
Abstract
Fat greenling (Hexagrammos otakii Jordan & Starks, 1895) is a valuable marine fish species, crucial for aquaculture in Northern China due to its high-quality meat and significant economic value. However, the aquaculture industry faces challenges such as trait degradation, early sexual maturity, and disease susceptibility, necessitating advanced genomic interventions for sustainable cultivation. This study presents the first chromosomal-level genome assembly of H. otakii, achieved using PacBio long-read sequencing and Hi-C technology. The assembly yielded a genome size of 682.43 Mb with a contig N50 size of 2.39 Mb and a scaffold N50 size of 27.83 Mb. The completeness of genome assessed by BUSCO is 96.99%. A total of 22,334 protein-coding genes were predicted, with 21,619 (96.80%) functionally annotated across various protein databases. This genomic resource is a step forward in supporting the breeding, germplasm conservation, and enhancement of H. otakii, facilitating genetic studies and the development of strategies for disease resistance and growth optimization in aquaculture.
Similar content being viewed by others

Background & Summary
Fat greenling (H. otakii) is a cold-water near-bottom reef fish (Fig. 1), mainly distributed in northern China such as Shandong, Hebei, Tianjin, Liaoning provinces. It also inhabits the seas of North Korea, Japan and the Far East of Russia1,2,3,4. Renowned for its high-quality meat and nutritional value, fat greenling is a cornerstone species in marine economic fisheries and is highly valued by both consumers and farmers. It is an ideal candidate for cage culture, proliferation, release, and resource restoration, with significant potential for broader application in aquaculture across Northern China. H. otakii prefers coastal and rocky reef habitats, typically found at depths up to 50 meters. This species is resilient to low temperatures, thriving in waters ranging from 2 to 26 °C, with an optimal growth temperature between 16 and 21 °C. The salinity range suitable for its survival is 16 to 32‰, allowing it to overwinter safely in northern sea areas5. As an omnivorous species, its diet is diverse, including over 40 different items such as shrimp, fish, clams, worms, and amphipods.

A picture of fat greenling (Hexagrammos otakii) used for the genome sequencing.
Reproduction in fat greenling is characterized by a single annual spawning event from mid-to-late October to late November, triggered when water temperatures drop to around 18 °C. Female fat greenling produce a relatively small number of eggs, typically between 2,000 and 9,000. Eggs from sexually mature female fishes (about 150 mm in length) range from 2,000 to 3,000, while the older females (aged 4-5 years and over 250 mm in length) can produce between 6,000 and 9,000 eggs. However, the wild population of H. otakii has been declining due to overfishing, nitrogen and phosphorus pollution, and increased industrial energy consumption, which has necessitated urgent measures to repair and conserve fishery resources. The development of fat greenling aquaculture has drawn significant attention due to these challenges. The main issues hindering the development and scaling of its market include the limited availability of resources and challenges in large-scale artificial seedling production, such as early sexual maturity, reduced ovarian egg production, and extended hatching periods. These issues create a bottleneck in the artificial breeding process.
The artificial breeding of fat greenling faces specific challenges due to the high viscosity of the eggs, which leads to the formation of agglomerates during artificial insemination and results in extremely low fertilization rates. Moreover, the prolonged incubation time of fertilized eggs renders them highly susceptible to pathogens, further causing hatching difficulties. Before the breakthrough in artificial breeding techniques, wild fry was used for cage culture, leading to instability in fry yields and a downward trend that could not meet the demands of high-volume production. Furthermore, harvesting wild seed contradicts the principles of fishery resource restoration and ecological protection.
The breakthrough in artificial breeding has alleviated the supply issues of fries, leading to an increase in cage breeding and marine stock enhancement and release, with a gradual recovery of natural resources. With the rapid development of fat greenling industry, the lag of breeding has been highlighted. Most of the fries for cage culture and marine stock enhancement and release come from the offspring of artificially bred wild broodstock. Issues such as lodging syndrome, skin ulcer disease, rotten tail disease, and bacterial rotten gill disease occur during the breeding process, with the underlying causes still unclear but potentially linked to genetic factors like individual physical disabilities, developmental deficiencies, and compromised immunity6.
Given the mixed germplasm, unknown parental history, and trait degradation, the primary challenges for fish fry include slow growth rate, weak disease resistance, and high mortality. High-quality and sufficient seed has become a key factor in the industrial chain of fat greenling. The current breeding situation highlights that it is urgent to conduct genome research to obtain genome sequence and study its immune regulation mechanism. It is worthwhile to conduct researches on wild population conservation and genetic improvement of fat greenling.
At present, the research on the fat greenling involves many aspects, including embryonic development7, genetic diversity8,9,10, nutritional requirements11,12,13, Breeding environmental conditions14,15,16,17,18,19, immunology20,21,22, behavioristics23,24 and so on. However, genomic resources for this species are limited, with only transcriptome and microsatellite marker analyses available, and no reference genome exists. This lack of genomic data hampers conservation and genetic studies of the species.
Advancements in sequencing technologies have paved the way for genome information acquisition25 and molecular breeding26, proving to be valuable methods. In this study, the chromosome-level genome of fat greenling was firstly constructed using PacBio sequencing and Hi-C technology. A 350 bp library was constructed and generated a total of 73.5 Gb of clean data on an Illumina HiSeq platform (Tables 1 and 2). The PacBio platform produced 113.58 Gb of high-quality clean reads, representing a 174.74-fold coverage of the fat greenling genome (Tables 1 and 4). This genomic data will benefit comprehensive conservation studies of fat greenling to implement better protection of wild populations, and it will facilitate researches on population genetics and the identification of functional genes related to important economic traits and the sex determination for fat greenling. Additionally, this data will provide crucial theoretical guidance for artificial and genetic engineering breeding, aiding in the screening of genetic variations related to rapid growth and disease resistance.
Methods
Sample collection and DNA extraction
To obtain high-quality genomic DNA for sequencing, the fresh muscle tissue was collected from a fat greenling in Lidao, Rongcheng city, Weihai city, Shandong province. The muscle tissue below the dorsal fin was taken and stored in liquid nitrogen until DNA extraction. The improved CTAB (Cetyltrimethylammonium bromide) method was used to extracted high-quality genomic DNA. The quality and concentration of the extracted genomic DNA were checked using NanoDrop 2000 spectrophotometer (NanoDrop Technologies, Wilmington, DE, USA), 0.8% agarose gel electrophoresis and a Qubit 3.0 fluorimeter (Life Technologies, Carlsbad, CA, USA).
Library construction, sequencing and data preparation
The second-generation DNA fragment library, PacBio CLR library and Hi-C data were obtained for generating a chromosome-level genome assembly of fat greenling. 1 μg DNA was used to construct the library according to the MGI DNA Library Kit (Vazyme, Nanjing) protocol. Then, the concentration and fragment size distribution of the samples in the constructed library were determined by Qubit 3.0 fluorometer (Life Technologies, Carlsbad, CA, USA) and Bioanalyzer 2100 system (Agilent Technologies, CA, USA) using the appropriate computer pooling program. After library validation, sequencing was performed on the MGISEQ-2000 platform (the sequencing service is provided by Wuhan Onemore Technology Co., Ltd.). A 350 bp library was constructed and generated a total of 73.5 Gb of clean data for the fat greenling on an Illumina NovaSeq 6000 platform, with Q20 and Q30 being 96.64% and 91.37% respectively (Tables 1 and 2). The SMRTbell Express Template Prep Kit 2.0 reagent (Pacific Biosciences) were used to constructed the SMRT Bell CLR Library. Approximately 5 μg of genomic DNA was used for the library construction. The library size and quality were evaluated using FEMTO Pulse and Qubit dsDNA HS assay kits. Sequencing primers and Sequel II DNA polymerases were annealed separately and combined with the final SMRTbell library. After library construction, sequencing was performed on the PacBio Sequel II platform (the sequencing service is provided by Wuhan Onemore Technology Co., Ltd.). In total, 113.58 Gb of clean data were obtained, which was 174.74-fold coverage of the genome assembly. Hi-C libraries were constructed using MboI restriction enzyme and sequenced on the Illumina NovaSeq 6000 platform in 150 bp paired-end mode. As a result, 101.7 Gb of Hi-C clean data were obtained, which covered 156.46-fold of the gene assembly (Table 1).
Additionally, muscle, heart, spleen, liver and stomach tissues were pooled to obtain the transcriptome of fat greenling. Transcriptome sequencing was performed on the Illumina NovaSeq 6000 platform, yielding a total of 7.8 G of clean data (Table 1).
Genome survey
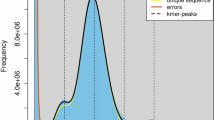
After obtaining the second-generation sequencing data PE150, HTQC v1.92.310 software27 was used to filter the raw data to obtain high-quality data. A survey of the fat greenling genome was performed using the k-mer (k = 17 in this case) method to estimate genome size, heterozygosity and repeat sequence information The software GCE(1.0.0)28 was utilized, and the total number of k-mers was 57,249,936,244, with a k-mer peak at a depth of 80 (Table 3, Fig. 2). the fat greenling genome size was estimated to be 689 Mb, revised to 678 Mb. The heterozygous ratio was 0.70%, and the repetitive rate was 37.52% (Table 3).

Distribution of k-mers of length 17 from the Illumina data.
De novo genome assembly
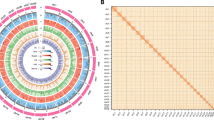
Genome assembly was performed by PacBio Sequel II CLR sequencing mode to obtain subreads data. 100 × random data were used for preliminary assembly by Mecat2 software29 with default parameters. After the initial assembly, the gcpp tool of SMRT Link v8.0 (https://github.com/PacificBiosciences/gcpp) was used to perform the third-generation error correction, and then used the second-generation data in the survey evaluation to perform the genome second-generation polish. After the polish was completed, we completed genome assembly at the contig level.The PacBio platform generated a total of 113.58 Gb high-quality clean reads from the long-read library, representing a 174.74-fold coverage of the fat greenling genome (Tables 1 and 4). These data were assembled using Canu(v2.2)30, followed by polishing with Plion(v1.22)31. For highly heterozygous genomes, initial assembly may assemble all the heterozygous fragments, resulting in larger genomes than expected. Purge_haplotigs(v1.0.4)32 was used to remove redundancy in the genome after initial assembly error correction. Redundant contigs were identified and removed according to the depth distribution and sequence similarity of reads. The genome fragment of 682.34 Mb was obtained, the length of which was close to the genome size estimated by k-mer analysis. The clean Hi-C data were aligned to the genome assembly using BWA (v0.7.12)33. During the Hi-C assisted assembly and error correction process, the original 1,026 contigs were broken and sorted based on the interaction map, and finally 24 chromosomes and 469 scaffolds were constructed. The sequence and direction of the H. otakii genome were determined. The final total length of the genome was 682.43 Mb, with a contig N50 of 2.39 Mb, scaffold N50 of 27.83 Mb, and a chromosome anchoring rate of 94.69% (Fig. 3; Tables 5 and 6). The genome size of H. otakii was smaller than that of Hexagrammos agrammus34.

Chromosomal Hi-C heatmap of the H. otakii genome assembly.
Assessment of the genome assemblies
To assess the completeness and accuracy of the genome assembly, Minimap2 (the default parameter of V2.5)35 was employed to align the CLR data from the third generation sequencing back to the assembled genome. The alignment rate, coverage degree and depth distribution of reads were counted to evaluate the integrity of assembly and uniformity of sequencing coverage. BWA software28 was used to map the second-generation sequencing data back to the assembled genome. The rate of successfully aligned reads was recorded to assess the quality of the assembly. Then GATK software36 was used to conduct SNP calling and filtering, and the results of SNPs (statistical heterozygous and homozygous) and InDels were analyzed. The Benchmarking Universal Single-Copy Orthologs (BUSCO v3.0.2)37 analysis was conducted to evaluate the assembly completeness. Using the OrthoDB database, BUSCO(actinopterygii_odb9) predicted the single-copy homologous genes and calculated their integrity, fragmentation, and possible loss rate. This assessment provided a quantitative measure of the integrity of the gene regions in the entire assembly.
The Illumina reads and PacBio-long reads were aligned using the software BWA and BLAST38 to evaluate the quality of initial assembly. The results showed that 99.27% of the Illumina reads and 98.37% of the PacBio long reads were successfully mapped to the assembled genome (Tables 6 and 7). This high mapping rate indicates a high-quality initial assembly.
Repeat sequence annotation
Both homology annotation and de novo annotation were used to identify repetitive sequences in the genome. Firstly, RepeatMasker (open-4.09)39 and RepeatProteinMask (open-4.09) were used to search for transposable element (TE) sequences from the Repbase (release 21.01)40 database based on homology. Secondly, RepeatModeler (open-1.0.11)41 and LTR-FINDER (v1 .0.5) Software42 were used to construct a repetitive sequence database of species from scratch. RepeatMasker (open-4.09) was utilized to identify repetitive sequences from the constructed database. In addition, Tandem Repeats Finder (TRF)43 was used to identify tandem repetitive sequences.
The results from both homology and de novo annotation methods were integrated, removing overlapping non-redundant parts to finalize the repeat sequence annotation. A total of 301.3 Mb of repeat sequences were detected, accounting for 44.16% of the assembly genome (Table 8). This repeat content was larger than the value (37.52%) obtained from k-mer analysis. The predominant repeats type was DNA (203.08 Mb; 29.76% of assembly), LINEs (65.5 Mb; 9.60%), and LTRs (42.2 Mb; 6.18%) (Table 9).
Coding gene structure and function annotation
Homologous annotation, de novo annotation and transcriptome assisted annotation were used to predict the structure and function of coding genes.
Related species at the Order level including Cottus gobio, Notothenia coriiceps, Perca flavescens, Acanthochromis polyacanthus, and Amphiprion ocellaris were selected as homolog for protein-coding gene annotation. TblastN software44 was used to compare protein sequences from these species to the H. otakii constructed reference genome. Following sequence comparison, the corresponding query proteins and their sequences were filtered and processed using Exonerate software45, Augustus (v3.3)46, Genscan, and GlimmerHMM (V3.0.4) to initiate gene structure annotations. For the second-generation transcriptome data (RNA-Seq), Tophat software47 was used to align the RNA-Seq reads to the constructed reference genome. The aligned sequences were then assembled into gene structures using Cufflinks software48. The open reading frames (ORFs) in the transcript were predicted using TransDecoder software49 to define the putative coding sequence (CDS). MAKER (V3.00) software50 integrated the gene sets predicted by homologous, de novo, and transcriptome-assisted methods into a comprehensive, non-redundant gene set. List of protein function databases (SwissProt, TrEMBL, KEGG, InterPro, GO, AnimalTFDB, etc.) were used to annotate the annotated gene set with information on protein function, metabolic pathways, structural domains, and other gene functions and metabolic pathways.
Overall, these combined approaches predicted 22,334 protein-coding genes, with average gene, exon, and intron lengths of 14,477.64 bp, 255.04 bp, and 1,312.62 bp, respectively (Table 10). The statistics of the predicted gene models were compared with those from five other teleost species, including C. gobio, A. polyacanthus, A. ocellaris, N. coriiceps, and P. flavescens, showing similar distribution patterns in mRNA length, CDS length, exon length, and intron length (Fig. 4). The summary of the genome characteristics of fat greenling is shown in Fig. 5. A total of 21,619 genes, accounting for 96.80% of the predicted genes, were successfully annotated by alignment to the nucleotide, protein, and annotation databases including InterPro, NR, Swissprot, TrEMBL, KOG, GO, and KEGG (Table 11).

Annotation quality comparison of protein-coding genes. The gene length, CDS length, exon length, and intron length were compared among 6 species: Hexagrammos otakii, Cottoperca gobio, Acanthochromis polyacanthus, Amphiprion ocellaris, Notothenia coriiceps, Perca flavescens.

Genome characteristics of fat greenling.
Non-coding RNA annotation
In the process of non-coding RNA annotation, tRNAscan-SE (V1.3.1) software51 was used to search for tRNA sequences in the genome according to the structural characteristics of tRNA. Since rRNA is highly conserved, rRNA sequences of closely related species were selected as reference sequences to search for rRNA in the genome by BLASTN (V2.6.0) alignment. In addition, Rfam (V14.1) family covariance model was used to predict miRNA and snRNA sequence information on the genome by INFERNAL software52 of Rfam. A total of 4,942 tRNAs, 3,409 rRNAs, and 707 microRNAs were identified by non-coding RNA prediction (Table 12).
Data Records
The raw data, including Illumina and Hi-C sequencing data have been deposited in a public database. Hi-C data of Hexagrammos otakii: SRR2763674853, Pacbio subreads data of Hexagrammos otakii: SRR2763674954, Whole Genome Sequencing (WGS) data of Hexagrammos otakii: SRR2763675055, RNA-seq data of Hexagrammos otakii: SRR3113230956, Assembly data of Hexagrammos otakii: GCA_043790635.157, Annotation data of Hexagrammos otakii: https://doi.org/10.6084/m9.figshare.2729984158.
Technical Validation
Evaluating quality of DNA and RNA
Prior to the genome sequencing, we used an improved CTAB method to extracted high-quality genomic DNA. The quality and concentration of the extracted genomic DNA was checked using NanoDrop 2000 spectrophotometer (NanoDrop Technologies, Wilmington, DE, USA), 0.8% agarose gel electrophoresis and a Qubit 3.0 fluorimeter (Life Technologies, Carlsbad, CA, USA).
Evaluating quality of genome assembly
The Illumina reads and PacBio long reads were aligned using BWA and BLAST to evaluate the quality of initial assembly. The results showed that 99.27% of the Illumina reads and 98.37% of the PacBio long reads were successfully mapped to the assembled genome (Tables 6 and 7). BUSCO analysis was conducted to evaluate the assembly quality based on the OrthoDB database. A total of 96.90% of the 4,584 single-copy orthologs in the assembled genome were determined as complete, including 4,308 single-copy (94.00%) and 134 duplicated (2.90%) orthologs, 1.10% and 2.00% of the total single-copy orthologs were fragmented and missing, respectively (Table 13).
Evaluating quality of the genome annotation
BUSCO analysis was conducted to evaluate the genome annotation quality based on the OrthoDB database. A total of 96.40% of the 4,854 single-copy ortholog genes in the assembled genome were determined as complete, including 4,245 single-copy genes (92.60%) and 174 duplicated genes (3.80%), 1.50% and 2.10% of the total genes were fragmented and missing, respectively (Table 13).
Code availability
No specific code was used in this study. The commands used in the processing were all executed according to the manuals and protocols of the corresponding bioinformatics software.
References
Habib, K. A. et al. Population genetic structure and demographic history of the fat greenling Hexagrammos otakii. Genes Genom. 33, 413–423, https://doi.org/10.1007/s13258-011-0059-4 (2011).
Hu, F. et al. Effect of temperature on incubation period and hatching success of fat greenling (Hexagrammos otakii, Jordan & Starks) eggs. Aquacult. Res. 48, 361–365, https://doi.org/10.1111/are.12853 (2017).
Diao, J. et al. Transcriptome analysis of immune response in fat greenling (Hexagrammos otakii) against Vibrio harveyi infection. Fish Shellfish Immunol. 84, 937–947, https://doi.org/10.1016/j.fsi.2018.10.067 (2019).
Liu, X. et al. Comparison between two GAMs in quantifying the spatial distribution of Hexagrammos otakii in Haizhou Bay, China. Fisheries Research. 218, 209–217, https://doi.org/10.1016/j.fishres.2019.05.019 (2019).
Cho, G. K. & Heath, D. D. Comparison of tricaine methanesulphonate (MS222) and clove oil anaesthesia effects on the physiology of juvenile chinook salmon Oncorhynchus tshawytscha (Walbaum). Aquac. Res. 31, 537–546, https://doi.org/10.1046/j.1365-2109.2000.00478.x (2000).
Valladão, G. M. R., Gallani, S. U. & Pilarski, F. Phytotherapy as an alternative for treating fish disease. J Vet Pharmacol Ther. 38(5), 417–428, https://doi.org/10.1111/jvp.12202 (2015).
Wu, Z. et al. Study on artificial induction and early development of gynogenetic fat greenling Hexagrammos otakii. Aquacult. Rep. 22, 100975, https://doi.org/10.1016/j.aqrep.2021.100975 (2022).
Wang, X. et al. Distribution characteristics of Fat Greenling (Hexagrammos otakii) inhabiting artificial reefs around Qiansandao island, Haizhou Bay. China. J. Ocean Univ. China. 18, 1227–1234, https://doi.org/10.1007/s11802-019-4010-7 (2019).
Gwak, W. S., Roy, A. & Nakayama, K. Contrasting Genetic Structures Among Populations of Two Hexagrammid Fish Species in the Northwestern Pacific. Ocean Sci J. 56, 275–286, https://doi.org/10.1007/s12601-021-00026-y (2021).
Zhang, R., Liu, H., Zhang, Q., Zhang, H. & Zhao, J. Trophic interactions of reef-associated predatory fishes (Hexagrammos otakii and Sebastes schlegelii) in natural and artificial reefs along the coast of North Yellow Sea, China. Sci Total Environ. 791, 148250, https://doi.org/10.1016/j.scitotenv.2021.148250 (2021).
Kwak, S. N., Baeck, G. W. & Klumpp, D. W. Comparative feeding ecology of two sympatric greenling species, Hexagrammos otakii and Hexagrammos agrammus in eelgrass Zostera marina beds. Environ Biol Fish. 74, 129–140, https://doi.org/10.1007/s10641-005-7429-1 (2005).
Fan, Y. et al. Gut microbiota changes in Hexagrammos otakii during development and association with diets based on metagenomics sequencing. Aquacult. Rep. 33, 101881, https://doi.org/10.1016/j.aqrep.2023.101881 (2023).
Gu, Y. et al. Effect of replacing fish meal with chicken gut meal on growth, immune parameters, and metabolism of juvenile Fat Greenling Hexagrammos otakii. N Am J Aquacult. 85(4), 339–354, https://doi.org/10.1002/naaq.10312 (2023).
Hu, F. et al. Effects of environmental cadmium on cadmium accumulation, oxidative response, and microelements regulation in the liver and kidney of Hexagrammos otakii. J. Ocean Univ. China. 21, 479–485, https://doi.org/10.1007/s11802-022-4969-3 (2021).
Li, L. et al. Transcriptomic Analysis of Liver Tissue in Fat Greenling (Hexagrammos otakii) Exposed to Elevated Ambient Ammonia. Front. Mar. Sci. 7, 418, https://doi.org/10.3389/fmars.2020.00418 (2020).
Wang, W. et al. Unveiling the hidden effects of hypoxia: Pituitary damage and hormonal imbalance in fat greenling (Hexagrammos otakii). Sci Total Environ. 927, 172381, https://doi.org/10.1016/j.scitotenv.2024.172381 (2024).
Shi, Y. et al. Developmental toxicity and potential mechanisms exposed to polystyrene microplastics and polybrominated diphenyl ethers during early life stages of fat greenling (Hexagrammos otakii). Aquat Toxicol. 271, 106933, https://doi.org/10.1016/j.aquatox.2024.106933 (2024).
Hu, F. et al. Effects of environmental cadmium on cadmium accumulation, oxidative response, and microelements regulation in the liver and kidney of Hexagrammos otakii. J. Ocean Univ. China. 21, 479–485, https://doi.org/10.1007/S11802-022-4969-3 (2022).
Zhou, Z. et al. Effects of salinity on growth, hematological parameters, gill microstructure and transcriptome of fat greenling Hexagrammos otakii. Aquaculture. 531, 1–11, https://doi.org/10.1016/j.aquaculture.2020.735945 (2021).
Diao, J. et al. Full-length transcriptome sequencing combined with RNA-seq analysis revealed the immune response of fat greenling (Hexagrammos otakii) to Vibrio harveyi in early infection. Microb Pathogenesis. 149, 104527, https://doi.org/10.1016/j.micpath.2020.104527 (2020).
Gu, Y. et al. Potential Targets and Signaling Mechanisms of Cinnamaldehyde Enhancing Intestinal Function and Nutritional Regulation in Fat Greenling (Hexagrammos otakii). Aquacult Nutr. 2024, 5566739, https://doi.org/10.1155/2024/5566739 (2024).
Wei, X. et al. Mucous cell histopathology and label-free quantitative proteomic analysis of skin mucus in fat greenling (Hexagrammos otakii) infected with Vibrio harveyi. Fish Shellfish Immun. 146, 109398, https://doi.org/10.1016/j.fsi.2024.109398 (2024).
Zhang, Y. et al. Reef effect of vertical relief on the shelter-seeking and foraging processes of juvenile fat greenling (Hexagrammos otakii) in a mesocosm experiment. Appl Anim Behav Sci. 272, 106213, https://doi.org/10.1016/j.applanim.2024.106213 (2024).
Zhang, Y. et al. Moderate relative size of covered and non-covered structures of artificial reef enhances the sheltering effect on reef fish. Front Mar Sci. 10, 1130626, https://doi.org/10.3389/fmars.2023.1130626 (2023).
Geng, Y. et al. A chromosome-level genome assembly of an avivorous bat species (Nyctalus aviator). Sci Data. 11, 480, https://doi.org/10.1038/s41597-024-03322-z (2024).
Xia, H. et al. MultiPrime: A reliable and efficient tool for targeted next-generation sequencing. iMeta. 2(4), e143, https://doi.org/10.1002/imt2.143 (2023).
Yang, X. et al. HTQC: a fast quality control toolkit for Illumina sequencing data. BMC bioinformatics 14, 33, https://doi.org/10.1186/1471-2105-14-33 (2013).
Liu, B. et al. Estimation of genomic characteristics by analyzing k-mer frequency in de novo genome projects. Quant. Biol. 35, 62-67, https://doi.org/10.48550/arXiv.1308.2012 (2013).
Xiao, C. et al. MECAT: fast mapping, error correction, and de novo assembly for single-molecule sequencing reads. Nat. Methods. 14, 1072, https://doi.org/10.1038/NMETH.4432 (2017).
Koren, S. et al. Canu: scalable and accurate long-read assembly via adaptive k-mer weighting and repeat separation. Genome Research. (2017).
Walker, B. J. et al. Pilon: an integrated tool for comprehensive microbial variant detection and genome assembly improvement[J]. PloS one 9(11), e112963 (2014).
Roach, M. J., Schmidt, S. A. & Borneman, A. R. Purge Haplotigs: allelic contig reassignment for third-gen diploid genome assemblies. BMC Bioinformatics 19, 460 (2018).
Li, H. & Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. https://doi.org/10.1093/bioinformatics/btp324 (2009).
Zhao, L. et al. Chromosomal assembly of the Hexagrammos agrammus genome via third-generation DNA sequencing and Hi-C technology. Front. Mar. Sci. 9, 923005, https://doi.org/10.3389/fmars.2022.923005 (2022).
Li, H. Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics 34, 3094–3100, https://doi.org/10.1093/bioinformatics/bty191 (2018).
McKenna, A. et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 20, 1297–1303, https://doi.org/10.1101/gr.107524.110 (2010).
Simão, F. A., Waterhouse, R. M., Ioannidis, P., Kriventseva, E. V. & Zdobnov, E. M. BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 31, 3210–3212, https://doi.org/10.1093/bioinformatics/btv351 (2015).
Altschul, S. F. et al. Basic local alignment search tool. Journal of molecular biology 215(3), 403–410 (1990).
Chen, N. S. Using RepeatMasker to identify repetitive elements in genomic sequences. Curr. Protoc. Bioinformatics 25, 4–10, https://doi.org/10.1002/0471250953.bi0410s05 (2004).
Jurka, J. et al. Repbase Update, a database of eukaryotic repetitive elements. Cytogenet. Genome Res. 110, 462–467, https://doi.org/10.1159/000084979 (2005).
Abrusán, G., Grundmann, N., DeMester, L. & Makalowski, W. TEclass—a tool for automated classification of unknown eukaryotic transposable elements. Bioinformatics 25, 1329–1330, https://doi.org/10.1093/bioinformatics/btp084 (2009).
Xu, Z. & Wang, H. LTR_FINDER: an efficient tool for the prediction of full-length LTR retrotransposons. Nucleic Acids Res. 35, W265–W268, https://doi.org/10.1093/nar/gkm286 (2007).
Benson, G. Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res. 27, 573–580, https://doi.org/10.1093/nar/27.2.573 (1999).
Gertz, E. M., Yu, Y. K., Agarwala, R., Schäffer, A. A. & Altschul, S. F. Composition-based statistics and translated nucleotide searches: improving the TBLASTN module of BLAST. BMC Biol. 4, 1–14, https://doi.org/10.1186/1741-7007-4-41 (2006).
Slater, G. S. & Birney, E. Automated generation of heuristics for biological sequence comparison. BMC Bioinformatics 6, 31, https://doi.org/10.1186/1471-2105-6-31 (2005).
Stanke, M. et al. AUGUSTUS: ab initio prediction of alternative transcripts. Nucleic Acids Res. 34, W435–W439, https://doi.org/10.1093/nar/gkl200 (2006).
Trapnell, C., Pachter, L. & Salzberg, S. L. TopHat: discovering splice junctions with RNA-Seq. Bioinformatics 25, 1105–1111, https://doi.org/10.1093/bioinformatics/btp120 (2009).
Trapnell, C. et al. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat. Protoc. 7, 562, https://doi.org/10.1038/nprot1014-2513a (2014).
Kim, H. S. et al. Identification of xenobiotic biodegradation and metabolism-related genes in the copepod Tigriopus japonicus whole transcriptome analysis. Mar. Genom. 24, 207–208, https://doi.org/10.1016/j.margen.2015.05.011 (2015).
Campbell, M. S., Holt, C., Moore, B. & Yandell, M. Genome annotation and curation using MAKER and MAKER-P. Curr. Protoc. Bioinformatics 48, 4–11, https://doi.org/10.1002/0471250953.bi0411s48 (2014).
Lowe, T. M. & Eddy, S. R. tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 25, 955–964, https://doi.org/10.1093/nar/25.5.955 (1997).
Nawrocki, E. P., Kolbe, D. L. & Eddy, S. R. Infernal 1.0: inference of RNA alignments. Bioinformatics 25, 1335–1337, https://doi.org/10.1093/bioinformatics/btp326 (2009).
NCBI Sequence Read Archive https://www.ncbi.nlm.nih.gov/sra/SRR27636748 (2024).
NCBI Sequence Read Archive https://www.ncbi.nlm.nih.gov/sra/SRR27636749 (2024).
NCBI Sequence Read Archive https://www.ncbi.nlm.nih.gov/sra/SRR27636750 (2024).
NCBI Sequence Read Archive https://www.ncbi.nlm.nih.gov/sra/SRR31132309 (2024).
NCBI GeneBank, https://identifiers.org/ncbi/insdc.gca:GCA_043790635.1 (2024).
Li, L. et al. A Chromosomal-level genome assembly and annotation of fat greenling (Hexagrammos otakii), figshare, https://doi.org/10.6084/m9.figshare.27299841 (2024).
Acknowledgements
This work was supported by Key R&D Plan of Shandong Province (2019GHY112071), Qingdao Science and Technology Benefiting the People Demonstration Project (24-1-8-xdny-3-nsh), Modern Agriculture Fish Industry Technology System of Shandong Province (SDAIT-12-01), Natural Science Foundation of Shandong Province (ZR2021MC089). We thank Dr. Qi Liu in Wuhan Onemore-tech Co., Ltd. for their assistance with genome sequencing and analysis.
Author information
Authors and Affiliations
Contributions
L.L., F.W.H. conceived the research project. L.L., D.L., X.L.W., J.D., Y.J.Z., L.P., Y.X.J. collected the samples. L.L., F.X.G., Y.F. performed the analyses. L.L., F.W.H., D.L., J.D., X.L.W., Y.F., X.L.W. wrote the manuscript. L.L., F.W.H., D.L., W.G. revised the manuscript. All authors read and approved the final version of the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Li, L., Hu, F., Liu, D. et al. A Chromosomal-level genome assembly and annotation of fat greenling (Hexagrammos otakii). Sci Data 12, 78 (2025). https://doi.org/10.1038/s41597-025-04368-3
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41597-025-04368-3
