
Abstract
Gelechioidea represents the most diverse superfamily of tiny boring pests in Lepidoptera that pose a serious threat to agricultural and forestry economic crops. However, the lack of high-quality genome of highly specialized species makes it difficult to draw general conclusions about the mechanism of the close binding relationship between pests and crops. In this study, based on second- and third-generation sequencing reads, we constructed a chromosome-level genome for the Atrijuglans aristata, a specialized boring pest, that specifically harms the green husk of cultivated walnuts. The genome is 480.99 Mb and spans 31 pseudo-chromosomes, including Z and a portion of W chromosome. Contig N50 and scaffold N50 of the genome are 2.68 Mb and 16.01 Mb respectively. The BUSCO completeness achieves 95.6% with a total of 22,542 protein-coding genes are annotated. As the first sequenced genome of Stathmopodidae family, this high-quality genome provides a genetic basis for the mining of genes for important functional traits in A. aristata, as well as an important reference for the study of host adaptation of the (insect) specialists.
Similar content being viewed by others


Background & Summary
Herbivorous insects are one of the most abundant groups in both natural and managed ecosystems, accounting for half of the metazoan species in the world1. As the most diverse superfamily of Lepidoptera, Gelechioidea comprises more than 18,400 described species2, which attack plants from Solanaceae, Juglandaceae, Rosaceae, Fabaceae and others3,4,5,6. For their host plants, loss rates to plants’ yield can reach 80–100%7, causing an economic loss of up to 1.1 billion US dollars per year in China alone, seriously impacting the development of agroforestry in the country8,9. Even so, Gelechioidea species have received only limited attention. For example, as of October 2024, only 30 species of the Gelechioidea have assembled genomes according to NCBI, far fewer than the Noctuoidea (212). The main reason for the lack of research is that they are small and easily overlooked, making them difficult to collect on a large scale in the field, despite their value as evolutionary models10,11.
The specialized moth Atrijuglans aristata, also known as Atrijuglans hetaohei, belongs to the family Stathmopodidae (Lepidoptera: Gelechioidea)2, a family whose larvae exhibit varied lifestyles, including feeding on fruits, buds, fern-spores, and galls11,12,13,14. It is mainly distributed in walnut-producing areas in China, and has been sporadically recorded in Japan and Korea according to the GBIF (https://www.gbif.org/) and BOLD Systems v415 databases. The larvae of A. aristata mainly attacks the green husk of Persian walnut (Juglans regia), and a small number of studies have also reported harm to Manchurian walnut (Juglans mandshurica)16,17. According to FAO (http://www.fao.org/, accessed 2005), China’s annual production of walnuts reaches 420,000 t, far surpassing the 322,000 t in the United States and becoming the world’s largest producer of walnuts18. However, A. aristata continues to damage walnut fruit during the growing season, with average infestation rates as high as 80%, resulting in substantial early fruit drop (Fig. 1e,f), severely reducing walnut yields by as much as 40–50% in China4,19,20. Unfortunately, the lack of a high-quality genome for A. aristata has severely hindered efforts to control and manage it.

Morphology and hazard of Atrijuglans aristata. (a) Egg. (b) Larva. (c) Pupa. (d) Adult. (e) The formation of a cavity in the walnut fruit by the A. aristata. (f) Walnut fruit drops early when it was damaged by A. aristata.
In this study, we assembled and annotated a chromosome-level genome for the specialist A. aristata based on PacBio CLR and HiFi reads, Nanopore reads, Hi-C reads, and Illumina sequencing reads, also further identified its Z and a portion of the W chromosome. This is also the first reference genome of the family Stathmopodidae. This genome provides a research foundation for the development of novel nano-pesticides for the A. aristata, as well as a basic data for the study of the adaptive evolution of specialists to host plants.
Methods
Sampling and sequencing
The adults of the A. aristata were collected in Jinan, Shandong Province in China (117°26′46″E, 36°32′6″N) from 2021–2022. After sex identification, the surface of these fresh samples was washed with PBS buffer and stored in a −80 °C refrigerator after liquid nitrogen flash freezing. The heads and thoraxes of female A. aristata used for different types of library construction and sequencing were separately executed for genomic DNA extraction according to the protocols, with different individual sets for most downstream procedures. 1% agarose gels were used to detect the extraction quality and contamination status. DNA purity was analyzed via OD 260/280 ratio using a Nanodrop and DNA concentration with a Qubit® 3.0 Flurometer (Invitrogen, USA). A 350 bp paired-end short fragment library was carried out with the NEB Next ®Ultra™ DNA Library Prep Kit (NEB, USA) and further sequenced on Illumina HiSeq platform (DNA from six individuals). 20 kb PacBio CLR (continuous long reads) library was constructed using the SMRTbell Express Template Preparation Kit 2.0 and sequenced on PacBio Sequel platform (The DNA comes from the remainder of the DNA of the six individuals above). 15 kb PacBio HiFi (high-fidelity) reads were generated on PacBio Sequel IIe sequencing platform (DNA from one individual). 20 kb Nanopore library was prepared with the Ligation Sequencing gDNA Kit (SQK-LSK109) and performed on PromethION platform (DNA from 30 individuals). The 150 bp Hi-C library was carried out on Illumina Hiseq platform (DNA from 204 individuals). The raw Illumina sequencing reads were processed with the non-open-source software pk_qc.v2 by Novogene to remove reads containing adapters and N’s proportions greater than 10%. Paired reads were removed when the number of low-quality (sequencing quality values less than 5) bases in a single read exceeded 20% of the read length. Other raw sequencing reads were filtered with default parameters. In the end, a total of 57 Gb Illumina paired-end reads, 18 Gb PacBio HiFi reads, 164 Gb PacBio CLR reads, 10 Gb nanopore reads, and 58 Gb Hi-C reads were generated in the clean data (Table 1).
In order to acquire transcriptome data, walnuts fruits damaged by A. aristata were collected at the sampling sites mentioned above, and the larvae within were raised in the laboratory under 25 ± 1 °C, 75 ± 10% RH (relative humidity), photoperiod 14 L:10 D. The heads, guts and Malpighian tube of the larvae and one cocoon pupa were taken and placed in liquid nitrogen for rapid freezing respectively. RNA was extracted according to the TRIzol protocol (Life Technologies, USA), the RNA integrity was detected using 1% agarose gel electrophoresis, while the concentration and purity were examined using a Nanodrop2000. library construction was performed with TruSeq RNA v2 Kit (Illumina), and finally 150 bp paired-end sequencing was performed on the Illumina NovaSeq 6000 platform. After filtering the raw reads using the default parameters of the fastp v0.23.221, 6 Gb clean reads are generated.
Genome assembly
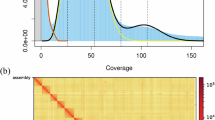
We used the paired-end reads from the Illumina sequencing to estimate the genome size of A. aristata based on the k-mer method. In brief, jellyfish v2.2.722 was used to generate the k-mer frequency distribution table (k = 21) and to further analyze genome size. Here, genome size was assessed by the total number of k-mer divided by the peak value of k-mer distribution. The output of the above analysis was further visualized using GenomeScope2.023. The results showed that the estimated genome size of A. aristata was 436.67 Mb, with a heterozygosity of 0.804% (Fig. 2).

Genome size estimation based on k-mer frequency distribution (k = 21).
Based on the previous evaluation of genome size, we assembled the frame of the draft genome with PacBio CLR reads in wtdbg2 v2.524. The first correction was performed using Illumina sequencing data and all PacBio sequencing data based on NextPolish v1.3.125. Heterozygous areas in the genome were removed using purge_haplotigs v1.0.2+ (https://bitbucket.org/mroachawri/purge_haplotigs/src/master/). To construct a reference genome at the chromosome level, high throughput chromosomal conformational capture (Hi-C) technology was used to anchor contigs onto chromosomes based on ALLHiC pipeline v0.9.1326 for anchoring and clustering, with results further corrected manually using Juicebox v1.9.827 based on the intensity of chromosome interaction. To increase N50 length, nanopore reads were added for gap filling using TGS-GapCloser v1.1.128. Further error correction was conducted using racon v1.4.2029. Finally, a reference genome with a long N50 and chromosome-level was obtained. To detect the accuracy and completeness of the genome assembly, an assessment was performed using the Insecta gene set (odb10, containing 1,367 core genes) with BUSCO v5.4.730. The final assembled reference genome is 480.99 Mb in length. Contig N50 and scaffold N50 of the genome are 2.68 Mb and 16.01 Mb respectively (Table 2). The contigs were anchored to 31 pseudo-chromosomes, accounting for 93.83% of the genome size, of which Chr01, at 37.57 Mb in length, is the longest chromosome (Fig. 3; Supplementary Table S1). The BUSCO completeness assessment was 95.6% (Table 2). In addition, we further compared the genome sizes of seven species of Gelechioidea and found that the genome sizes of Gelechioidea varied greatly, with the acer sober Anarsia innoxiella having a genome of only 302.93 Mb and the dotted grey groundling Athrips mouffetella having a genome of 869.73 Mb (Supplementary Table S2). This large genome difference may be related to their lifestyles31.

Heatmap of chromosomes anchored via Hi-C technology.
Genome annotation
Repetitive element detection was conducted using the EDTA pipeline v2.0.032 with the main parameter “-species others -sensitive 1 -anno 1”. The non-redundant repeat database generated in the previous steps was passed to RepeatMasker v4.1.233 to identify the content and proportion of repeat sequences in the genome. Three different lines of evidence were used for gene prediction: ab initio, homologous protein, and transcriptome alignment. For ab initio annotation, SNAP v2013-02-1634 and Augustus v3.4.035 were used for gene prediction. For homologous annotation, more than 100,000 protein-coding genes from model insect species (Apis mellifera GCF_003254395.2, Bombyx mori GCF_000151625.1, Drosophila melanogaster GCF_000001215.4, Tribolium castaneum GCF_000002335.3, Helicoverpa armigera GCF_002156985.1, Plutella xylostella GCF_932276165.1 and Spodoptera litura GCF_002706865.1) were downloaded from NCBI. These protein sequences were aligned onto the reference genome using GenBlastA v1.0.136. After extracting high-scoring pairs (HSPs) from the results, the aligned region was extended 2 kb to both sides and gene prediction was performed using GeneWise v2.4.137. For transcriptome alignment-based gene prediction, we used the transcriptome data sequenced in this study (SRR2346268638, SRR31891377-SRR3189137938) and the open access transcriptome data in SRA under SRR10321778 and SRR10242502-SRR1024250739. These data were assembled using StringTie v2.2.140 and the genome-guided mode of trinity v2.1.141, respectively. PASApipeline v2.5.242 was used to align the transcript to the reference genome and obtain gene prediction results based on the transcriptome data. The EVidenceModeler v1.1.143 software was used to integrate the three types of evidence and obtain the final annotations. To verify the completeness of the annotations, BUSCO assessment was conducted on the protein-coding genes. At the same time, Circos v0.6944 was used to visualize the GC content and gene distribution on each chromosome of the genome.
After our analysis, nearly half of the genome (48.17%; 231.68 Mb in length) is repetitive sequences (Table 2). Among them, DNA transposons were the most abundant repeats, accounting for 26.03% of the genome, followed by retroelements (15.70%; 75.49 Mb). A total of 22,542 protein-coding genes were predicted in the genome. BUSCO assessment with protein-coding genes showed 94.3% completeness, indicating a well-annotated genome. Overall, both the density of repeated sequences and genes are more evenly represented on each chromosome of the genome (Fig. 4).

Circular diagram of the genomic features on each chromosome of Atrijuglans aristata. After splitting each chromosome into windows every 200 kb, the following statistics were collected, displayed from outer to inner ring: a: Chromosome ideograms. b: Gene density. c: GC content. The lighter to darker green color in the heat map represents a gradual increase in the GC content value. d: Repeat density.
For functional annotation, we searched genes to the eggNOG 5.0 database using eggNOG-mapper v245. At the same time, InterProScan v5.52–86.046 was also used for annotations based on its own database. Finally, 14,966 genes were annotated based on eggNOG and 18,128 genes were annotated based on InterProScan, for a total of 19,167 genes annotated by combining the two methods (Table 2).
Whole-genome collinearity and sex chromosomes identification
We performed whole-genome collinearity analysis for three species: A. aristata, the rice leaffolder Cnaphalocrocis medinalis and tobacco cutworm S. litura. The latter two species are well-studied species and have high-quality reference genomes47,48. After the amino acid sequences of three species were aligned with blastp (E-value ≤ 1e-5), collinearity analysis between two species was performed by MCScanX49, and collinearity blocks of gene pairs greater than 5 were plotted using MCscan Python v1.1.1250. To characterize the sex chromosomes of A. aristata, we reused the sequencing reads used for genome assembly; in brief, we first randomly selected 1,000,000 reads from the PacBio CLR reads used for genome assembly. These PacBio CLR reads and all nanopore reads were then mapped to the reference genome of A. aristata using Minimap2 v2.17-r94151. Finally, the coverage on each chromosome was calculated using the flagstat function of SAMtools v1.16.152. In general, the W chromosome has female-biased coverage, while the coverage of the Z chromosome in the female is about half of that in the male, and the autosomes remain roughly the same between the sexes. This difference was often used to identify the sex chromosomes of species48,53,54.
The collinearity analysis indicated that the chromosomes of A. aristata showed substantial collinearity with those of the rice leaf-folder C. medinalis and the tobacco cutworm S. litura. We observed a chromosome fusion event on Chr01, which was formed by the fusion of a Z chromosome and an autosome (Fig. 5a). In addition, the W chromosome of the rice leaf-folder C. medinalis has strong collinearity with Chr31 of A. aristata, although only 1.23 Mb was detected on this chromosome (Supplementary Table S1). In addition, we see that the Chr01 coverage (15.78×) based on PacBio CLR reads is lower than the average coverage of autosomes (25.70×), while Chr31 (58.56×) is higher than that, and the Nanopore reads produced a similar result (Fig. 5b). Therefore, these results indicated that Chr01 and Chr31 of A. aristata may be Z and a portion of the W chromosomes, respectively.

Chromosome analysis of the Atrijuglans aristata. (a) Collinearity relationship between three species (Cnaphalocrocis medinalis, Atrijuglans aristata, and Spodoptera litura). Each long bar represents a chromosome. Regions of the genomes of the two species that are aligned with each other are connected with gray lines. (B) 1,000,000 PacBio CLR reads and all nanopore reads were mapped to the reference genome of A. aristata separately, and the coverage of each chromosome was counted.
Data Records
The raw genome and transcriptome sequencing reads of A. aristata have been deposited as BioProject PRJNA93337838. The corresponding SRA accession numbers for the genomic sequencing reads are SRR23818966, SRR23796504, SRR23795132, SRR23693017 and SRR23622219. The corresponding SRA accession numbers for the transcriptomic sequencing reads are SRR23462686, SRR31891377-SRR31891379. The genome assembly of A. aristata has been released to NCBI GenBank with the accession number JAQYUT00000000055. Moreover, all the genome assembly and annotation results have been made available in the Figshare repository56.
Technical Validation
Although haplotype duplication is inevitable, we have minimized genomic heterozygosity by collecting samples with consistent genetic backgrounds and removing heterozygous regions during the assembly process. The predicted genome size based on k-mer analysis was 436.67 Mb, which is 44.32 Mb smaller than the final genome. 93.83% (451.31 Mb) of genome contigs were anchored to chromosomes. Clean reads from Pacbio HiFi data and nanopore data were mapped to the reference genome using Minimap2 v2.17-r94151, and the mapping rates were 99.92% and 94.53%, respectively. A. aristata showed substantial collinearity with both C. medinalis and S. litura. The BUSCO completeness based on genome and protein-coding gene analysis was 95.6% and 94.3%, respectively, demonstrating the completeness and accuracy of the genome assembly and annotation.
Code availability
Most of analysis methods used in this study were performed using default parameters according to manual and protocols of the bioinformatic tools, and the use of a small number of parameters has been noted in the Methods section. In addition, the information on the versions of any software has been fully indicated in the method.
References
Hardy, N. B., Peterson, D. A. & Normark, B. B. Nonadaptive radiation: pervasive diet specialization by drift in scale insects? Evolution 70, 2421–2428, https://doi.org/10.1111/evo.13036 (2016).
Wang, Q. Y. & Li, H. H. Phylogeny of the superfamily Gelechioidea (Lepidoptera: Obtectomera), with an exploratory application on geometric morphometrics. Zool Scr 49, 307–328, https://doi.org/10.1111/zsc.12407 (2020).
Biondi, A., Guedes, R. N. C., Wan, F. H. & Desneux, N. Ecology, worldwide spread, and management of the invasive south American tomato pinworm, Tuta absoluta: past, present, and future. Annu Rev Entomol 63, 239–258, https://doi.org/10.1146/annurev-ento-031616-034933 (2018).
Wang, Q. Q., Shaheen, T., Rong, L. & Tang, G. H. Phylogeography of walnut pest (Lepidoptera: Gelechioidea) reveals comprehensive influence of geographic barriers and human activities. J Asia Pac Entomol 25, 101962, https://doi.org/10.1093/jee/55.1.67 (2022).
Shiller, I., Noble, L. W. & Fife, L. C. Host plants of the pink bollworm. J Econ Entomol 55, 67–70, https://doi.org/10.1093/jee/55.1.67 (1962).
Kim, S., Lee, W. & Lee, S. Estimation of a new molecular marker of the genus Stathmopoda (Lepidoptera: Stathmopodidae): Comparing EF1a and COI sequences. J Asia Pac Entomol 20, 269–280, https://doi.org/10.1016/j.aspen.2016.12.002 (2017).
Desneux, N. et al. Biological invasion of european tomato crops by Tuta absoluta: ecology, geographic expansion and prospects for biological control. J Pest Sci 83, 197–215, https://doi.org/10.1007/s10340-010-0321-6 (2010).
Weng, Y. M. et al. Evolutionary genomics of three agricultural pest moths reveals rapid evolution of host adaptation and immune-related genes. GigaScience 13, giad103, https://doi.org/10.1093/gigascience/giad103 (2024).
Xi, M. et al. Assessment of the economic loss to the tomato industry caused by Tuta absoluta in China based on @RISK. J Biosaf 31, 300–308, https://doi.org/10.3969/j.issn.2095-1787.2022.04.002 (2022).
Sohn, J. C. et al. Phylogeny and feeding trait evolution of the mega‐diverse Gelechioidea (Lepidoptera: Obtectomera): new insight from 19 nuclear genes. Syst Entomol 41, 112–132, https://doi.org/10.1111/syen.12143 (2015).
Shen, Z. Y. et al. Systematics and evolutionary dynamics of insect-fern interactions in the specialized fern-spore feeding Cuprininae (Lepidoptera, Stathmopodidae). Mol Phylogenet Evol 194, 108040, https://doi.org/10.1016/j.ympev.2024.108040 (2024).
Kaila, L., Mutanen, M. & Nyman, T. Phylogeny of the mega-diverse Gelechioidea (Lepidoptera): Adaptations and determinants of success. Mol Phylogenet Evol 61, 801–809, https://doi.org/10.1016/j.ympev.2011.08.016 (2011).
Abe, Y. Well-developed gall tissues protecting the gall wasp, Andricus mukaigawae (MUKAIGAWA) (Hymenoptera: Cynipidae) against the gall-inhabiting moth, Oedematopoda sp. (Lepidoptera: Stathmopodidae). Appl Entomol Zool 32, 135–141, https://doi.org/10.1303/aez.32.135 (1997).
Park, K. T., Cho, S., Na, S., Shin, Y. M. & Kim, S. Genus Stathmopoda Herrich-Shäffer (Lepidoptera, Stathmopodidae) from the Korean Peninsula with two new species. J Asia Pac Biodivers 11, 259–266, https://doi.org/10.1016/j.japb.2018.04.004 (2018).
Ratnasingham, S. & Hebert, P. D. bold: The Barcode of Life Data System (http://www.barcodinglife.org). Mol Ecol Notes 7, 355–364, 10.1111/j.1471-8286.2007.01678.x (2007).
Zhang, Y. C. et al. Primary biological characteristics of an emerging major boring pest, Conogethes punctiferalis (Guenée) (Lepidoptera: Crambidae), on Juglans regia (Juglandales: Juglandaceae) in Taihang Mountains. Entomol News 130, 296–307, https://doi.org/10.3157/021.130.0310 (2022).
Chang, W. L. Characteristics and control of Atrijuglans hetaohei. Beijing Agric 21, 63 (2013).
Thakur, M. & Singh, K. Walnut (Juglan regia L.) a complete health and brain food. Asian J Biol Sci 8, 276–288 (2013).
NanGong, Z. et al. Potential of different entomopathogenic nematode strains in controlling Atrijuglans hetaohei Yang (Lepidoptera: Heliodinidae). Egypt J Biol Pest Co 32, 108, https://doi.org/10.1186/s41938-022-00591-x (2022).
Deng, L. L., Ma, Z., Zhang, Y. Z. & Zhao, B. X. Investigation on the pest species of Juglans regia in Ankang. Shaanxi Forest Sci Technol 48, 65–67+82 (2020).
Chen, S., Zhou, Y., Chen, Y. & Gu, J. fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 34, i884–i890, https://doi.org/10.1093/bioinformatics/bty560 (2018).
Marçais, G. & Kingsford, C. A fast, lock-free approach for efficient parallel counting of occurrences of k-mers. Bioinformatics 27, 764–770, https://doi.org/10.1093/bioinformatics/btr011 (2011).
Ranallo-Benavidez, T. R., Jaron, K. S. & Schatz, M. C. GenomeScope 2.0 and Smudgeplot for reference-free profiling of polyploid genomes. Nat Commun 11, 1432, https://doi.org/10.1038/s41467-020-14998-3 (2020).
Ruan, J. & Li, H. Fast and accurate long-read assembly with wtdbg2. Nat Methods 17, 155–158, https://doi.org/10.1038/s41592-019-0669-3 (2020).
Hu, J., Fan, J., Sun, Z. & Liu, S. NextPolish: a fast and efficient genome polishing tool for long-read assembly. Bioinformatics 36, 2253–2255, https://doi.org/10.1093/bioinformatics/btz891 (2020).
Zhang, X., Zhang, S., Zhao, Q., Ming, R. & Tang, H. Assembly of allele-aware, chromosomal-scale autopolyploid genomes based on Hi-C data. Nat Plants 5, 833–845, https://doi.org/10.1038/s41477-019-0487-8 (2019).
Robinson, J. T. et al. Juicebox.js provides a cloud-based visualization system for Hi-C data. Cell Syst 6, 256–258.e1, https://doi.org/10.1016/j.cels.2018.01.001 (2018).
Xu, M. et al. TGS-GapCloser: A fast and accurate gap closer for large genomes with low coverage of error-prone long reads. GigaScience 9, giaa094, https://doi.org/10.1093/gigascience/giaa094 (2020).
Vaser, R., Sović, I., Nagarajan, N. & Šikić, M. Fast and accurate de novo genome assembly from long uncorrected reads. Genome Res 27, 737–746, https://doi.org/10.1101/gr.214270.116 (2017).
Simon, J. C. et al. Genomics of adaptation to host-plants in herbivorous insects. Brief Funct Genomics 14, 413–423, https://doi.org/10.1093/bfgp/elv015 (2015).
Heckenhauer, J. et al. Genome size evolution in the diverse insect order Trichoptera. GigaScience 11, giac011, https://doi.org/10.1093/gigascience/giac011 (2022).
Ou, S. et al. Benchmarking transposable element annotation methods for creation of a streamlined, comprehensive pipeline. Genome Biol 20, 275, https://doi.org/10.1186/s13059-019-1905-y (2019).
Tarailo-Graovac, M. & Chen, N. Using RepeatMasker to identify repetitive elements in genomic sequences. Curr Protoc Bioinformatics Chapter 4, 4.10.11–14.10.14, https://doi.org/10.1002/0471250953.bi0410s25 (2009).
Korf, I. Gene finding in novel genomes. BMC Bioinformatics 5, 59, https://doi.org/10.1186/1471-2105-5-59 (2004).
Stanke, M. & Morgenstern, B. AUGUSTUS: a web server for gene prediction in eukaryotes that allows user-defined constraints. Nucleic Acids Res 33, W465–W467, https://doi.org/10.1093/nar/gki458 (2005).
She, R., Chu, J. S. C., Wang, K., Pei, J. & Chen, N. genBlastA: Enabling BLAST to identify homologous gene sequences. Genome Res 19, 143–149, https://doi.org/10.1101/gr.082081.108 (2009).
Birney, E., Clamp, M. & Durbin, R. GeneWise and Genomewise. Genome Res 14, 988–995, https://doi.org/10.1101/gr.1865504 (2004).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRP422682 (2025).
Li, F. et al. Identification and expression profiling of neuropeptides and neuropeptide receptor genes in Atrijuglans hetaohei. Gene 743, 144605, https://doi.org/10.1016/j.gene.2020.144605 (2020).
Pertea, M. et al. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat Biotechnol 33, 290–295, https://doi.org/10.1038/nbt.3122 (2015).
Grabherr, M. G. et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat Biotechnol 29, 644–652, https://doi.org/10.1038/nbt.1883 (2011).
Haas, B. J. et al. Improving the genome annotation using maximal transcript alignment assemblies. Nucleic Acids Res 31, 5654–5666, https://doi.org/10.1093/nar/gkg770 (2003).
Haas, B. J. et al. Automated eukaryotic gene structure annotation using EVidenceModeler and the Program to Assemble Spliced Alignments. Genome Biol 9, R7, https://doi.org/10.1186/gb-2008-9-1-r7 (2008).
Krzywinski, M. et al. Circos: An information aesthetic for comparative genomics. Genome Res 19, 1639–1645, https://doi.org/10.1101/gr.092759.109 (2009).
Cantalapiedra, C. P., Hernández-Plaza, A., Letunic, I., Bork, P. & Huerta-Cepas, J. Eggnog-mapper v2: functional annotation, orthology assignments, and domain prediction at the metagenomic scale. Mol Biol Evol 38, 5825–5829, https://doi.org/10.1093/molbev/msab293 (2021).
Zdobnov, E. M. & Apweiler, R. InterProScan–an integration platform for the signature-recognition methods in InterPro. Bioinformatics 17, 847–848, https://doi.org/10.1093/bioinformatics/17.9.847 (2001).
Cheng, T. et al. Genomic adaptation to polyphagy and insecticides in a major east Asian noctuid pest. Nat Ecol Evol 1, 1747–1756, https://doi.org/10.1038/s41559-017-0314-4 (2017).
Zhao, X. et al. A chromosome‐level genome assembly of rice leaffolder, Cnaphalocrocis medinalis. Mol Ecol Resour 21, 561–572, https://doi.org/10.1111/1755-0998.13274 (2020).
Wang, Y. et al. MCScanX: a toolkit for detection and evolutionary analysis of gene synteny and collinearity. Nucleic Acids Res 40, e49, https://doi.org/10.1093/nar/gkr1293 (2012).
Tang, H. et al. Synteny and collinearity in plant genomes. Science 320, 486–488, https://doi.org/10.1126/science.1153917 (2008).
Li, H. Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics 34, 3094–3100, https://doi.org/10.1093/bioinformatics/bty191 (2018).
Li, H. & Durbin, R. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 25, 1754–1760, https://doi.org/10.1093/bioinformatics/btp324 (2009).
Wan, F. et al. A chromosome-level genome assembly of Cydia pomonella provides insights into chemical ecology and insecticide resistance. Nat Commun 10, 4237, https://doi.org/10.1038/s41467-019-12175-9 (2019).
Mongue, A. J., Nguyen, P., Voleníková, A. & Walters, J. R. Neo-sex Chromosomes in the monarch butterfly, Danaus plexippus. G3 7, 3281–3294, https://doi.org/10.1534/g3.117.300187 (2017).
NCBI GenBank https://identifiers.org/ncbi/insdc:JAQYUT000000000 (2025).
Feng, D. D. et al. Chromosome-level genome assembly of a specialist walnut pest Atrijuglans aristata. Figshare. https://doi.org/10.6084/m9.figshare.27290562 (2025).
Acknowledgements
We would like to thank the teachers from Genek (Ying Wang and Xudong Zhang) for their help in the process of genome annotation. This research was supported by the Natural Science Foundation of China (Grant No. 32200343, 32170421), Beijing Municipal Natural Science Foundation (5232001), Support Project of High-level Teachers in Beijing Municipal Universities in the Period of 14th Five-year Plan (BPHR20220114), and Academy for Multidisciplinary Studies, Capital Normal University.
Author information
Authors and Affiliations
Contributions
D.D.F. and A.B.Z. were responsible for the topic selection and experimental design. D.D.F. is responsible for sample collection, data analysis and manuscript writing. C.S. was responsible for manuscript revision, and data analysis guidance. Y.C.L. and Q.F.G. were involved in sample collection and processing, and G.F.W. participated in the construction of analysis platform. H.H.L. was involved in the project guidance, and M.C.O. was responsible for grammar and manuscript revision. C.Q.Y. and A.B.Z. were responsible for the manuscript revision and project guidance. All authors approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Feng, Dd., Sun, C., Li, Yc. et al. Chromosome-level genome assembly of a specialist walnut pest Atrijuglans aristata. Sci Data 12, 434 (2025). https://doi.org/10.1038/s41597-025-04754-x
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41597-025-04754-x
