
Abstract
Chinese cherry belongs to the family Rosaceae, genus Prunus, and has high nutritional and economic value. ‘Duiying’ is a Chinese cherry variety local to Beijing, and has better performance than sweet cherry in terms of disease resistance. However, disease resistance resources of ‘Duiying’ have not been fully exploited partially due to the lack of a high-quality genome. In this study, we report a high-quality chromosome-scale genome assembly for Chinese cherry ‘Duiying’, by combining PacBio HiFi, Bionano and Hi-C sequencing data. The assembled genome has a size of 1035.19 Mb, with a scaffold N50 of 28.99 Mb, and 978.61 Mb (94.54%) assembled into 32 pseudochromosomes. A total of 547.16 Mb (52.86%) sequences were identified as repetitive sequences, and 114,451 protein-coding genes were annotated. Moreover, a total of 1635 microRNA (miRNA), 6637 transfer RNA (tRNA), 38,258 ribosomal RNA (rRNA), and 169 small nuclear RNAs (snRNA) genes were identified. The genome assembly presented here provides valuable genomic resources to enhance our understanding of genetic and molecular basis of Chinese cherry.
Similar content being viewed by others

Background & Summary
Chinese cherry (Prunus pseudocerasus (Lindl.)) belongs to the family Rosaceae, genus Prunus, and subgenus Cerasus. It originates from Southwest China and is distributed in the temperate zone of the Northern Hemisphere1. Chinese cherry has been cultivated for more than 3000 years2. Most Chinese cherries are tetraploid, with a main karyotype formula of 2n = 4x = 32 = 28 m + 4sm3. Karyotype analysis and rDNA distribution have shown that the Chinese cherry is more likely an autopolyploid rather than an allopolyploid4. And this is further demonstrated by the phylogenetic and comparative genomic analyses5.
Chinese cherry fruit contains rich nutritional ingredients and trace elements, such as proteins, carotene, Vitamin C, saccharides, iron, and phosphorus1. Among 60 representative accessions, the soluble solids content ranged from 10.97% to 34.00%; about 70% of these accessions had a high yield ability3. In addition, the flowers, leaves, roots, bark, and core of Chinese cherry are of high medicinal value. Chinese cherries have a good affinity, developed roots, and soil salinity tolerance; thus, they have also been used as the root stock for sweet cherry6.
‘Duiying’, a Chinese cherry variety local to Beijing, is distributed in the valleys and on slopes. It has better performance than sweet cherry because of its leaf spot and crown gall disease resistance and adaptability to the Chinese soil and climate7. By crossing ‘Duiying’ with sweet cherry and sour cherry, serials of sweet cherry rootstocks have been released that present resistance to crown gall and leaf spot diseases8. It possesses great application potential for transferring resistance genes to sweet or sour cherry. However, the genomic features that underlie these important biological characteristics remain unclear. Several draft genomes or high-quality genomes have been assembled and released for sweet cherry varieties (2n = 2x = 16)9,10,11,12,13,14, while no high-quality reference genomes are available for Chinese cherry ‘Duiying’ to date.
To understand the genetic and molecular basis of Chinese cherry and to promote genomic-associated breeding studies in cherry and Prunus crops, we present a high-quality chromosome-level genome assembly for Chinese cherry ‘Duiying’. The high-quality genome of ‘Duiying’ was obtained using Illumina, Pacific Biosciences (PacBio), high-fidelity (HiFi), and BioNano sequencing combined with 10 × genomic and high-throughput/resolution chromosome conformation capture (Hi-C) technologies. The genome sequence of P. pseudocerasus ‘Duiying’ reported here will be a valuable resource for genetic studies and breeding programs on cherry plants, both for exploring the genome evolution and functional genomic studies of Rosaceae/Prunus and for its excellent trait gene resources.
Methods
Sampling and whole genome sequencing
Leaf samples of ‘Duiying’ were collected from the cherry orchard of the Institute of Pomology and Forestry, Beijing Academy of Agriculture and Forestry Sciences, in Tongzhou District, Beijing. Genomic DNA of ‘Duiying’ was extracted from leaf samples using a plant genomic DNA extraction kit (TIANGEN, Beijing, China). The quality and quantity of the extracted DNA were assessed using NanoDrop 2000 (Thermo Fisher Scientific, Boston, MA, USA).
For Illumina paired-end sequencing, 1.5 μg of genomic DNA was used to construct a 350-bp DNA library using an Illumina TruSeq® Nano DNA library preparation kit (Illumina, San Diego, CA, USA). The refined library was subsequently sequenced using the Illumina Novaseq 6000 platform (Illumina, San Diego, CA, USA), generating 42.76 Gb of raw sequences. Fastp software (v0.23.4)15 was employed to filter out low-quality paired reads. The remaining 42.68 Gb (99.81%) of high-quality data, with 97.44% and 93.88% of the bases having a quality score of ≥Q20 and ≥Q30, respectively, was utilized for genome survey and assessment.
For long-read sequencing, a 40-kb SMRTbell library was constructed based on the PacBio protocol. PacBio polymerase reads were obtained using the PacBio Sequel II System (PacBio, Menlo Park, CA, USA) in circular consensus sequencing (CCS) mode. After the adapter sequences were removed from the raw polymerase reads, we derived subreads, with the parameter set to ‘Filtering subreads by minimum length = 50’. We then utilized ccs software (https://github.com/PacificBiosciences/ccs) to generate HiFi reads, using ‘min-passes = 3 and min-rq = 0.99’ parameters. This process yielded 39.21 Gb of HiFi data, with a contig N50 of 15,530 bp, which was then used for genome assembly (Table 1).
To generate Bionano optical mapping data, the Bionano official extraction kit16 was initially used to isolate long fragment molecules exceeding 150 Kb in length from high-quality DNA. Then, a single-enzyme cutting technique was applied with the DLE-1 (CTTAAG) endonuclease for digestion. Following standard Bionano protocols, the DNA molecules were labeled and subsequently imaged using the Bionano Irys system (Bionano Genomics, San Diego, CA, USA). The raw imaging data were transformed into BNX files, with the basic labeling and DNA length information converted via AutoDetect in the Bionano Solve package (v3.5.1) (https://bionanogenomics.com/support/software-downloads/). Following filtration based on molecule length and label density, we successfully produced optical mapping data for ‘Duiying’. We generated 584.546 Gb of data, with an average label density of 22.64 per 100 Kb and an N50 value of 366.4 Kb (Table 1).
Hi-C libraries were constructed using leaf cells from ‘Duiying’. The process started with cell fixation using formaldehyde, followed by cell lysis. The cross-linked DNA was then digested with the DpnII enzyme. The resulting sticky ends were biotinylated and proximity ligated to form chimeric junctions. We then enriched DNA fragments of 300–500 bp using a physical shearing process. These chimeric fragments, which are indicative of the original long-distance physical interactions within the cross-linked DNA, were converted into paired-end sequencing libraries. The paired-end reads were then sequenced using the Illumina NovaSeq platform (Illumina, San Diego, CA, USA), resulting in 145 Mb of read pairs. To ensure data quality, we employed fastp software17 to filter out low-quality reads from the raw sequencing data. After removing duplicate reads, we obtained 127 Mb of read pairs to assemble the chromosome-level genome.
Transcriptome sequencing and analysis
Total RNA was extracted from three tissues (leaf, stem, and root) using an RNA extraction kit (QIAGEN China(Shanghai) Co., Ltd., Shanghai, China). High-quality cDNA libraries were prepared using the TruSeq Stranded mRNA Sample Preparation Kit and sequenced on the Novaseq 6000 platform by Novogene (Beijing, China). Quality control was performed using fastp software15. An average of 6.94 Gb of high-quality RNA-seq data was used per tissue for transcript evidence analysis to determine the gene structure annotation for the ‘Duiying’ genome (Table 1).
Genome survey
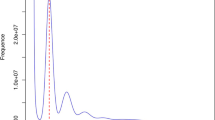
Before genome assembly, we conducted a genome survey using k-mer spectrum analysis. Specifically, we used Jellyfish (v2.3.0)18 to count the k-mer frequency from high-quality paired-end reads by setting k to 17. We removed k-mers with a low frequency of 3, which occur due to sequencing errors. The genome size was calculated by dividing the total k-mers by their coverage depth, and the distribution of the k-mer frequency reflected that of this genome.
The k-mer frequency distribution graph displayed three distinct peaks (Fig. 1), suggesting that the ‘Duiying’ genome is a homologous tetraploid. Our analysis identified 30.08 billion k-mers, with a significant majority of 30.02 billion (98.05%) categorized as high frequency (≥3). The primary peak in the k-mer frequency distribution was observed at a depth of 27×. As a result, the genome size was estimated to be approximately 1118.42 Mb (Table 2).

Frequency distribution of 17-mers.
In addition, we aligned the high-quality paired-end reads of ‘Duiying’ to the genome sequence of its closely related diploid species, Prunus avium ‘Tieton’ (GCA_014155035.1), using the BWA-MEM algorithm (v0.7.17-r1188)19. Of ‘Duiying”s reads, 82.15% covered 95.78% of the P. avium genome (Table 2), supporting that the ‘Duiying’ genome is a homologous tetraploid.
Genome assembly of Chinese cherry ‘Duiying’
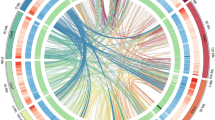
PacBio HiFi reads were used to assemble the initial contigs in the hifiasm (0.19.5-r587) package20 with default parameters. This process yielded a 1013.46 Mb assembly for Chinese cherry ‘Duiying’, with a contig N50 value of 4.18 Mb (Table 3). We then conducted hybrid scaffolding analysis using Bionano optical maps by mapping the Bionano data to the initial contigs using RefAligner in the Bionano Solve software package (v3.5.1). The alignment results were visualized using IrysView within the Bionano Solve software package (v3.5.1). We combined the genome maps with the initial contigs to generate hybrid scaffold genome maps using the Bionano Solve software package (v.3.5.1), with the parameters set to ‘-B 2 -N 2’. We obtained a scaffold-level assembly with a genome size of 1023.26 Mb and a scaffold N50 value of 11.68 Mb (Table 3). Pseudochromosome construction was then performed to obtain the ‘Duiying’ assembly, and the single-ended model in Bowtie2 software (v2.4.1)21 was used to map the Hi-C data onto the previously established scaffold-level assembly. After discarding the invalid self-ligated and unligated fragments within the uniquely mapped pairs using the HiCUP pipeline (version 0.8.0)22, 91,274,501 interaction pairs were used to calculate the linkage frequency among all scaffolds via an agglomerative hierarchical clustering algorithm implemented in ALLHiC software (v0.9.8)23 (Table 1). We manually rectified any placement and orientation errors that exhibited distinct chromatin interaction patterns. As a result, we produced a final assembly for ‘Duiying’ with a genome size of 1035.19 Mb and a scaffold N50 value of 28.99 Mb. A total of 978.61 Mb (94.54%) assembled sequences were anchored onto 32 pseudochromosomes (Tables 3, 4; Fig. 2). All chromosomes were grouped into eight clusters based on their sequence similarity, indicating that our assembly effectively distinguished the sequences of the four haplotypes in the ‘Duiying’ genome (Fig. 2). The synteny analysis indicated that the four haplotype sequences exhibited very high synteny, with a synteny rate exceeding 85%, which is significantly higher than the synteny between the ‘Duiying’ genome and its closely related species, P. avium ‘Tieton’ (68.15%) (Fig. 3).

Chromatin interactions in each chromosome of the ‘Duiying’ genome at a resolution of 1 Mb. The dark red dots show a high probability of interaction, and the light dots show a low probability of interaction.

Synteny plot. Align the other three haplotype sequences of P. pseudocerasus ‘Duiying’ (Hap-b, Hap-c, Hap-d) and its diploid relative species P. avium ‘Tieton’ to the P. pseudocerasus ‘Duiying’ Hap-a sequence.
Genome assessment
We evaluated the genome assembly quality from two perspectives: completeness and accuracy. For assembly completeness, complete Benchmarking Universal Single-Copy Orthologs (BUSCOs) were evaluated in the assembled genome by searching against the 1614 BUSCOs in embryophyta_odb10 (version 5.4.2)24, and the mapping ratio and coverage depth were calculated when the Illumina pairs were realigned to the assembled genome using BWA software19. For assembly accuracy, we detected homozygous SNPs from the realignment results, which represent single base errors in the assembly.
Genome structure annotation for Chinese cherry ‘Duiying’
Repetitive sequences
We utilized both homologous searching and ab initio prediction techniques to annotate repeated sequences within the ‘Duiying’ genome. For ab initio prediction, we concurrently utilized four transposable element (TE) prediction software packages—LTR_FINDER v1.0.725, PILER v3.3.017, RepeatScout v1.0.526, and RepeatModeler v1.0.827—to build a candidate de novo library within the ‘Duiying’ genome. All software was run using their default parameters. Following this, the de novo libraries and the Repbase database were used to annotate repeated sequences in the ‘Duiying’ assembly with RepeatMasker (v4.0.5)27. For homologous searching, we used RepeatProteinMask (v4.0.5) with default parameters to predict TEs. We then amalgamated these results, identifying 547.16 Mb (equivalent to 52.86%) of the ‘Duiying’ assembly as repeat sequences (Table 5). Notably, among these repeat sequences, long terminal repeat (LTR) sequences were the most abundant, accounting for 46.46% of the whole genome sequences.
Protein-coding genes
We utilized homologous-, de novo-, and transcriptome-based approaches to predict protein-coding genes within the ‘Duiying’ genome. For homologous-based gene prediction, the protein sequences from eight Prunus genomes, namely P. avium ‘Bigstar’ (GCA_013416215.1)10, P. avium ‘Tieton’11, P. persica28, P. mume29, P. yedoensis30, P. armeniaca31, P. salicina32, and P. armeniaca33, were aligned against the ‘Duiying’ genome using TBLASTN (version 2.2.29 +) with an e-value cut-off of 1e−534. All remaining blast hits were concatenated using Solar software (version 0.9.6). We extracted the corresponding genomic region, including 1000 bp upstream and downstream of each candidate gene, to predict the precise gene structure using wise2 (v2.4.1)35. The resulting predictions were designated as the ‘Homology set’. For transcriptome-based prediction, RNA-seq data were assembled and transcript sequences were generated using Trinity (v2.1.1)36. We aligned the transcript sequences against the ‘Duiying’ genome using the Program to Assemble Spliced Alignment (PASA)37, in which effective alignments were clustered based on their genome mapping location and assembled into gene structures. The gene models created by PASA were labeled as the PASA Trinity set. RNA-seq reads were also directly mapped to the ‘Duiying’ genome using TopHat (v2.0.13)38, and the mapped reads were assembled into gene models (Cufflinks-set) using Cufflinks (v2.1.1)39. For de novo gene prediction, we employed Augustus (v2.5.5)40, GeneID (v1.4)41, GeneScan (v1.0)42, GlimmerHMM (v3.0.1)43, and SNAP (version 2013-11-29)44 to predict genes in the repeat-masked genome. The specific parameters used in Augustus, SNAP, and GlimmerHMM were trained with the gene models from the PASA Trinity set. All gene models from these sets were integrated using EVidenceModeler (v1.1.1), with the following weights assigned to each type of evidence: PASA-T-set > Homology-set = Cufflinks-set > Augustus > GeneID = SNAP = GlimmerHMM = GeneScan. In addition, we filtered out genes that were less than 50 amino acids in length, supported only by ab initio evidence, and with an expression value of less than 1. As a result, 114,451 protein-coding genes were obtained in the ‘Duiying’ genome (Table 6). The length distribution of each element type in the gene structure annotated for ‘Duiying’ was similar to that of gene elements in other species within the Prunus genus (Fig. 4), reflecting the accuracy of the ‘Duiying’ gene structure annotation.

Length comparison chart of gene elements in closely related species within the Prunus genus.
We annotated the function of protein-coding genes within the ‘Duiying’ genome using SwissProt45, Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway46, Non-Redundant Protein Sequence Database (NR, from NCBI), and InterPro databases, leveraging a homologous searching method. We obtained Pfam domain and Gene Ontology (GO) information from the InterPro database and predicted these using the InterProScan tool47, based on conserved protein domains and functional sites. For the other databases, we used BLATP with an e-value cut-off of 1e−434. Consequently, 99.24% of the protein-coding genes were supported by functional databases (Table 7).
Noncoding RNA gene
We predicted the gene structures of noncoding RNAs in the ‘Duiying’ genome, using the t-RNAscan-SE tool (v1.3.1) to predict tRNAs48. We predicted ribosomal RNA (rRNA) sequences by searching against the invertebrate rRNA database using BLAST, with an E-value cut-off of 1e−1049. We also annotated small nuclear and nucleolar RNAs, as well as miRNAs using Infernal (v1.1rc4) based on the Rfam database8. As a result, we identified 1635 microRNA (miRNA), 6637 transfer RNA (tRNA), 38,258 ribosomal RNA (rRNA), and 169 small nuclear RNAs (snRNA) genes (Table 8).
Data Records
The raw data (Illumina reads, PacBio HiFi reads, and Hi-C sequencing reads) used for genome assembly were deposited in the SRA at the National Center for Biotechnology Information (NCBI)50. The RNA-seq data were deposited in the SRA at NCBI with accession numbers SRR2966054551 and SRR2966054652. The assembled genome was deposited in the DDBJ/ENA/GenBank databases under the accession number JBFBPF00000000053, and the genome annotation files are available on figshare repository54.
Technical Validation
Assembly assessment of Chinese cherry ‘Duiying’
The analysis results of the genome showed that the Chinese cherry genome was homologous tetraploid (Figs. 1, 2), supporting the previous karyotype research results on Chinese cherry chromosomes4. Our assembled ‘Duiying’ genome exhibited exceptional completeness, as evidenced by the coverage of 98.52% of Illumina paired reads across 99.82% of the genome. In addition, it recovered 99.4% of BUSCOs in the 1614 conserved Embryophyta genes from the embryophyta_odb10 database9 (Table 9). This assembled genome also demonstrated superior accuracy, with a single base error ratio of 9.08 × 10−8, indicating that there were only 9 assembly error sites per 100 Mb genome region.
Code availability
There were no custom scripts or codes used in this study. The version and parameters have been mentioned in the Methods section.
References
Yu, D. & Li, C. in Flora of China Vol. 38 (Science Press (Beijing), 1986).
Luo, G. Approach upon history of cultivation of apricot and Chinese cherry. Ancient and modern agriculture 2, 38–46 (2013).
Wang, Y. et al. Ploidy level of Chinese cherry (Cerasus pseudocerasus Lindl.) and comparative study on karyotypes with four Cerasus species. Sci Hortic-Amsterdam 232, 46–51, https://doi.org/10.1016/j.scienta.2017.12.065 (2018).
Li X. Study on chromosomal homology of polyploid Chinese cherry (Cerasus pseudocerasus). (Sichuan Agricultural University, 2019).
Jiu, S. et al. Haplotype-resolved genome assembly for tetraploid Chinese cherry (Prunus pseudocerasus) offers insights into fruit firmness. Hortic Res 11, uhae142, https://doi.org/10.1093/hr/uhae142 (2024).
Zhang, Q. & Gu, D. Genetic relationships among 10 Prunus rootstock species from China, based on simple sequence repeat markers. J Amer Soc Hort Sci 141, 520–526 (2016).
Zhang, X. et al. Identification of cherry crown gall by hydroponics. China Fruits 51–52 (2005).
Nawrocki, E. P. & Eddy, S. R. Infernal 1.1: 100-fold faster RNA homology searches. Bioinformatics 29, 2933–2935, https://doi.org/10.1093/bioinformatics/btt509 (2013).
Shirasawa, K. et al. The genome sequence of sweet cherry (Prunus avium) for use in genomics-assisted breeding. DNA Res 24, 499–508, https://doi.org/10.1093/dnares/dsx020 (2017).
Pinosio, S. et al. A draft genome of sweet cherry (Prunus avium L.) reveals genome‐wide and local effects of domestication. Plant J 103, 1420–1432, https://doi.org/10.1111/tpj.14809 (2020).
Wang, J. et al. Chromosome-scale genome assembly of sweet cherry (Prunus avium L.) cv. Tieton obtained using long-read and Hi-C sequencing. Hortic Res 7, 122, https://doi.org/10.1038/s41438-020-00343-8 (2020).
Xanthopoulou, A. et al. Whole genome re-sequencing of sweet cherry (Prunus avium L.) yields insights into genomic diversity of a fruit species. Hortic Res 7, 60, https://doi.org/10.1038/s41438-020-0281-9 (2020).
Wang, J. et al. A de novo assembly of the sweet cherry (Prunus avium cv. Tieton) genome using linked-read sequencing technology. PeerJ. 8, e9114, https://doi.org/10.7717/peerj.9114 (2020).
Sharpe, R. M. et al. Draft genome data of Prunus avium cv ‘Stella. ’. Data Brief 45, 108611, https://doi.org/10.1016/j.dib.2022.108611 (2022).
Chen, S., Zhou, Y., Chen, Y. & Gu, J. fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 34, i884–i890, https://doi.org/10.1093/bioinformatics/bty560 (2018).
Lam, E. T. et al. Genome mapping on nanochannel arrays for structural variation analysis and sequence assembly. Nat Biotechnol 30, 771–776, https://doi.org/10.1038/nbt.2303 (2012).
Edgar, R. C. & Myers, E. W. PILER: identification and classification of genomic repeats. Bioinformatics 21(Suppl 1), i152–i158, https://doi.org/10.1093/bioinformatics/bti1003 (2005).
Marcais, G. & Kingsford, C. A fast, lock-free approach for efficient parallel counting of occurrences of k-mers. Bioinformatics 27, 764–770, https://doi.org/10.1093/bioinformatics/btr011 (2011).
Li, H. & Durbin, R. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics 26, 589–595, https://doi.org/10.1093/bioinformatics/btp698 (2010).
Cheng, H., Concepcion, G. T., Feng, X., Zhang, H. & Li, H. Haplotype-resolved de novo assembly using phased assembly graphs with hifiasm. Nat Methods 18, 170–175, https://doi.org/10.1038/s41592-020-01056-5 (2021).
Langmead, B. & Salzberg, S. L. Fast gapped-read alignment with Bowtie 2. Nat Methods 9, 357–359, https://doi.org/10.1038/nmeth.1923 (2012).
Wingett, S. et al. HiCUP: pipeline for mapping and processing Hi-C data. F1000Res 4, 1310, https://doi.org/10.12688/f1000research.7334.1 (2015).
Zhang, X., Zhang, S., Zhao, Q., Ming, R. & Tang, H. Assembly of allele-aware, chromosomal-scale autopolyploid genomes based on Hi-C data. Nat Plants 5, 833–845, https://doi.org/10.1038/s41477-019-0487-8 (2019).
Simao, F. A., Waterhouse, R. M., Ioannidis, P., Kriventseva, E. V. & Zdobnov, E. M. BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 31, 3210–3212, https://doi.org/10.1093/bioinformatics/btv351 (2015).
Xu, Z. & Wang, H. LTR_FINDER: an efficient tool for the prediction of full-length LTR retrotransposons. Nucleic Acids Res 35, W265–W268, https://doi.org/10.1093/nar/gkm286 (2007).
Price, A. L., Jones, N. C. & Pevzner, P. A. De novo identification of repeat families in large genomes. Bioinformatics 21(Suppl 1), i351–i358, https://doi.org/10.1093/bioinformatics/bti1018 (2005).
Smit, A. & Hubley, R. R. Open-1.0. Available from. http://www.repeatmasker.org (2008).
Tan, Q. et al. Chromosome-level genome assemblies of five Prunus species and genome-wide association studies for key agronomic traits in peach. Hortic Res 8, 213, https://doi.org/10.1038/s41438-021-00648-2 (2021).
Zhang, Q. et al. The genome of Prunus mume. Nat Commun 3, https://doi.org/10.1038/ncomms2290 (2012).
Baek, S. et al. Draft genome sequence of wild Prunus yedoensis reveals massive inter-specific hybridization between sympatric flowering cherries. Genome Biol 19, https://doi.org/10.1186/s13059-018-1497-y (2018).
Jiang, F. The apricot (Prunus armeniaca L.) genome elucidates Rosaceae evolution and beta-carotenoid synthesis. Hortic Res 6, 128, https://doi.org/10.1038/s41438-019-0215-6 (2019).
Huang, Z. et al. Chromosome-scale genome assembly and population genomics provide insights into the adaptation, domestication, and flavonoid metabolism of Chinese plum. Plant J 108, 1174–1192, https://doi.org/10.1111/tpj.15482 (2021).
Smit, A., Hubley, R. & Green, P. RepeatMasker Open-4.0. 2013–2015 (2015).
Mount, D. W. Using the Basic Local Alignment Search Tool (BLAST). CSH Protoc 2007, pdb.top17, https://doi.org/10.1101/pdb.top17 (2007).
Birney, E., Clamp, M. & Durbin, R. GeneWise and Genomewise. Genome Res 14, 988–995, t https://doi.org/10.1101/gr.1865504 (2004).
Grabherr, M. G. et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat Biotechnol 29, 644–652, https://doi.org/10.1038/nbt.1883 (2011).
Haas, B. J. et al. Improving the Arabidopsis genome annotation using maximal transcript alignment assemblies. Nucleic Acids Res 31, 5654–5666, https://doi.org/10.1093/nar/gkg770 (2003).
Kim, D. et al. TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol 14, R36, https://doi.org/10.1186/gb-2013-14-4-r36 (2013).
Trapnell, C. et al. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat Protoc 7, 562–578, https://doi.org/10.1038/nprot.2012.016 (2012).
Stanke, M. & Waack, S. Gene prediction with a hidden Markov model and a new intron submodel. Bioinformatics 19(Suppl 2), ii215–ii225, https://doi.org/10.1093/bioinformatics/btg1080 (2003).
Guigo, R. Assembling genes from predicted exons in linear time with dynamic programming. J Comput Biol 5, 681–702, https://doi.org/10.1089/cmb.1998.5.681 (1998).
Burge, C. & Karlin, S. Prediction of complete gene structures in human genomic DNA. J Mol Biol 268, 78–94, https://doi.org/10.1006/jmbi.1997.0951 (1997).
Majoros, W. H., Pertea, M. & Salzberg, S. L. TigrScan and GlimmerHMM: two open source ab initio eukaryotic gene-finders. Bioinformatics 20, 2878–2879, https://doi.org/10.1093/bioinformatics/bth315 (2004).
Korf, I. Gene finding in novel genomes. BMC Bioinformatics 5, https://doi.org/10.1186/1471-2105-5-59 (2004).
UniProt, C. T. UniProt: the universal protein knowledgebase. Nucleic Acids Res 46, https://doi.org/10.1093/nar/gky092 (2018).
Kanehisa, M. et al. Data, information, knowledge and principle: back to metabolism in KEGG. Nucleic Acids Res 42, D199–205 https://doi.org/10.1093/nar/gkt1076 (2014).
Quevillon, E. et al. InterProScan: protein domains identifier. Nucleic Acids Res 33, W116–W120, https://doi.org/10.1093/nar/gki442 (2005).
Schattner, P., Brooks, A. N. & Lowe, T. M. The tRNAscan-SE, snoscan and snoGPS web servers for the detection of tRNAs and snoRNAs. Nucleic Acids Res 33, W686–W689, https://doi.org/10.1093/nar/gki366 (2005).
Altschul, S. F., Gish, W., Miller, W., Myers, E. W. & Lipman, D. J. Basic local alignment search tool. J Mol Biol 215, 403–410, https://doi.org/10.1016/S0022-2836(05)80360-2 (1990).
NCBI Bioproject https://identifiers.org/ncbi/bioproject:PRJNA1125168 (2024).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR29660545 (2024).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR29660546 (2024).
Yan, J. et al. Prunus pseudocerasus cultivar Duiying, whole genome shotgun sequencing project. GenBank https://identifiers.org/ncbi/insdc:JBFBPF000000000 (2024).
Zhang, W. Chromosome-level genome assembly of tetraploid Chinese cherry (Prunus pseudocerasus). figshare https://doi.org/10.6084/m9.figshare.26170204 (2024).
Acknowledgements
This work was supported by Beijing Academy of Agriculture and Forestry Sciences (KJCX20210403, KJCX20240326 and KJCX20240403).
Author information
Authors and Affiliations
Contributions
Jiye Yan, Kaichun Zhang and Wei Zhang conceived and designed this study. Jing Wang, Xuwei Duan, Xiaoming Zhang collected and prepared the sequencing samples. Xuncheng Wang, Junbo Peng and Qikai Xing performed bioinformatic analyses. Wei Zhang wrote the manuscript. Jiye Yan and Jing Wang revised it. All authors reviewed and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Zhang, W., Wang, J., Wang, X. et al. Chromosome-level genome assembly of tetraploid Chinese cherry (Prunus pseudocerasus). Sci Data 12, 136 (2025). https://doi.org/10.1038/s41597-025-04462-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41597-025-04462-6
