
Abstract
Tritrichomonas musculus is a commensal protist colonizing the large intestine of laboratory mice. Parasite colonization reshapes the gut microbiome and modulates mucosal immunity. This parasite is refractory to axenic culture. In order to facilitate functional genomic investigations we assembled a 193.49 Mbp high quality reference genome from FACS-purified parasites recovered from monocolonized mice using an integrated approach that combined long-read (PacBio and Oxford Nanopore) sequencing technologies for the draft genome assembly. The genome assembled into 756 contigs and RNA-Seq data was used to support the gene models for 46,131 annotated genes. Of these, 24,215 genes had an InterPro, Enzyme Commission and/or a Gene Ontology annotation. BUSCO analyses established that 53% of the genome annotations matched with available BUSCO genes in the eukaryote_odb10 database. This high quality reference genome will serve as a valuable resource to develop a metabolic and genetic model to grow T. musculus axenically and study genes relevant to its biology, life cycle transmission, and pathogenesis.
Similar content being viewed by others


Background & Summary
Commensal bacteria, fungi, viruses and protists are common within the mammalian intestine, interacting with each other and contributing to gut homeostasis1. Protozoa are increasingly being recognised as essential to the gut microenvironment2,3 by their ability to reshape the constituent bacteriome4,5 and mucosal immune responses6,7,8 with minimal signs of disease. Despite the biological significance of these commensal protists, reference genome assemblies with annotation to facilitate genetic diversity and comparative genomic studies are often lacking.
Tritrichomonas musculus, also known as Tmu, is an extracellular anaerobic commensal protist that frequently colonizes the mouse large intestine. The trophozoites have an ovoid shape, are 10 to 16 µm long, possess an anterior and posterior flagella and one undulating membrane2 (Fig. 1A). Mice colonized with T. musculus do not present with disease, including symptoms such as diarrhea and weight loss, but do exhibit a mild goblet cell hyperplasia, host epithelial cell inflammasome activation, including the release of IL-1β and IL-18, and a profound shift in the 16S bacterial community structure during T. musculus colonization5. This reprogramming of the constituent microbiome and host immune potential is sufficient to protect mice from a lethal challenge with the pathogenic bacteria Salmonella typhimurium6. However, the change in the immune status increases the risk of colitis and colorectal cancer6,7.

(A) Representative T. musculus trophozoite colonizing mouse ceca surrounded by bacteria. In situ cecum scanning electron microscopy image (methods available in Tuzlak & Alves-Ferreira, 2023). (B) Schematic workflow for T. musculus purification and whole genome sequencing. Created with BioRender.com.
Here, we describe a high quality, annotated T. musculus genome assembly. We purified T. musculus from the cecal content of laboratory mice monocolonized with the EAF2021 isolate. The protists were purified by Percoll gradient centrifugation followed by flow cytometry prior to DNA and RNA extraction. Sequencing libraries were prepared using purified DNA or RNA and Illumina short-read, PacBio and Oxford Nanopore (ONT) long-read sequencing was performed to facilitate genome assembly and annotation (Fig. 1B). The genome is 193.5 Mb-long with an N50 scaffold length of 3.5 Mb that assembled into 756 contigs. A total of 46,131 protein-coding genes were identified. This annotated genome will be a useful resource to develop a genetic model system to study the biology, evolution and diversity of T. musculus and to discover the metabolic pathways that are essential for colonization and survival within the host.
Methods
Sample collection, whole genome DNA and RNA sequencing
Tritrichomonas musculus trophozoites of the EAF2021 isolate were purified from laboratory mice cecal content using flow cytometry as described previously2. High molecular weight (HMW) genomic DNA (gDNA) necessary for long-read sequencing was extracted using the Blood & Cell Culture DNA Mini Kit-G20 (Qiagen) and 700 ng and 1 µg of DNA was used for PacBio and Nanopore library preparation, respectively (Fig. 1B). HMW PacBio libraries were generated following the Pacific Biosciences protocol “Preparing HiFi SMRTbell® Libraries using SMRTbell Express Template Prep Kit 2.0”. The libraries were run on an 8 M SMRTCell using sequencing reagents version 2.0. Sequencing was performed on a Sequel II sequencer (Pacific Biosciences) with control software version 9.0.0.9223 and a movie collection time of 15 hours per SMRTCell. ONT libraries were generated using the Ligation Sequencing SQK-LSK109 kit protocol per manufacturer’s instructions (Nanopore-MinION) and sequencing was performed on a FLO-MIN106 flow cell using the operation software MinkNOW (version 21.06.9) for 60 hours. In total 947,266 and 2,631,769 raw reads were generated from PacBio and ONT sequencing, respectively (Table 1). PacBio reads were used for genome assembly and ONT reads larger than 10 kb were used for scaffolding. Total RNA was extracted using RNeasy Micro Kit (Qiagen) and submitted to CD Genomics (New York, USA) for library preparation and sequencing. RNA libraries were prepared using SMART-Seq v4 Ultra Low Input RNA Kit (Takara) and sequencing was performed using Hi-Seq X Ten (Illumina) by CD Genomics. A total of 55,879,424 cleaned reads were used for genome annotation (Table 1).
Genome assembly
PacBio HiFi reads were assembled using canu (v2.1)9. MinION reads >10 kb were used to correct and scaffold the contigs using LongStitch (v 1.0.1)10 with the parameters k = 32, w = 250, with the gap fill option selected. Next, the assembly was polished using the PacBio reads with racon (v1.4.13)11. Genomic summary statistics were determined using Galaxy12. Forty contigs were removed from the assembly due to being <1 kb in length or having either zero or only one MinION read mapping to the PacBio assembly. Only contigs that had similar coverage from both long-read sequencing platforms were included. The genome had a GC content of 29.47% (Table 2). In summary, the 193.49 Mbp genome was assembled in 756 contigs with N50 of ~ 3.5 Mb, the shortest contig size was 7,256 bp and the longest was ~11 Mbp (Table 2).
Gene and functional predictions
Gene annotation was done using funannotate (v1.8.11)13 which removed 298 duplicate contigs from the assembly. Repeat masking was done using redmask (v0.0.2)13 which masked 42.67% of the genome, indicating that it is a highly repetitive genome, which likely explains its larger size compared to other trichomonads (Table 3). NCBI’s Foreign Contamination Screen14 identified 18 contigs as contaminants that were removed from the assembly and an additional 4 contigs as chimeric that were separated into multiple contigs with the intervening sequence removed. RNA-Seq data used by funannotate was assembled with Trinity (v2.12.0)15. Funannotate ran the following programs: PASA (v2.5.2)16, GeneMark (v4)17, Augustus (v3.3.3)18, eggnog-mapper (v2.1.2)19, signalp (v6)20, and interproscan (v5.52–86.0)21. Ribosomal RNA genes were identified by RNAmmer (v1.2)22. Relatively few genes possessed introns (5.66%) and the gene function annotation predicted that 36.6% of the genome was covered by 46,131 genes. BUSCO statistical analysis showed that the genome assembly captured 100% of the expected trichomonad genes, indicating that the genome assembly was complete, but only 53% of available BUSCO genes within the eukaryote_odb10 database (Table 4). Accordingly, 44,152 genes were identified as protein coding genes and 39,744 as hypothetical genes. Of these, 24,105 of the hypothetical genes have an InterPro number, 13,990 have a GO (Gene Ontology) term, 4,671 have an EC (Enzyme Commission) number and 24,215 have Interpro, GO and EC numbers (Table 3).
Ploidy analysis
Ploidy analysis was performed using a combination of GenomeScope 2.0 and Smudgeplot23 using T. musculus Illumina Hi-Seq reads (150 bp, two biological replicates). GenomeScope was run to fit a mixed model of negative binomial distributions to the k-mer spectrum of sequencing data as measured by FASTK (Fig. 2). The k-mer spectra was also visualized using Smudgeplot to estimate organism ploidy. The ploidy analysis by GenomeScope was consistent with T. musculus being haploid, with a homozygosity rate of 99.7% and heterozygosity rate of 0.345% (Fig. 1). However, the ploidy analysis using Smudgeplot (Sup Fig. 1), a software suite trained to recognize polyploid states, rather supported an organism that was diploid with highly homozygous sister chromosomes.

GenomeScope plot visualization of T.mu genome presented a single k-mer peak, indicated haploid genome. K-mer analysis (k = 21) estimated genome size is 334 Mb with coverage of 20.3X, 50.5% unique sequences, homozygous rate of 99.7% and heterozygosity rate of 0.345%.
Comparative functional genomic analysis
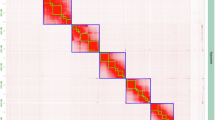
A gene functional comparison of T. musculus was performed by comparing predicted Pfam domains against other representative genomes, including protozoa that colonize the mammalian gut mucosa and are described as pathogenic or opportunistic, as well as Saccharomyces cerevisiae. Predicted T. musculus proteins were submitted to InterProScan v.5.30–69.021 for functional annotation (minimum E-value 1e-05). Pfam domains were retrieved from available annotated genomes for Trichomonas vaginalis G3 2022, Tritrichomonas foetus strain K, Giardia Assemblage A isolate WB 2019 (GiardiaDB release 62), Histomonas meleagridis 2922-C6/04-10x (TrichDB release 62) and Entamoeba dispar SAW760 (AmoebaDB release 62). For Blastocystis sp. ST1 (UP000078348) and Saccharomyces cerevisiae (UP000002311), Pfam annotations were retrieved from proteomes in UniProt release 2023_01 (Fig. 3A). Overall frequencies of domain-containing proteins were tabulated and ranked for each organism.

Functional protein domain comparison between T. musculus and other protists colonizing the mammalian gut mucosa. (A) Total number of Pfam domains identified for each annotated protist genome analyzed. Saccharomyces cerevisiae genome was used as a model organism. (B) Top 20 most abundant Pfam domains detected for all annotated T. musculus protein sequences, ranked by their frequency of occurrence compared to other protists and yeast (left panel). Each unique Pfam domain was ranked by the frequency of its occurrence. The number indicated in each box represents the rank order within each species from most abundant to least abundant (right panel). Shading was dependent on rank order, with darker shades indicating a lower rank.
We examined the frequency of the 20 most abundant Pfam domains identified in the T. musculus proteome against other infectious protists and S. cerevisiae (Fig. 3B; Suppl. Table 1). The most prevalent domain in T. musculus, the protein kinase domain (PF00069), with 3411 copies in the genome, was consistent with other organisms. As expected, the transcription-initiator DNA-binding domain IBD (PF10416), first reported to bind a DNA element unique to T. vaginalis, was present only in the trichomonads24. Of particular interest was the BspA-type Leucine rich repeat region (LRR_5 PF13306) found in 2,217 T. musculus proteins. BspA-like proteins are known to mediate interactions with the host extracellular matrix and were previously reported in large numbers in T. vaginalis and Entamoeba spp25,26. We also identified an expansion in the multi-antimicrobial extrusion protein domain (MatE; PF01554) in trichomonads (87 genes in T. musculus, 36th most abundant domain; 48 and 44 genes in T. vaginalis and T. foetus; 14 genes in H. meleagridis), whereas the domain was detected in only three or fewer genes among the remaining protists, and yeast.
Data Records
Technical Validation
Benchmarking Universal Single-Copy Orthologs (BUSCO) is a comprehensive tool to evaluate the quality of the genome assemblies and transcriptomes for eukaryotes and prokaryotes30,31. To estimate the completeness of the T. musculus assembly, we conducted BUSCO (version 5.4.2) analyses using mode euk_genome_met mode and metaeuk as gene predictor (eukaryote_odb10). BUSCO reported 53% total complete BUSCO genes representing 132 out of 225 BUSCOs and 104 complete and single-copy BUSCOs (41.6%) (Table 4). BUSCOs scores lower than 50% have been observed in published protists genomes32,33,34 and they are acceptable as satisfactory and indicate a good completeness. The low number detected is likely due to the lack of complete or partial BUSCO datasets for protists, especially for T. musculus related species.
Code availability
No custom codes were used in this study. All bioinformatics tools were used following the public access instructions detailed in the methods section.
References
Lukes, J., Stensvold, C. R., Jirku-Pomajbikova, K. & Wegener Parfrey, L. Are Human Intestinal Eukaryotes Beneficial or Commensals? PLoS Pathog 11, e1005039, https://doi.org/10.1371/journal.ppat.1005039 (2015).
Tuzlak, L. et al. Fine structure and molecular characterization of two new parabasalid species that naturally colonize laboratory mice, Tritrichomonas musculus and Tritrichomonas casperi. J Eukaryot Microbiol, e12989 https://doi.org/10.1111/jeu.12989 (2023).
Gerrick, E. R. et al. Metabolic diversity in commensal protists regulates intestinal immunity and trans-kingdom competition. Cell 187, 62–78 e20, https://doi.org/10.1016/j.cell.2023.11.018 (2024).
Wei, Y. et al. Commensal Bacteria Impact a Protozoan’s Integration into the Murine Gut Microbiota in a Dietary Nutrient-Dependent Manner. Appl Environ Microbiol 86 https://doi.org/10.1128/AEM.00303-20 (2020).
Popovic, A. et al. The commensal protist Tritrichomonas musculus exhibits a dynamic life cycle that induces B cell-modulated remodeling of the gut microbiota. bioRxiv https://doi.org/10.1101/2023.03.06.528774 (2023).
Chudnovskiy, A. et al. Host-Protozoan Interactions Protect from Mucosal Infections through Activation of the Inflammasome. Cell 167, 444–456 e414, https://doi.org/10.1016/j.cell.2016.08.076 (2016).
Escalante, N. K. et al. The common mouse protozoa Tritrichomonas muris alters mucosal T cell homeostasis and colitis susceptibility. J Exp Med 213, 2841–2850, https://doi.org/10.1084/jem.20161776 (2016).
Howitt, M. R. et al. Tuft cells, taste-chemosensory cells, orchestrate parasite type 2 immunity in the gut. Science 351, 1329–1333, https://doi.org/10.1126/science.aaf1648 (2016).
Koren, S. et al. Canu: scalable and accurate long-read assembly via adaptive k-mer weighting and repeat separation. Genome Res 27, 722–736, https://doi.org/10.1101/gr.215087.116 (2017).
Coombe, L. et al. LongStitch: high-quality genome assembly correction and scaffolding using long reads. BMC Bioinformatics 22, 534, https://doi.org/10.1186/s12859-021-04451-7 (2021).
Vaser, R., Sovic, I., Nagarajan, N. & Sikic, M. Fast and accurate de novo genome assembly from long uncorrected reads. Genome Res 27, 737–746, https://doi.org/10.1101/gr.214270.116 (2017).
Galaxy, C. The Galaxy platform for accessible, reproducible and collaborative biomedical analyses: 2022 update. Nucleic Acids Res 50, W345–W351, https://doi.org/10.1093/nar/gkac247 (2022).
J, P. Funannotate: Fungal genome annotation scripts, https://github.com/nextgenusfs/funannotate (2017).
Astashyn, A. et al. Rapid and sensitive detection of genome contamination at scale with FCS-GX. Genome Biol 25, 60, https://doi.org/10.1186/s13059-024-03198-7 (2024).
Grabherr, M. G. et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat Biotechnol 29, 644–652, https://doi.org/10.1038/nbt.1883 (2011).
Haas, B. J. et al. Improving the Arabidopsis genome annotation using maximal transcript alignment assemblies. Nucleic Acids Res 31, 5654–5666, https://doi.org/10.1093/nar/gkg770 (2003).
Besemer, J. & Borodovsky, M. GeneMark: web software for gene finding in prokaryotes, eukaryotes and viruses. Nucleic Acids Res 33, W451–454, https://doi.org/10.1093/nar/gki487 (2005).
Stanke, M., Diekhans, M., Baertsch, R. & Haussler, D. Using native and syntenically mapped cDNA alignments to improve de novo gene finding. Bioinformatics 24, 637–644, https://doi.org/10.1093/bioinformatics/btn013 (2008).
Cantalapiedra, C. P., Hernandez-Plaza, A., Letunic, I., Bork, P. & Huerta-Cepas, J. eggNOG-mapper v2: Functional Annotation, Orthology Assignments, and Domain Prediction at the Metagenomic Scale. Mol Biol Evol 38, 5825–5829, https://doi.org/10.1093/molbev/msab293 (2021).
Teufel, F. et al. SignalP 6.0 predicts all five types of signal peptides using protein language models. Nat Biotechnol 40, 1023–1025, https://doi.org/10.1038/s41587-021-01156-3 (2022).
Jones, P. et al. InterProScan 5: genome-scale protein function classification. Bioinformatics 30, 1236–1240, https://doi.org/10.1093/bioinformatics/btu031 (2014).
Lagesen, K. et al. RNAmmer: consistent and rapid annotation of ribosomal RNA genes. Nucleic Acids Res 35, 3100–3108, https://doi.org/10.1093/nar/gkm160 (2007).
Ranallo-Benavidez, T. R., Jaron, K. S. & Schatz, M. C. GenomeScope 2.0 and Smudgeplot for reference-free profiling of polyploid genomes. Nat Commun 11, 1432, https://doi.org/10.1038/s41467-020-14998-3 (2020).
Schumacher, M. A., Lau, A. O. & Johnson, P. J. Structural basis of core promoter recognition in a primitive eukaryote. Cell 115, 413–424, https://doi.org/10.1016/s0092-8674(03)00887-0 (2003).
Noel, C. J. et al. Trichomonas vaginalis vast BspA-like gene family: evidence for functional diversity from structural organisation and transcriptomics. BMC Genomics 11, 99, https://doi.org/10.1186/1471-2164-11-99 (2010).
Davis, P. H. et al. Identification of a family of BspA like surface proteins of Entamoeba histolytica with novel leucine rich repeats. Mol Biochem Parasitol 145, 111–116, https://doi.org/10.1016/j.molbiopara.2005.08.017 (2006).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRP446471 (2024).
Alves-Ferreira, E., Grigg, M., Lorenzi, H. & Galac, M. Tritrichomonas musculus strain EAF2021, whole genome shotgun sequencing project. Genbank. https://identifiers.org/ncbi/insdc:JAPFFF000000000.1 (2024).
Alves-Ferreira, E., Grigg, M., Lorenzi, H. & Galac, M. Genbank https://identifiers.org/ncbi/insdc.gca:GCA_039105265.1 (2024).
Seppey, M., Manni, M. & Zdobnov, E. M. BUSCO: Assessing Genome Assembly and Annotation Completeness. Methods Mol Biol 1962, 227–245, https://doi.org/10.1007/978-1-4939-9173-0_14 (2019).
Manni, M., Berkeley, M. R., Seppey, M., Simao, F. A. & Zdobnov, E. M. BUSCO Update: Novel and Streamlined Workflows along with Broader and Deeper Phylogenetic Coverage for Scoring of Eukaryotic, Prokaryotic, and Viral Genomes. Mol Biol Evol 38, 4647–4654, https://doi.org/10.1093/molbev/msab199 (2021).
Onut-Brannstrom, I. et al. A Mitosome With Distinct Metabolism in the Uncultured Protist Parasite Paramikrocytos canceri (Rhizaria, Ascetosporea). Genome Biol Evol 15 https://doi.org/10.1093/gbe/evad022 (2023).
Zarsky, V. et al. Contrasting outcomes of genome reduction in mikrocytids and microsporidians. BMC Biol 21, 137, https://doi.org/10.1186/s12915-023-01635-w (2023).
Abdel-Glil, M. Y., Solle, J., Wibberg, D., Neubauer, H. & Sprague, L. D. Chromosome-level genome assembly of Tritrichomonas foetus, the causative agent of Bovine Trichomonosis. Sci Data 11, 1030, https://doi.org/10.1038/s41597-024-03818-8 (2024).
Acknowledgements
This study was supported by the Division of Intramural Research, National Institute of Allergy and Infectious Diseases (NIAID) at the National Institutes of Health. This project has been funded in part with Federal funds from the National Institute of Allergy and Infectious Diseases (NIAID), National Institutes of Health, Department of Health and Human Services under BCBB Support Services Contract HHSN316201300006W/75N93022F00001 to Guidehouse, Inc. This study used the Office of Cyber Infrastructure and Computational Biology (OCICB) High Performance Computing (HPC) cluster at the National Institute of Allergy and Infectious Diseases (NIAID), Bethesda, MD, the NIH Intramural Sequencing Center (NISC), as well as the computational resources of the NIH HPC Biowulf cluster. This project was also funded by a Canadian Institutes for Health Research grant (MRT-168043) to J.P. and M.E.G. The authors would like to thank Cindi Schwartz for the scanning electron microscopy image and Beth Gregg for maintaining the mice colony.
Funding
Open access funding provided by the National Institutes of Health.
Author information
Authors and Affiliations
Contributions
M.E.G. and E.V.C.A.F. designed the study, discussed all bioinformatics analyses, and wrote the manuscript; E.V.C.A.F. and E.T. prepared the samples for sequencing; M.R.G., H.L. and M.C.W.H. performed the assembly and annotation; J.P. and A.P. performed the comparative genomic analysis. All authors reviewed and approved the final version of this manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Alves-Ferreira, E.V.C., Galac, M.R., Lorenzi, H.A. et al. Whole Genome Sequence of the gut commensal protist Tritrichomonas musculus isolated from laboratory mice. Sci Data 12, 590 (2025). https://doi.org/10.1038/s41597-025-04921-0
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41597-025-04921-0
This article is cited by
-
Chromosome-level genome assembly of Trichomonas vaginalis strain IR-78 (ATCC 50138)
Scientific Data (2025)
