 76.84 Mb, respectively. A total of 77.9% of the repeats sequence in the P. tinctoria genome was annotated by using homolog-based and ab initio approaches. A total of 76,742 protein-coding genes were annotated by homology-based, transcriptome-based and de novo prediction, and functional annotations were obtained for 72,352 protein-coding genes by BLAST with various functional databases. An overview of the genome survey, chromosome-level genome assembly and genome annotation in P. tinctoria is shown in Fig. 1. Expanding the available P. tinctoria genome information provides significant genetic resources for elucidating the biosynthetic pathways and evolutionary mechanisms of indigoids. Meanwhile, the genome sequence can be used as a fundamental reference for further study on traditional Chinese medicine and Polygonaceae plants.

Overview of the genome survey, chromosome-level genome assembly and genome annotation in P. tinctoria. The bioinformatics tools utilized in each step were marked near the arrows.
Methods
Plant materials
P. tinctoria collected in a previous ethnobotanical investigation was tissue cultured, planted and preserved in Kunming City, Yunnan Province, China (24.991918°N, 102.662224°E). The leaves of a single fresh plant were collected for genome and Hi-C sequencing, and the inflorescence, flowers, roots, stems and leaves of the same plant were collected for second-generation transcriptome sequencing. The collected materials to be sequenced were frozen in liquid nitrogen and stored in a −80 °C freezer. The P. tinctoria plant specimen was identified by the Plant Resources Investigation, Evaluation and Identification Center of Kunming Institute of Botany, Chinese Academy of Sciences, and the voucher specimen with the specimen number 1585760 is stored in the Herbarium of Kunming Institute of Botany.
Estimation of genome size in P. tinctoria
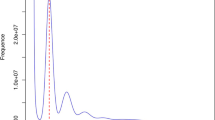
In this study, two methods, flow cytometry and K-mer analysis, were used to estimate the genome size of P. tinctoria. The genome size of a plant was estimated using the C value for P. tinctoria calculated using tomato (Solanum lycopersicum)32 as an internal reference. P. tinctoria seeds were collected and stored in a −20 °C refrigerator and planted in a greenhouse (Fig. 2a). Fresh and tender leaves of P. tinctoria and S. lycopersicum were collected and immediately placed in 0.8 mL of precooled MGb dissociation solution (45 mM MgCl2·6H2O, 20 mM MOPS, 30 mM sodium citrate, 1% (w/v) PVP 40, 0.2% (v/v) Triton X-100). In 10 mM Na2EDTA, pH 7.5 with 20 μL/mL β-mercaptoethanol, leaves were quickly chopped vertically with a sharp blade and left in the dissociation solution on ice for 10 min and then filtered with a 40 micron aperture filter to assemble a nuclear suspension. Precooled propidium iodide (PI) (50 μg/mL) and RNase solution (50 μg/mL) were added to the nuclear suspension, which was placed on ice and stained for 0.5–1 h away from light. Using a BD FACScalibur flow cytometer to measure the fluorescence intensity of the sample, the genome size of P. tinctoria was estimated to be 1.62 Gb, with tomato as the internal standard (Fig. 2b).

Morphology and genome size estimation of P. tinctoria. (a) Morphology of P. tinctoria. (b) Estimation of genome size by flow cytometry with S. lycopersicum as the internal standard. (c) Estimation of genome size by 17-kmer distribution. (d) The karyotype of P. tinctoria.
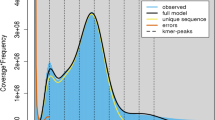
K-mer analysis, derived from low-depth sequencing of small fragment libraries, can map essential genomic information for P. tinctoria, including genome size, heterozygosity, and repeat sequence ratio33. In this study, P. tinctoria cultivated in a greenhouse was selected as the material for DNA extraction and library construction, and paired end sequencing was performed using the BGI sequencing platform to produce the original data. The raw data generated by sequencing were 176.68 Gbp (Table 1), and clean data of 169.127 Gbp were obtained after quality control by SOAPnuke software (v2.1.0, major parameter:–lowQual = 20, –nRate = 0.005, –qualRate = 0.5, Other parameters default)34. The sequencing quality was evaluated by FastQC software (v0.11.7)35, and both the sequencing quality and the sequencing error rate were determined to be normal. A total of 10,000 pairs of read data were randomly selected from the filtered high-quality data and then compared with the NCBI nucleotide database (NT library) with BLAST software (v0.7.1)36. The results of NT library comparison showed that the sample libraries in P. tinctoria were most similar to the DNA of related species, which suggested that the sample libraries in P. tinctoria did not have significant exogenous contamination. The library was constructed and sequenced successfully. Then, GCE software (v1.0.2)37 was used to analyze the K-mer counts, and K-mer analysis showed that the genome size of P. tinctoria was approximately 1.77 Gb, the rate of heterozygosity was 0.16%, the repeat sequence ratio was 76.51%, and the genome GC content was 39.6% (Fig. 2c).
Investigation of karyotype in P. tinctoria
The root tips of tissue cultured P. tinctoria derived from genome sequencing material were used for karyotypic investigations. The roots were rinsed with flowing water, transferred to a centrifuge tube filled with cold water, immersed in 0.002 mol/L 8-hydroxyquinoline solution and pretreated at 25 °C for 3 h in the dark. After pretreatment, the root tips were cleaned with ultrapure water five times for two minutes each and fixed with Carnoy’s solution (anhydrous ethanol:acetic acid = 3:1) for 2 h at 4 °C. After fixation, the root tips were washed 5 times with ultrapure water and then acid hydrolyzed with 45% acetic acid (v/v) and 1 mol/L HCl at a volume ratio of 1:1 in a water bath at 60 °C for 2 min. After acid hydrolysis, the root tips were washed with ultrapure water 5 times for 2 min each, soaked in ultrapure water for 2 h, and dyed with improved modified carbol-fuchsin solution for 30–45 min. After processing, clear chromosome images were obtained under a Leica DM1000 optical microscope objective (20×) and photographed under a 100× objective. Karyotype results showed that the chromosome number of P. tinctoria is 40 (Fig. 2d); consistent with literature reports38,39.
Genome sequencing and assembly
High-quality P. tinctoria genomic DNA was extracted using the CTAB method40 from young leaves of plants cultivated in a greenhouse. The quality and quantity of the extracted DNA were examined using a NanoDrop 2000 spectrophotometer and Qubit dsDNA HS Assay Kit on a Qubit 3.0 Fluorometer and electrophoresis on a 0.8% agarose gel. The PacBio Sequel II sequencer developed by Pacific Biotechnology (http://www.pacb.com), which uses single-molecule real-time (SMRT) technology, was used to sequence the whole genome of P. tinctoria. The original genome data for P. tinctoria were generated by the PacBio Sequel II sequencing platform of Wuhan Frasergen Co., Ltd. (http://www.frasergen.cn) using 3 SMRT cells. Finally, a total of 67.76 Gb of raw sequencing data were generated (Table 1). Preliminary assembly results drafted from Hifiasm software (v0.16.1)41 show that the total length of the P. tinctoria genome was 1.68 Gb, comprising a total of 322 contigs with a contig N50 of 31,049,992 bp.
Chromosome-level genome assembly
The young and tender leaves of P. tinctoria, from the same plants as the genome sequencing material, were treated with paraformaldehyde to fix the DNA conformation of the cells. After the cells were lysed, restriction endonuclease was applied to the cross-linked DNA to form sticky ends. Then, the sticky ends of DNA were repaired, and biotin-labeled oligonucleotide ends were added. DNA ligases were used to connect DNA fragments. The DNA cross-links were removed by protease digestion, and the DNA was purified and randomly digested to form 300–500 bp fragments. Finally, the biotin-labeled DNA was captured by adsorption to avidin magnetic beads, the DNA fragments were end-repaired and A-tailed, sequencing adapters were ligated, the number of PCR amplification cycles was evaluated, and the entire library was purified42,43. After library construction was completed, Qubit2.0 was used for preliminary quantification, and the library was diluted to 1 ng/μl. Then, an Agilent 2100 instrument was used to confirm that the insert size of the library was as expected, and Q-PCR was used to determine that the effective concentration of the library was greater than 2 nM. BGI MGISEQ-2000 PE150 sequencing was performed to produce raw reads. Finally, a total of 180.39 Gb of raw sequencing data were generated (Table 1). Trimmomatic software (v0.39)44 was used to remove sequencing adapters and low-quality fragments from the original data. The clean data were mapped to the draft genome using Juicer software (v1.6.2)45, after low-quality and redundant reads were filtered out; the remaining sequences were used for auxiliary assembly. 3D-DNA software (v201008)46 was used to cluster the data, construct an interaction matrix and draw an interaction map, and Juicebox (v2.15.08)47 was then used to allow visual inspection and manual error correction. The sequence and direction of contigs in the assembly process or assembly errors within contigs that need to be corrected could be found through the interaction graph. The assembled genome was obtained by Hi-C technology, and the final genome assembly result was produced after dehybridization. The color range from light to dark indicates an increase in the intensity of the interaction, with darker indicating stronger interactions. The horizontal and vertical coordinates indicate the N*bin position on the genome.
The 20 squares in this image are the 20 chromosomes of P. tinctoria. In the process of assembly and error correction, the original 322 contigs were rearranged cording to the interaction map, and a total of 322 contigs were formed and sequenced. Finally, 20 chromosomes were constructed, including 84 contigs with a total length of 1.66 Gb and a contig N50 = 34.06 Mb, scaffold N50 = 82.35 Mb, representing 98.55% of the original genome length. We divided the assembled genome into two subgenomes, with subgenome A containing 10 chromosomes named A01 to A10, with subgenome B containing 10 chromosomes named B01 to B10 (Fig. 3). The genome size of subgenome A and subgenome B was 888.67 Mb and 771.58 Mb, respectively. In subgenome A, the longest chromosome was A01 with sequence length of 100,276,464 bp, and the shortest chromosome was A10 with sequence length of 72,367,963 bp. In subgenome B, the longest chromosome was B01 with sequence length of 85,563,471 bp, and the shortest chromosome was B10 with sequence length of 67,428,849 bp. The scaffold N50 length of subgenome A and subgenome B were 90.56 Mb and 76.84 Mb, respectively (Table 2).

Hi-C interaction heatmap with a resolution of 500 kb. The strength of Hi-C interaction within chromosomes was represented by color, ranging from red (indicating high interaction strength) to yellow (indicating low interaction strength).
In the 20 chromosomes of subgenomes A and B, genes and repetitive sequences were unevenly distributed on the chromosomes. The gene density was higher at the terminal positions with fewer repetitive sequences, while the gene density was lower in the intermediate positions with a higher proportion of repetitive sequences. The chromosomes exhibited the highest number of transfer RNAs (tRNAs) and the lowest number of microRNAs (miRNAs). The GC content ranged from 29% to 57%, with an average GC content of 39.25% for the subgenome A and 38.91% for the subgenome B. Additionally, a large number of synteny blocks demonstrated high synteny between the two subgenomes of P. tinctoria (Fig. 4).

Overview of the P. tinctoria genome assembly. (a) The chromosomes of subgenome A and B (scale is 1 M for each), the subgenome A comprised chromosomes A01 to A10, while the subgenome B encompassed chromosomes B01 to B10. (b) The Y-axis represents the gene density of subgenome A and B, measured in 1 Mb windows, with a range from 0 to 136. (c) The Y-axis represents the repeat sequence of subgenome A and B with 1 Mb windows, with values ranging from 0% to 100%. (d) Distribution of ncRNA in subgenome A and B with 1 Mb (ribosomal RNA: purple, small nuclear RNA: red, transfer RNA: green, microRNA: blue). (e) The Y-axis represents the GC content of subgenome A and B, using 1 Mb windows, and ranges from 29% to 57%. (f) Intersubgenome syntenic gene pairs of P. tinctoria in subgenome A and B.
Transcriptome sequencing
The P. tinctoria tisssue was originally collected from inflorescences, flowers, roots, stems and leaves. Total RNA was extracted using Trizol reagent (Invitrogen, CA, USA). Sequencing libraries were generated using the VAHTS Universal V6 RNA-seq Library Kit for MGI (Vazyme, Nanjing, China) following the manufacturer’s recommendations. Subsequently, sequencing was performed on a MGI-SEQ. 2000 platform by Frasergen Bioinformatics Co., Ltd. (Wuhan, China). Finally, a total of 23.43 Gb RNA-seq clean data were obtained for genome annotation (Table 1).
Genome annotation
After the assembly of the P. tinctoria genome, multiple software programs were used to annotate the repeat sequences throughout the whole genome. TRF software (v4.09)48 was used to annotate the tandem repeat sequences in P. tinctoria and explained 7.37% of the repeat sequences of the whole genome. RepeatMasker software (v4.1.2)49 and RepeatProteinMask software were used to predict sequences similar to those in the known repeat sequence database Repbase, which annotated 23.85% repeat sequences of the whole genome. De novo prediction of repeat sequences in P. tinctoria accounted for 74.92% of the repeats in the whole genome. Finally, the results obtained by above annotation methods were compared, and the redundant results were removed. The proportion of total repeat sequences in the genome has been conclusively determined to be 77.9%. In the P. tinctoria genome, the transposable element (TE) type had the highest content of repetitive sequences, accounting for 77.31%, and we further categorized them. A total of 5.25% of the repeats sequences were DNA transposons, 8.36% were long interspersed nuclear elements (LINE)(>1000 bp), and 0.02% were short interspersed nuclear elements (SINE)(approximately 300 bp). 64.30% repeat sequences were long terminal repeat sequences (LTR) (1.5 kbp-10 kbp), among which 28.33% were Copia, and 28.67% were Gypsy. 4.7% of repeat sequences could not be annotated (Table 3).
The gene structure of the P. tinctoria genome was annotated by three methods. The first method applied was to perform homologous alignment of encoded proteins with annotated proteins from related species, such as Fagopyrum tataricum31, Rumex hastatulus29, Beta vulgaris50 and Arabidopsis thaliana51. Second, assisted annotation was carried out to predict gene structure in P. tinctoria by comparison with RNA-seq data obtained from inflorescences, flowers, roots, stems and leaves and performed on the MGI-SEQ. 2000 platform. The third was to use Augustus (v3.3)52 and GlimmerHMM software (v3.0.4)53 to make de novo predictions of coding gene structures. MAKER software (v3.00)54 was used to integrate the gene sets predicted by the above methods into a nonredundant and more complete gene set. Then, PASA (v2.4.1)55 combined with transcriptome data was used to update the gene structure. A total of 76,742 protein-coding genes were annotated (Table 4). The average length of all predicted genes was 3,378.74 bp, each gene had an average of 4.07 exons, and the average exon length was 316.03 bp.
The protein sequences encoded by the P. tinctoria genome were analyzed by homology searches in SwissProt56, TrEMBL57, InterPro58, NR59, GO60 and KEGG61 databases to obtain functional annotation information62. Finally, 94.28% (72,352) of the 76,742 protein-coding genes obtained by gene structure annotation were functionally annotated in the P. tinctoria genome (Table 4).
Data Records
The sequencing raw data, final genome assembly data reported in this paper had been deposited in NCBI database under BioProject number PRJNA1055428. The sequencing raw data were deposited in the SRA database with accession number SRR2731368963, SRR2731369064, SRR2731369165, SRR2731369266, SRR2731369367, SRR2731369468. And the final genome assembly has been deposited at GenBank under the accession JBANSL00000000069. Genome annotation data has been deposited at the figshare database70.
Technical Validation
Evaluation of the assembled genome
First, the Hi-C interaction heatmap revealed no obvious sequence or contig direction errors in the P. tinctoria genome assembly and had characteristics suggesting intrachromosomal interaction enrichment, distance-dependent interaction decay and local interaction smoothness. The Hi-C results showed a high-quality P. tinctoria genome (Fig. 3).
Moreover, BUSCO (v5.2.2)71 (Benchmarking Universal Single-Copy Orthology) analysis using the single-copy homologous gene set in OrthoDB was used to evaluate the predicted genes and their integrity, fragmentation degree and possible loss rate and thus evaluate the integrity of the gene regions in the entire assembly. The BUSCO gene set used in this evaluation was embryophyta_odb10. The results showed that the P. tinctoria genome contained complete gene sequences for 97.6% of the 1614 BUSCO groups, indicating high integrity of the gene regions in the P. tinctoria genome assembly (Table 5).
Finally, to evaluate the integrity of the genome assembly and the uniformity of sequencing coverage in P. tinctoria, the second- and third-generation sequencing data were selected and compared with the assembled genome using the comparison tool minimap2 (v2.12), and the mapping rate, extent of genome coverage and depth distribution of reads were analyzed. The mapping rate and coverage between the final assembled genome and sequencing data are more than 99%, indicating that the P. tinctoria genome assembly has decent quality, integrity and full coverage. Overall, all of the above results confirmed that both the A and B subgenome assemblies of P. tinctoria were of high quality.
Evaluation of the gene annotation
The annotated genes were evaluated using BUSCO with the dataset embryophyta_odb10. The proportion of complete BUSCOs was 98.3%, Fragmented BUSCOs was 0.3% and Missing BUSCOs was 1.4% (Table 5). The results from the BUSCO assessment indicate that genome annotation of P. tinctoria genome was of high quality.
Code availability
All bioinformatics software and databases used to analyze the data were used according to the manuals and default protocols, and no custom code was used in this study.
References
Sturgeon, D. Chinese text project. https://ctext.org/da-dai-li-ji/xia-xiao-zheng/zhs (2023).
Lopes, H. D. F., Tu, Z. H., Sumi, H., Furukawa, H. & Yumoto, I. Indigofera tinctoria leaf powder as a promising additive to improve indigo fermentation prepared with sukumo (composted Polygonum tinctorium leaves). World J. Microbiol. Biotechnol. 37, 179 (2021).
Tu, Z., Lopes, H. D. S., Narihiro, T. & Yumoto, I. The mechanism underlying of long-term stable indigo reduction state in indigo fermentation using sukumo (composted Polygonum tinctorium leaves). Front. Microbiol. 12, 698674 (2021).
Commission, C. P. Chinese Pharmacopoeia 2020 (China Medical Science Press, 2020).
Iwaki, K., Koya-Miyata, S., Kohno, K., Ushio, S. & Fukuda, S. Antimicrobial activity of Polygonum tinctorium Lour: extract against oral pathogenic bacteria. J. Nat. Med. 60, 121–125 (2006).
Kataoka, M. et al. Antibacterial action of tryptanthrin and kaempferol, isolated from the indigo plant (Polygonum tinctorium Lour.), against Helicobacter pylori-infected Mongolian gerbils. J. Gastroenterol. 36, 5–9 (2001).
Zhong, Y. et al. Highly potent anti-HIV-1 activity isolated from fermented Polygonum tinctorium Aiton. Antivir. Res. 66, 119–128 (2005).
Tokuyama-Nakai, S., Kimura, H., Ishihara, T., Jisaka, M. & Yokota, K. In vitro anti-inflammatory and antioxidant activities of 3,5,4′-trihydroxy-6,7-methylenedioxyflavone-O-glycosides and their aglycone from leaves of Polygonum tinctorium lour. Appl. Biochem. Biotechnol. 184, 414–431 (2018).
Jeong, H. J., Oh, H. A., Lee, B. J. & Kim, H. M. Inhibition of IL-32 and TSLP production through the attenuation of caspase-1 activation in an animal model of allergic rhinitis by Naju Jjok (Polygonum tinctorium). Int. J. Mol. Med. 33, 142–150 (2014).
Kim, D. H., Kim, C. S., Subedi, L., Kim, S. Y. & Lee, K. R. Alkaloids of NIRAM, natural dye from Polygonum tinctorium, and their anti-inflammatory activities. Tetrahedron Lett. 60, 151130 (2019).
Jiang, Q., Yuan, Z., Li, J. & Hou, Y. Extraction of beta-sitosterol from Polygonum tinctorium Aiton and study on its antioxidation. Sci. Technol. Food Ind. 36, 108–112 (2015).
Tokuyama-Nakai, S. et al. Constituents of flavonol O-glycosides and antioxidant activities of extracts from seeds, sprouts, and aerial parts of Polygonum tinctorium Lour. Heliyon 5, e01317 (2019).
Iwaki, K. & Kurimoto, M. Cancer preventive effects of the indigo plant, Polygonum tinctorium. Recent Res. Dev. Cancer 4, 429–437 (2002).
Jang, H.-G. et al. Chemical composition, antioxidant and anticancer effects of the seeds and leaves of indigo (Polygonum tinctorium Ait.) plant. Appl. Biochem. Biotechnol. 167, 1986–2004 (2012).
Koya-Miyata, S. et al. Prevention of azoxymethane-induced intestinal tumors by a crude ethyl acetate-extract and tryptanthrin extracted from Polygonum tinctorium lour. Anticancer Res. 21, 3295–3300 (2001).
Yang, Q. Y. et al. Exploring the mechanism of Indigo naturalis in the treatment of ulcerative colitis based on TLR4/MyD88/NF-κB signaling pathway and gut microbiota. Front. Pharmacol. 12, 674416 (2021).
Yokote, A. et al. Ferroptosis in the colon epithelial cells as a therapeutic target for ulcerative colitis. J. Gastroenterol. 58, 868–882 (2023).
Lin, Y.-K. et al. Anti-psoriatic effects of Indigo naturalis on the proliferation and differentiation of keratinocytes with indirubin as the active component. J. Dermatol. Sci. 54, 168–174 (2009).
Cheng, H.-M. et al. Clinical efficacy and IL-17 targeting mechanism of Indigo naturalis as a topical agent in moderate psoriasis. BMC Complement. Altern. Med. 17, 439 (2017).
Wu, X. et al. Characterization of anti-leukemia components from Indigo naturalis using comprehensive two-dimensional K562/cell membrane chromatography and in silico target identification. Sci. Rep. 6, 30103 (2016).
Jiang, Z. et al. Dissection of scientific compatibility of Chinese medicinal formula Realgar-Indigo naturalis as an effective treatment for promyelocytic leukemia from the perspective of toxicology. J. Ethnopharmacol. 317, 116895 (2023).
Wang, L. et al. Dissection of mechanisms of Chinese medicinal formula Realgar-Indigo naturalis as an effective treatment for promyelocytic leukemia. Proc. Natl. Acad. Sci. USA. 105, 4826–4831 (2008).
Lin, Y.-K. et al. Anti-inflammatory effects of the extract of Indigo naturalis in human neutrophils. J. Ethnopharmacol. 125, 51–58 (2009).
Feng, J. et al. Isatis indigotica: from (ethno) botany, biochemistry to synthetic biology. Mol Hortic. 1, 17 (2021).
Mohn, T., Potterat, O. & Hamburger, M. Quantification of active principles and pigments in leaf extracts of Isatis tinctoria by HPLC/UV/MS. Planta Med. 73, 151–156 (2007).
Otto, S. P. & Whitton, J. Polyploid incidence and evolution. Annu Rev Genet. 34, 401–437 (2000).
Kim, S. T., Sultan, S. E. & Donoghue, M. J. Allopolyploid speciation in Persicaria (Polygonaceae): insights from a low-copy nuclear region. Proc. Natl. Acad. Sci. USA. 105, 12370–12375 (2008).
Zhang, Y. et al. Assembly and annotation of a draft genome of the medicinal plant Polygonum cuspidatum. Front. Plant Sci. 10, 15 (2019).
Rifkin, J. L. et al. Widespread recombination suppression facilitates plant sex chromosome evolution. Mol. Biol. Evol. 38, 1018–1030 (2021).
Zhang, H. et al. The haplotype-resolved genome assembly of autotetraploid rhubarb Rheum officinale provides insights into its genome evolution and massive accumulation of anthraquinones. Plant Commun. https://doi.org/10.1016/j.xplc.2023.100677 (2023).
Lin, H. et al. Haplotype-resolved genomes of two buckwheat crops provide insights into their contrasted rutin concentrations and reproductive systems. BMC Biol. 21, 87 (2023).
Sato, S., Serra, R. G., Cámara, F. & Gianese, S. The tomato genome sequence provides insights into fleshy fruit evolution. Nature 485, 635–641 (2012).
Huang, H., Wang, C., Pei, S. & Wang, Y. A chromosome-level reference genome of an aromatic medicinal plant Adenosma buchneroides. Sci. Data 10, 660 (2023).
Ameria, S. P. L. et al. Characterization of a flavin-containing monooxygenase from Corynebacterium glutamicum and its application to production of Indigo and indirubin. Biotechnol. Lett. 37, 1637–1644 (2015).
Brown, J., Pirrung, M. & McCue, L. A. FQC dashboard: integrates FastQC results into a web-based, interactive, and extensible FASTQ quality control tool. Bioinformatics 33, 3137–3139 (2017).
Altschul, S. F., Gish, W., Miller, W., Myers, E. W. & Lipman, D. J. Basic local alignment search tool. J. Mol. Biol. 215, 403–410 (1990).
Liu, B. et al. Estimation of genomic characteristics by analyzing k-mer frequency in de novo genome projects. Quant. Biol. 35, 62–67 (2013).
Kawakami, S. M., Fujisawa, I., Murai, K., Kawakami, T. & Kato, J. Characteristics of established hexaploid plants derived from an octoploid plant induced by colchicine treatment in Persicaria tinctoria. Cytologia 87, 49–54 (2022).
Doida, Y. Cytological studies in Polygonum and related genera. Bot. Mag. Tokyo 73, 337–340 (1960).
Porebski, S., Bailey, L. G. & Baum, B. R. Modification of a CTAB DNA extraction protocol for plants containing high polysaccharide and polyphenol components. Plant Mol. Biol. Report. 15, 8–15 (1997).
Cheng, H., Concepcion, G. T., Feng, X., Zhang, H. & Li, H. Haplotype-resolved de novo assembly using phased assembly graphs with hifiasm. Nat. Methods 18, 170–175 (2021).
Lieberman-Aiden, E. et al. Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science 326, 289–293 (2009).
Naumova, N., Smith, E. M., Zhan, Y. & Dekker, J. Analysis of long-range chromatin interactions using Chromosome Conformation Capture. Methods 58, 192–203 (2012).
Bolger, A. M., Lohse, M. & Usadel, B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120 (2014).
Durand, N. C. et al. Juicer provides a one-click system for analyzing loop-resolution Hi-C experiments. Cell Syst. 3, 95–98 (2016).
Dudchenko, O. et al. De novo assembly of the Aedes aegypti genome using Hi-C yields chromosome-length scaffolds. Science 356, 92–95 (2017).
Durand, N. C. et al. Juicebox provides a visualization system for Hi-C contact maps with unlimited zoom. Cell Syst. 3, 99–101 (2016).
Benson, G. Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res. 27, 573–580 (1999).
Bailly-Bechet, M., Haudry, A. & Lerat, E. “One code to find them all”: a perl tool to conveniently parse RepeatMasker output files. Mob. DNA 5, 13 (2014).
Lehner, R., Blazek, L., Minoche, A. E., Dohm, J. C. & Himmelbauer, H. Assembly and characterization of the genome of chard (Beta vulgaris ssp. vulgaris var. cicla). J. Biotechnol. 333, 67–76 (2021).
Sloan, D. B., Wu, Z. & Sharbrough, J. Correction of persistent errors in arabidopsis reference mitochondrial genomes. Plant Cell 30, 525–527 (2018).
Stanke, M. et al. AUGUSTUS: ab initio prediction of alternative transcripts. Nucleic Acids Res. 34, W435–W439 (2006).
Majoros, W. H., Pertea, M. & Salzberg, S. L. TigrScan and GlimmerHMM: two open source ab initio eukaryotic gene-finders. Bioinformatics 20, 2878–2879 (2004).
Holt, C. & Yandell, M. MAKER2: an annotation pipeline and genome-database management tool for second-generation genome projects. BMC Bioinform. 12, 491 (2011).
Haas, B. J. Improving the Arabidopsis genome annotation using maximal transcript alignment assemblies. Nucleic Acids Res. 31, 5654–5666 (2003).
Bairoch, A. The SWISS-PROT protein sequence database and its supplement TrEMBL in 2000. Nucleic Acids Res. 28, 45–48 (2000).
Junker, V. et al. The role SWISS-PROT and TrEMBL play in the genome research environment. J. Biotechnol. 78, 221–234 (2000).
Hunter, S. et al. InterPro: the integrative protein signature database. Nucleic Acids Res. 37, D211–D215 (2009).
Yu, K. & Zhang, T. Construction of customized sub-databases from NCBI-nr database for rapid annotation of huge metagenomic datasets using a combined BLAST and MEGAN approach. PLoS One 8, e59831 (2013).
Ashburner, M. et al. Gene ontology: tool for the unification of biology. Nat. Genet. 25, 25–29 (2000).
Ogata, H. et al. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 27, 29–34 (1999).
Xu, C. et al. Chromosome level genome assembly of oriental armyworm Mythimna separata. Sci. Data 10, 597 (2023).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR27313689 (2024).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR27313690 (2024).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR27313691 (2024).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR27313692 (2024).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR27313693 (2024).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR27313694 (2024).
NCBI GenBank https://identifiers.org/ncbi/insdc.gca:GCA_037127255 (2024).
Li, Q. genome annotation of Persicaria tinctoria. Figshare https://doi.org/10.6084/m9.figshare.25321858 (2024).
Simão, F. A., Waterhouse, R. M., Ioannidis, P., Kriventseva, E. V. & Zdobnov, E. M. BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 31, 3210–3212 (2015).
Acknowledgements
This work was supported by the Second Tibetan Plateau Scientific Expedition and Research (STEP) program (2019QZKK0502). We sincerely thank Professor Pei Shengji for collecting and providing the original seed of P. tinctoria.
Author information
Authors and Affiliations
Contributions
Y.W., C.Z. and H.H. conceived the project and designed the experiments. Q.L., R.F., Q.Y., and Z.W. prepared plant samples and conducted the experiments. Q.L. and Y.H. performed the data analysis. Q.L. drafted the manuscript. All the authors have read, edited, and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Li, Q., Huang, H., Fan, R. et al. Chromosome-level genome assembly of the tetraploid medicinal and natural dye plant Persicaria tinctoria. Sci Data 11, 1440 (2024). https://doi.org/10.1038/s41597-024-04317-6
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41597-024-04317-6
This article is cited by
-
TCMP-300: A Comprehensive Traditional Chinese Medicinal Plant Dataset for Plant Recognition
Scientific Data (2025)
