
Abstract
Reynoutria japonica, commonly known as Huzhang or Japanese knotweed, is a perennial herbaceous plant belonging to the family Polygonaceae and order Caryophyllales. This plant is valued for its traditional medicinal uses in China. In this study, we present a high-quality, chromosome-scale reference assembly for R. japonica using a combination of PacBio long-read sequencing, Hi-C reads, and Illumina short-read sequencing. The final assembled genome spans approximately 3.30 Gb, with a contig N50 of 1.39 Mb. Notably, 99.22% of the assembled sequences were anchored to 22 pseudo-chromosomes, and 74.79% of the genome is composed of repetitive elements. Genome annotation revealed 68,646 protein-coding genes and 14,788 non-coding RNAs. This genomic resource provides a robust foundation for comparative genomics and will enable deep insights into the evolutionary relationships across related species.
Similar content being viewed by others


Background & Summary
Reynoutria japonica, also known as Polygonum cuspidatum, and commonly referred to Huzhang in Chinese and Japanese knotweed in Japan1, is a perennial herbaceous species belonging to the family Polygonaceae. In Europe, it is recognized as one of the most invasive alien plant species and is currently prevalent across nearly all European countries2. However, in the Qinba mountain region of central China, it is valued as a traditional medicinal plant and a vegetable crop3. Over the past five years, rapid advancements in sequencing technologies have significantly expanded our understanding of the complete genomes of medicinal plants4. To date, the genomes of approximately 126 key Chinese herbs have been described4, including Artemisia argyi5, Dendrobium officinale6, Taxus wallichiana7, Coptis chinensis8,9, and Andrographis paniculata10. Among these efforts, a previous study utilizing next-generation short-read Illumina sequencing and transcriptome-assisted annotation produced a draft genome assembly for P. cuspidatum, revealing a genome size of 2.56 Gb and identifying 55,075 functional genes11. Due to the high abundance of transposable elements (TEs) in the R. japonica genome, however, this draft genome remains incomplete11, hindered by technological limitations inherent to the sequencing platform. These TEs complicates de novo assembly, leading to numerous gaps and errors, particularly in complex genomic regions. These challenges emphasize the necessity of further refinement to achieve a more accurate and complete genome representation in R. japonica.
In this study, to overcome the difficulties associated with assembling the R. japonica genome, we employed a combination of Illumina sequencing, high-throughput chromosome conformation capture (Hi-C) sequencing, and single molecule real-time (SMRT) sequencing. Subsequently, the completeness and contiguity of the assembled genome were evaluated. The final assembled genome spans approximately 3.30 Gb with a contig N50 of 1.39 Mb. 99.22% of the assembled sequences were anchored to 22 pseudo-chromosomes, and 74.79% of the genome consisting of repeat elements. Genome annotation revealed 68,646 protein-coding genes and 14,788 non-coding RNAs. The present high-resolution genome of R. japonica provides a valuable reference for the entire Polygonum genus, offering insights into comparative genomics and advancing our understanding of evolutionary relationships and gene functions across closely related species.
Methods
Sample collection and DNA/RNA extraction
R. japonica plants were cultivated in the Qinba Mountains of Shiyan, Hubei Province, China12. Fresh young leaves from one-year-old plants were harvested and immediately frozen in liquid nitrogen (Fig. 1a,b). Genomic DNA was extracted using an improved CTAB method13. Five tissues types (leaves, stems, flowers, roots, and fruits) were collected from a single individual for RNA extraction (Fig. 1c–e). The samples were promptly frozen in liquid nitrogen and stored at −80 °C until extraction. Total RNA was extracted using the TruSeq Stranded mRNA preparation kit, according to the manufacturer’s instructions.

Photographs taken from the sampled plant of Reynoutria japonica. (a) Cultivation field; (b) One-year-old plants of P. japonica; (c) Inflorescences with floral buds; (d) Fruits; (e) Underground tubers.
Genome sequencing
The sequencing library (DNBSEQ) was constructed and detected by MGIEasy Universal DNA Library Prep Set (MGI), Qubit™ dsDNA BR Assay Kit (Invitrogen) and Qubit® ssDNA Assay Kit (Invitrogen), and the sequencing was conducted on the MGISEQ-2000, generating 150-bp paired-end reads (PE150). For PacBio sequencing, DNA libraries were prepared using the SMRTbell® prep kit 2.0, following the manufacturer’s instructions. Sequencing was performed on the PacBio Sequel II platform. For Hi-C sequencing, DNA was purified using the QIAamp DNA Mini Kit (CAT#51306, Qiagen) according to the manufacturer’s protocol. The Hi-C library was subsequently sequenced on the MGISEQ-2000 platform. All genome sequencing and Hi-C sequencing data were derived from a single plant. Details of the data from each platform are provided in Table 1. Raw reads from transcriptome sequencing were processed sequenced using the Illumina NovaSeq. 6000 platform to generate 5.24–6.91 Gb of paired-end reads. These transcriptomic data were utilized for subsequent gene structure annotation.
In total, we generated 276.14 Gb (~84 × coverage) Illumina reads with a Q20 rate at 96.18%, 322.36 Gb (~98 × coverage) Hi-C reads with a Q20 rate at 92.57%, 252.45 Gb (~77 × coverage) PacBio reads, and 123.30 Gb RNA data with GC content was stable at 39.60%. These controls ensure the reliability of our sequencing data (Table 1).
Genomic survey
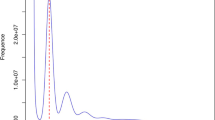
The generated Illumina sequencing data were processed using Fastp software (v0.23.3)14 with default parameters. This process included discarding reads with adapter contamination, trimming low-quality bases from both the 5′ and 3′ ends using a sliding window approach, and correcting mismatched base pairs in the overlapping regions of paired-end reads. Then the clean data were then used for K-mer analysis with GCE software (v1.0.2)15. Based on the 17-mer distribution (Fig. 2), information on the peak depth (86) and the number of 17-mers (241,952,873,067) was obtained and used to estimate genome size (2,813 Mb) (Table 2). The estimation was carried out using the following formula: Genome size = K-mer num/Peak depth16. Additionally, based on K-mer analysis, the heterozygosity rate (0.35%) and proportion of repeat sequence (81.28%) were calculated according to the methods described by Liu et al.16.

Frequency distribution of depth and K-mer Species.
Genome assembly and quality assessment
The 252.45 Gb PacBio subreads were initially assembled using Canu v2.1.117. The primary assembled genome was polished using PacBio long reads processed with Arrow (Arrow: https://github.com/PacificBiosciences/GenomicConsensus) and short reads processed with Pilon18 with default parameters. Based on this primary genome assembly, Hi-C short reads were subsequently employed to construct chromosomes for elongate loach. Approximately 84,647,123 of valid paired reads, filtered from the total pool of 331.31 Gb of clean Hi-C reads (Table 1), were utilized for assembly and error correction in scaffold extension and chromosome assembly. Quality control measures were applied to the Hi-C reads using Juicer (v1.6)19. The contig assembly was subsequently organized into a chromatin scaffold utilizing 3D-DNA20 (v180922, parameter -r2). The visualization of Hi-C interactions was conducted with 3D-DNA and further examined through Juicebox (v1.11.08)21. The de novo genome assembly generated a draft genome of approximately 3,297.29 Mb, consisting of 9,085 contigs with an N50 of about 1.39 Mb and a scaffold N50 of roughly 158.33 Mb (Table 3). Finally, a total of 22 pseudochromosomes were obtained (Figs. 3 and 4), encompassing 99.22% (3,271.86 Mb) of the assembled contigs (Table 3). The GC content of these pseudochromosomes was approximately 38.40% (Table 3), ranging from 38.10% to 38.58% (Table 4).

The circos diagram of P. japonica genome. Circles (a) to (g) represent 22 pseudochromosomes of the P. japonica pseudochromosomes (a), GC content (b), gene density (c), repeat density (d), copia elements density (e), gypsy elements density (f) and collinearity (g) between the pseudochromosomes (g), respectively. All calculations were done within 1 Mb windows.

Hi-C interactive heatmap (bin size = 100 kb). Genome-wide chromatin interactions in the R. japonica genome at 100-kb resolution. Color blocks represent interaction strength, ranging from white (low) to red (high).
The assessment of genome assembly’s completeness was conducted using the Benchmarking Universal Single-Copy Orthologs (BUSCO v5.4.3) assessment22. After searching against the eudicots_odb10 database, R. japonica genome was validated to 95.20% of 2,326 BUSCO groups (Table 5). These findings collectively demonstrate the high sequence integrity, continuity, and accuracy of the R. japonica assembly, meeting reference-quality standards.
Repeat annotation
A combination strategy of homology-based and de novo prediction methods was used to identify the repeat elements (REs) in the R. japonica genome. In the homology-based approach, RepeatMasker v4.0.6 (-e rmblast) and RepeatProteinMask v4.0.6 (-pvalue 0.0001)23 were employed to identify repeats at both the DNA and protein levels by searching against the RepBase library24 and the TE protein database. Tandem repeats were characterized using Tandem Repeats Finder (TRF, v4.07)25. Additionally, LTR_FINDER v1.0.626 with default parameters were utilized for the de novo prediction of novel repetitive elements.
In this study, the annotated 2,465.7 Mb of repetitive sequences accounted for 74.79% of the assembled R. japonica genome (Table 6). Among these sequences, Long Terminal Repeats (LTR) constituted the greatest proportion (47.918%, consist of 6.831% Copia, 16.958% Gypsy, and 24.129% Other LTRs), followed by DNA transposons (DNA) (3.750%), Long Interspersed Nuclear Elements (LINE) (2.707%), and Short Interspersed Nuclear Elements (SINE) (0.004%) (Table 6). The repetitive regions of the genome were then masked before proceeding with further gene prediction.
Non-coding gene annotation
In this study, we examined the gene structures of tRNAs, rRNAs, and other non-coding RNAs. tRNAs were predicted using the t-RNAscan-SE v1.427 program (http://lowelab.ucsc.edu/tRNAscan-SE/). Given the high conservation of rRNAs, we chose reference rRNA sequences from closely related species and used BLAST (blastn, evalue 1e-05) for rRNA sequences prediction. We also identified additional ncRNAs such as miRNAs and snRNAs by searching the Rfam28 database with Infernal v1.129 using default parameters. This analysis result in the annotation of 14,788 noncoding genes, which include 339 miRNAs, 7,508 tRNAs, 1355 rRNAs, and 5,586 snRNAs (Table 7).
Protein-coding genes prediction and functional annotation
To ensure precise gene prediction, a comprehensive approach combining de novo prediction, homology-based prediction, and transcriptome-based prediction. First, it predicted the de novo gene structure with AUGUSTUS v3.2.130 and GlimmerHMM v.3.0.431. Second, homologous protein sequences of three other plants in the Caryophyllales order, including Fagopyrum tataricum, Beta vulgaris, and Spinacia oleracea obtained from NCBI were aligned with the R. japonica genome with TBLASTN. Third, the RNA-seq data from five tissues were mapped onto the assembled genomes with HISAT2 v.2.2.17832. RNA-seq data were filtered using SOAPnuke software (v2.1.0)33 with the following parameters: -lowQual = 20, -nRate = 0.005, and -qualRate = 0.5. The data were processed by removing paired reads containing adapters, discarding those with more than 0.5% Ns, and eliminating low-quality reads where over 50% of bases had a quality score (Q) ≤ 20. Subsequently, StringTie v.2.1.67934 identified potential exon regions, and ORFs were predicted via TransDecoder v.5.1.0 using the transcript sequences. Finally, the gene sets were integrated by braker v2.1.535.
In this study, we identified 68,646 protein-coding genes in the R. japonica genome. The gene structure and gene elements, including average transcript length, average CDS length, and average exon and intron length, were compared with the above three related species in the order Caryophyllales (Table 8).
Gene functions were assigned aligned all predicted protein-coding genes against multiple publicly available databases such as Nr (http://www.ncbi.nlm.nih.gov/protein/), Uniprot, InterPro, Pfam, Swissprot, GO, and KEGG. Overall, 65,774 protein-coding genes were functionally annotated in at least one database (Fig. 5, Table 9). Among these annotated genes, 65,441 genes were annotated in the Nr database36, 65,312 genes were annotated in the Uniport database37, 58,797 genes were annotated in the InterPro database38, 54,309 genes were annotated in the Pfam database39, 48,078 in the Swiss-Prot database40, 37,217 in the GO database41, and 32,456 in the KEGG database42 (Fig. 5, Table 9).

The UpSet plot of Gene function annotations. The intersection size of genes with functional annotation using multiple public databases.
Data Records
The raw sequence data reported in this paper have been deposited in the Genome Sequence Archive (GSA43) in National Genomics Data Center44. Access to this data is available to the public under the accession number PRJCA030379, which can be found with the following GSA IDs: CRA01925145, CRA01918246, CRA01918347, CRA01945148. The assembled genome sequence has been made available in GenBank with JBLJBX00000000049. Additionally, the annotation data has been deposited at the Figshare repository50.
Technical Validation
DNA quality was assessed using 1% agarose gel electrophoresis, and DNA concentration was measured with Qubit 3.0 Fluorometer, achieving an absorbance ratio of around 2.0 at 260/280. We used Fastp14 to assess the quality scores of all bases in the raw sequencing data. Additionally, the 17-mer distribution analysis was performed on the clean data to estimate the target genome size. The genome size estimated by the survey closely matched the assembled genome size, further supporting the reliability of the evaluation results.
The genome-wide Hi-C interaction heatmap was generated using Juicerbox. In the heatmap, the coordinates represent bins across individual chromosomes, with the color of each point reflecting the logarithmic value of the interaction strength between corresponding bin pairs (Fig. 4). Notably, regions with higher interaction strength are represented by deeper colors, and the diagonal shows significantly stronger interactions compared to the ends.
The scaffold N50, the length at which half of the genome assembly is represented in scaffolds of that size, improved significantly to 158.33 Mb, indicating high assembly quality (Table 3). For the genome evaluation, 95.20% of BUSCOs were classified as complete, with 19.20% being single-copy and 76.00% being duplicated. Fragmented BUSCOs made up only 1.30%, while 3.50% were missing. The gene set evaluation similarly shows a high percentage of completeness at 94.30%, with 22.80% single-copy and 71.50% duplicated BUSCOs. Fragmented BUSCOs were slightly lower at 0.60%, and missing BUSCOs were higher at 5.10%. The BUSCO analysis indicates excellent sequencing quality, with over 94% of BUSCOs complete in both the genome and gene set, suggesting minimal fragmentation and high completeness in the assembly. The presence of a higher proportion of duplicated BUSCOs may indicate some degree of redundancy, but the low percentage of missing and fragmented BUSCOs further confirms the robustness of the assembly (Table 5).
Usage Notes
The final assembled R. japonica genome spans approximately 3.30 Gb, larger than the 2.56 Gb genome of P. cuspidatum11. Although both genomes contain a high proportion of repetitive sequences, R. japonica has a slightly higher percentage (74.79% compared to 71.54%). However, R. japonica exhibits superior assembly quality, with an N50 of 1.39 Mb, and 99.22% of the sequences are anchored to 22 pseudo-chromosomes, demonstrating a high level of assembly integrity. Future research could explore gene functions in R. japonica that are linked to its invasiveness and pharmacological properties, as well as utilize this reference genome for selective breeding initiatives.
Code availability
All software and pipelines were executed following the manuals and protocols of the published bioinformatics tools. The software versions and parameters are detailed in the Methods section. No custom programming or coding was used.
References
Peng, W., Qin, R., Li, X. & Zhou, H. Botany, phytochemistry, pharmacology, and potential application of Polygonum cuspidatum Sieb.et Zucc.: a review. J Ethnopharmacol 148, 729–745, https://doi.org/10.1016/j.jep.2013.05.007 (2013).
Nentwig, W., Bacher, S., Kumschick, S., Pyšek, P. & Vilà, M. More than “100 worst” alien species in Europe. Biological Invasions 20, 1611–1621, https://doi.org/10.1007/s10530-017-1651-6 (2017).
Zheng, L. et al. Global transcriptome analysis reveals dynamic gene expression profiling and provides insights into biosynthesis of resveratrol and anthraquinones in a medicinal plant Polygonum cuspidatum. Industrial Crops and Products 171 https://doi.org/10.1016/j.indcrop.2021.113919 (2021).
Cheng, Q. Q. et al. Review on the Development and Applications of Medicinal Plant Genomes. Front Plant Sci 12, 791219, https://doi.org/10.3389/fpls.2021.791219 (2021).
Miao, Y. et al. Genome sequencing reveals chromosome fusion and extensive expansion of genes related to secondary metabolism in Artemisia argyi. Plant Biotechnol J 20, 1902–1915, https://doi.org/10.1111/pbi.13870 (2022).
Niu, Z. et al. The chromosome-level reference genome assembly for Dendrobium officinale and its utility of functional genomics research and molecular breeding study. Acta Pharm Sin B 11, 2080–2092, https://doi.org/10.1016/j.apsb.2021.01.019 (2021).
Cheng, J. et al. Chromosome-level genome of Himalayan yew provides insights into the origin and evolution of the paclitaxel biosynthetic pathway. Mol Plant 14, 1199–1209, https://doi.org/10.1016/j.molp.2021.04.015 (2021).
Chen, D. X. et al. The chromosome-level reference genome of Coptis chinensis provides insights into genomic evolution and berberine biosynthesis. Hortic Res 8, 121, https://doi.org/10.1038/s41438-021-00559-2 (2021).
Liu, Y. et al. Analysis of the Coptis chinensis genome reveals the diversification of protoberberine-type alkaloids. Nat Commun 12, 3276, https://doi.org/10.1038/s41467-021-23611-0 (2021).
Sun, W. et al. The genome of the medicinal plant Andrographis paniculata provides insight into the biosynthesis of the bioactive diterpenoid neoandrographolide. Plant J 97, 841–857, https://doi.org/10.1111/tpj.14162 (2019).
Zhang, Y. et al. Assembly and Annotation of a Draft Genome of the Medicinal Plant Polygonum cuspidatum. Front Plant Sci 10, 1274, https://doi.org/10.3389/fpls.2019.01274 (2019).
Zhang, T. et al. A chromosome-level genome reveals genome evolution and molecular basis of anthraquinone biosynthesis in Rheum palmatum. BMC Plant Biol 24, 261, https://doi.org/10.1186/s12870-024-04972-2 (2024).
Hills, P. N. & van Staden, J. An improved DNA extraction procedure for plant tissues with a high phenolic content. South African Journal of Botany 68, 549–550, https://doi.org/10.1016/s0254-6299(15)30384-7 (2002).
Chen, S., Zhou, Y., Chen, Y. & Gu, J. fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 34, i884–i890, https://doi.org/10.1093/bioinformatics/bty560 (2018).
Liu, B. et al. Estimation of genomic characteristics by analyzing k-mer frequency in de novo genome projects. arXiv preprint arXiv:1308.2012 (2013).
Liu, B. H. et al. Estimation of genomic characteristics by analyzing k-mer frequency in de novo genome projects (2013).
Koren, S. et al. Canu: scalable and accurate long-read assembly via adaptive k-mer weighting and repeat separation. Genome Res 27, 722–736, https://doi.org/10.1101/gr.215087.116 (2017).
Walker, B. J. et al. Pilon: an integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS One 9, e112963, https://doi.org/10.1371/journal.pone.0112963 (2014).
Durand, N. C. et al. Juicer Provides a One-Click System for Analyzing Loop-Resolution Hi-C Experiments. Cell Syst 3, 95–98, https://doi.org/10.1016/j.cels.2016.07.002 (2016).
Dudchenko, O. et al. De novo assembly of the Aedes aegypti genome using Hi-C yields chromosome-length scaffolds. Science 356, 92–95, https://doi.org/10.1126/science.aal3327 (2017).
Durand, N. C. et al. Juicebox Provides a Visualization System for Hi-C Contact Maps with Unlimited Zoom. Cell Syst 3, 99–101, https://doi.org/10.1016/j.cels.2015.07.012 (2016).
Simão, F. A., Waterhouse, R. M., Ioannidis, P., Kriventseva, E. V. & Zdobnov, E. M. BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 31, 3210–3212, https://doi.org/10.1093/bioinformatics/btv351 (2015).
Tarailo-Graovac, M. & Chen, N. Using RepeatMasker to identify repetitive elements in genomic sequences. Curr Protoc Bioinformatics Chapter 4, 4.10.11–14.10.14, https://doi.org/10.1002/0471250953.bi0410s25 (2009).
Jurka, J. et al. Repbase Update, a database of eukaryotic repetitive elements. Cytogenet Genome Res 110, 462–467, https://doi.org/10.1159/000084979 (2005).
Benson, G. Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res 27, 573–580, https://doi.org/10.1093/nar/27.2.573 (1999).
Xu, Z. & Wang, H. LTR_FINDER: an efficient tool for the prediction of full-length LTR retrotransposons. Nucleic Acids Res 35, W265–268, https://doi.org/10.1093/nar/gkm286 (2007).
Lowe, T. M. & Eddy, S. R. tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res 25, 955–964, https://doi.org/10.1093/nar/25.5.955 (1997).
Griffiths-Jones, S. et al. Rfam: annotating non-coding RNAs in complete genomes. Nucleic Acids Res 33, D121–124, https://doi.org/10.1093/nar/gki081 (2005).
Nawrocki, E. P. & Eddy, S. R. Infernal 1.1: 100-fold faster RNA homology searches. Bioinformatics 29, 2933–2935, https://doi.org/10.1093/bioinformatics/btt509 (2013).
Stanke, M. et al. AUGUSTUS: ab initio prediction of alternative transcripts. Nucleic Acids Res 34, W435–439, https://doi.org/10.1093/nar/gkl200 (2006).
Majoros, W. H., Pertea, M. & Salzberg, S. L. TigrScan and GlimmerHMM: two open source ab initio eukaryotic gene-finders. Bioinformatics 20, 2878–2879, https://doi.org/10.1093/bioinformatics/bth315 (2004).
Kim, D., Langmead, B. & Salzberg, S. L. HISAT: a fast spliced aligner with low memory requirements. Nat Methods 12, 357–360, https://doi.org/10.1038/nmeth.3317 (2015).
Chen, Y. et al. SOAPnuke: a MapReduce acceleration-supported software for integrated quality control and preprocessing of high-throughput sequencing data. GigaScience 7, 1–6, https://doi.org/10.1093/gigascience/gix120 (2018).
Pertea, M. et al. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nature biotechnology 33, 290–295 (2015).
Brůna, T., Hoff, K. J., Lomsadze, A., Stanke, M. & Borodovsky, M. BRAKER2: automatic eukaryotic genome annotation with GeneMark-EP+ and AUGUSTUS supported by a protein database. NAR Genom Bioinform 3, lqaa108, https://doi.org/10.1093/nargab/lqaa108 (2021).
Sayers, E. W. et al. Database resources of the National Center for Biotechnology Information. Nucleic Acids Res 49, D10–d17, https://doi.org/10.1093/nar/gkaa892 (2021).
UniProt, C. UniProt: a worldwide hub of protein knowledge. Nucleic Acids Res 47, D506–D515, https://doi.org/10.1093/nar/gky1049 (2019).
Biswas, M. et al. Applications of InterPro in protein annotation and genome analysis. Brief Bioinform 3, 285–295, https://doi.org/10.1093/bib/3.3.285 (2002).
Mistry, J. et al. Pfam: The protein families database in 2021. Nucleic Acids Res 49, D412–d419, https://doi.org/10.1093/nar/gkaa913 (2021).
Bairoch, A. & Apweiler, R. The SWISS-PROT protein sequence database and its supplement TrEMBL in 2000. Nucleic Acids Res 28, 45–48, https://doi.org/10.1093/nar/28.1.45 (2000).
Harris, M. A. et al. The Gene Ontology (GO) database and informatics resource. Nucleic Acids Res 32, D258–261, https://doi.org/10.1093/nar/gkh036 (2004).
Kanehisa, M., Furumichi, M., Sato, Y., Kawashima, M. & Ishiguro-Watanabe, M. KEGG for taxonomy-based analysis of pathways and genomes. Nucleic Acids Res 51, D587–d592, https://doi.org/10.1093/nar/gkac963 (2023).
Chen, T. et al. The Genome Sequence Archive Family: Toward Explosive Data Growth and Diverse Data Types. Genomics Proteomics Bioinformatics 19, 578–583, https://doi.org/10.1016/j.gpb.2021.08.001 (2021).
Members, C.-N. & Partners. Database Resources of the National Genomics Data Center, China National Center for Bioinformation in 2023. Nucleic Acids Res 51, D18–D28, https://doi.org/10.1093/nar/gkac1073 (2023).
National Genomics Data Center https://ngdc.cncb.ac.cn/gsa/browse/CRA019251 (2024).
National Genomics Data Center https://ngdc.cncb.ac.cn/gsa/browse/CRA019182 (2024).
National Genomics Data Center https://ngdc.cncb.ac.cn/gsa/browse/CRA019183 (2024).
National Genomics Data Center https://ngdc.cncb.ac.cn/gsa/browse/CRA019451 (2024).
NCBI GenBank https://identifiers.org/ncbi/insdc:JBLJBX010000000 (2025).
Zhou, C., Zhang, Y. & Xu, Q. Chromosome-scale genome assembly and annotation of Huzhang (Reynoutria japonica). figshare https://doi.org/10.6084/m9.figshare.28498157.v1 (2025).
Acknowledgements
This study was supported by the Principal Investigator Program [HBMUPI202104] and the Advantages Discipline Group (Biology and Medicine) Project in Higher Education of Hubei Province (2021–2025) [2022BMXKQT4] at Hubei University of Medicine; the National Nature Science Foundation of China [32200680, 32402391]; the Key Project at Central Government Level: The ability establishment of sustainable use for valuable Chinese medicine resources [2060302]; the Science Research Program of Hubei Provincial Department of Education [T2023016]; the Joint supported by Hubei Provincial Natural Science Foundation and Shiyan-of China [2024AFD105, 2024AFD106] and the Young Top-notch Talent Cultivation Program of Hubei Province.
Author information
Authors and Affiliations
Contributions
Y. Z., L. Y., and J. Z. designed the study and wrote the manuscript. J. Z., Q. X., B. L., W. L, and X. L. performed the experiments and analysis. Z. Z., Z. Y., and L. Z. contributed to the materials. C. L., N. J., G. W., H. H., and C. Z. revised the manuscript and provided valuable comments.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Zhang, J., Xu, Q., You, L. et al. Chromosome-scale genome assembly and annotation of Huzhang (Reynoutria japonica). Sci Data 12, 474 (2025). https://doi.org/10.1038/s41597-025-04773-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41597-025-04773-8
