
Abstract
Bougainvillea glabra is an ornamental tree or shrub with nearly 200 years of application in gardening and landscapes globally. Recently, the growing research interest in the applications of B. glabra extracts, such as medicinal applications, and synthetic materials for nutraceuticals, has led to the development of new techniques to be utilized for studying B. glabra. Moreover, the formations of polymorphic coloration and the mechanism of metamorphic bracts in B. glabra cultivars are worthy of study. However, the multi-omics information for B. glabra cultivars is lacking which hinders the progress of gene-level research and genetic applications. We sequenced the bracts transcriptomes of 22 B. glabra accessions and generated more than 80 Gb clean data. After de novo assembly and optimization, 174,758 unigenes (E90N50 = 2,473 bp) and annotation data were obtained. In addition, a total of 100,115 CDSs were detected. On average, each variety has 69,990 unigenes containing SNPs, among which 35,682 were annotated per variety. These transcriptome data are valuable for gene mining and expression experiments or other scientific areas.
Similar content being viewed by others


Background & Summary
Bougainvillea glabra, an important horticultural plant over the world, is a popular ornamental tree for tropical or subtropical gardens and landscapes due to its large, colorful bracts and free flowering habit. Although a total of 18 species in the Bougainvillea genus, there are only four species including B. glabra that have specialized large, colorful, and ornamental bracts. With nearly 200 years of cultivation, there are about 600 cultivars all over the world. Because of their colorful bracts in a wide range of colors viz. white, yellow, pink, red, mauve, bi-colored and multi-colored easy-bloom habit, fewer diseases and insects, strong growth vigor, B. glabra are used extensively in the bonsai, gardens, urban landscaping1. The portable potted plant of B. glabra in unique shape and size has made the nursery business a successful entrepreneurship and millions dollar business in China recently2.
B. glabra were not only popular in ornamental applications. In the recent decade, plenty of research on biochemistry, stress physiology, and biosynthesis area arisen. They are widely explored for nutritive, medicinal, and photovoltaic purposes3,4,5,6,7,8,9,10. Regarding the perianth tube and colorful bracts of B. glabra, it may mostly attract moths and hummingbirds to help pollinate in the tropical area in Brazil11,12. The bracts of B. glabra have a distinct variation in color range and shape which is rare in other plant bracts, it is a wonderful resource to study the molecular mechanism of wide-range color change and functions in plant ecological interaction, adaptive evolution, and reproductive innovations.
Recently, a Bougainvillea x.buttiana ‘Mrs Butt’ genome was published, which will speed up the Bougainvillea developmental and systematic research13. There were some transcriptomic resources available but limited to a few cultivars14,15. Transcriptome sequencing is the way to identify novel genes, reveal genetic functions, and explore molecular mechanisms of development and color variation of B. glabra bracts. However, there is no transcriptome data on multiple cultivars or populations. In this study, ribonucleic acid sequencing (RNA-Seq) was used to sequence the floral bracts for 22 B. glabra accessions at the same blooming period using the Illumina Novaseq 6,000 platform. A total of 80.32 Gb filtered data from samples of 22 accessions were obtained, with at least 3.18 Gb for each sample. After de novo assembly and optimization, 174,758 unigenes with E90N50 of 2,473 bp were obtained. The unigene dataset can help explore the potential genes towards color variation in bracts of B. glabra.
Methods
Sample collection
Tissue samples were collected from cuttings (3–5 years old) of Bougainvillea glabra of 22 different cultivars or accessions grown in the nursery of the Yuanshan Institute of Bougainvillea in Longhai, in Zhangzhou city, China. Bract surface color features of 21 B. glabra accessions was measured for the apex, middle and base part of bract (Table 1). Mature bracts tissues were sampled on several branches from one individual for each of 22 accessions. Fresh materials were frozen in liquid nitrogen immediately and stored in dry ice during transportation and stored at −80 °C eventually. A total of 22 samples from 22 accessions were used for RNA extraction.
Illumina sequencing and data processing
Total RNA was extracted using MJzol reagent [Majorbio (Shanghai) Co., Ltd.] following the manufacturer’s instructions. Adequate RNA quality and quantity of RNA samples were ensured by Nanodrop2000 [Thermo Fish (Shanghai) Co., Ltd.], RNA integrity and RIN (RNA Integrity Number) were measured by agarose gel electrophoresis analysis and Agilent Bioanalyzer 2100 [Agilent Technologies (Beijing) Co., Ltd.]16. Only the RNA sample with 260/280 ratio ≥1.8, 260/230 ratio ≥1.0 were used for library preparation.
mRNA was isolated using Oligo (dT) magnetic beads [Majorbio (Shanghai) Co., Ltd.] from total RNA and broke into short fragments of nearly 300 bp in fragmentation buffer17. Under the action of reverse transcriptase, one-strand cDNA was synthesized using mRNA as a template with random hexamers. The first-strand cDNA and second-strand cDNA were synthesized into a stable double-stranded cDNA and then filled the cohesive end with End Repair Mix and added an “A” base for connecting the Y-type adapter. Then the cDNA libraries were generated for 15 cycles by PCR amplification and the cDNA libraries were sequenced with a PE mode of 2 × 150 bp on an Illumina Novaseq 6000 platform18.
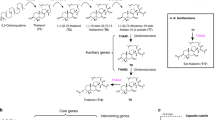
To obtain high-quality reads, raw RNA-seq reads were pre-processed using the software SeqPrep19 and Sickle20 with default parameters according to the following steps: (i) adaptors clips on reads and self-linked reads with adaptors were removed using SeqPrep, (ii) low-quality 3′-end sequences (<Q20) were trimmed and then reads with low quality (<Q10) still were removed using Sickle, (iii) reads with N base (unclear information base) rate >10% were removed, (iv) reads base less than 30 bp after trimming were removed. The major process of the study is listed in Fig. 1.

Flowchart of the study. The study comprised the following steps: RNA extraction from bracts for RNA-seq, filtering of raw data to obtain high-quality clean reads, de novo assembly of transcripts, annotation of assemblies against six databases, and quality evaluation and gene structures and variations detection.
De novo assembly and annotation
All high-quality sequence data obtained from the 22 libraries were assembled by the software Trinity program, including Inchworm, Chrysalis, and Butterfly modules, with default parameters21,22. The usual error (chimerism, structure error, incomplete assembly, base error), assembly length, and quality score of assembly were evaluated by using the software TransRate23 with default parameters. By integrating scores for contigs that are longer than 200 bp, providing an overall score for the transcriptome assembly, a filtered de novo assembly was outputted by TransRate. Furthermore, non-redundant assembly was obtained by clustering the sequence to remove redundant and similar ones by software CD-HIT24 with default parameters. After getting the filtered non-redundant transcripts, the unigenes were then derived by picking up the longest transcript using Trinity22. Then the transcriptome assembly was compared against the BUSCO consensus sequence in tBLASTn mode using the software BUSCO25.
After the assembly, annotations for the transcripts and unigenes were carried out via BLAST in NCBI NR database (https://ftp.ncbi.nlm.nih.gov/blast/db/FASTA/nr.gz (accessed on 1st Oct. 2021)), Pfam protein database (https://ftp.ebi.ac.uk/pub/databases/Pfam/releases/Pfam34.0/), Swiss-Prot protein database (ftp://ftp.uniprot.org/pub/databases/uniprot/current_release/knowledgebase/complete/uniprot_sprot.fasta.gz (accessed on 1st June. 2021)), eggNOG (Protein orthogenesis cluster database, http://eggnog5.embl.de/download/emapperdb-5.0.2/ /), KEGG (Kyoto Encyclopedia of Genes and Genomes database, http://www.genome.jp/kegg/ (accessed on 1st Sep. 2021)), GO (Gene Ontology Consortium gene database, https://www.geneontology.org/ (accessed on 18th Sep. 2021)) respectively.
Gene structure analysis
Coding Sequence (CDS) prediction were performed by following two steps. 1) Unigenes were blasted using BlastX (-E < = 1e-5) against NR and Swissprot protein database with the command blastx -query sample.fasta -out nr.result -db nr_db -max_target_seqs1 -evalue 1e-5 -outfmt 6; blastx -query sample.fasta -out swiss-prot.result -db uniprot_sprot_db -max_target_seqs1 -evalue 1e-5 -outfmt 6. If a match was identified, the Open Reading Frames (ORFs) for the transcript was extracted from the alignment results by command bedtools getfasta -fi sample.fasta -bed regions.bed -fo orf1.fasta, resulting in an accurate ORFs, essentially the CDS sequence. 2) For the sequences which did not match NR and Swissprot, candidate CDS were then identified using software TransDecoder26. The module TransDecoder.LongOrfs with default parameters was used to define the longest ORF using TransDecoder.LongOrfs -t sample.fasta. Then, a HMMER search were used to identify common protein domains using hmmsearch -cpu 8 –domtblout pfam.domtblout Pfam-A.hmm transdecoder_dir/longest_orfs.pep -E 0.001–domE 0.001 > pfam.log. Then, the coding region predictions were carried out by TransDecoder.Predict -t sample.fasta–retain_pfam_hits pfam.domtblout -T 3000. After above two steps, the ORFs sequences surpassing the threshold were outputted together resulting in final ORFs and CDS file.
SNP variations including transition and transversion were also detected. Raw sequences of 22 accessions were aligned to the transcriptome assembly by software Bowtie227, and the SNPs were called from mapped BAM files using the GATK pipeline (https://github.com/broadinstitute/gatk).
SSRs are repeat sequences that consist of several nucleotides (1–6) as repeat units and are widely distributed in the eukaryotic genome. In this study, SSRs were detected from unigenes by software MISA28 with default parameters. Specifically, Mono-nucleotide repeats were considered if a nucleotide sequence was repeated 10 or more times consecutively. Di-nucleotide repeats were included if the sequence was repeated at least 6 times. For tri-nucleotide to hexa-nucleotide repeats, the minimum repeat count was 5. We then conducted distribution statistics to analyze the frequency of these different repeat counts across single-nucleotide to six-nucleotide repeats.
Data Records
Raw reads of transcriptome sequences have been submitted to the National Genomics Data Center (NGDC) (https://ngdc.cncb.ac.cn) under BioProject PRJCA011746 with GSA number CRA00862429. The de novo assemblies of transcript and unigene have been submitted to NGDC under BioProject PRJCA011746 with OMIX number OMIX00207030, and also have been uploaded to NCBI GenBank under BioProject PRJNA950320 with TSA number GLCL00000000.131. The annotations of transcript and unigene are provided on Figshare (https://doi.org/10.6084/m9.figshare.26489128)32. The SNP data has been submitted to NGDC under BioProject PRJCA011746 with GVM number GVM00098433, and also have been uploaded to EVA database at EMBL-EBI under accession number PRJEB8774334. The repeat data (SSR data) has been provided on Figshare (https://doi.org/10.6084/m9.figshare.26489128)32.
Technical Validation
RNA quality
The density and purity of the total RNA were assessed using Nanodrop2000. The integrity of the RNA was assessed using agarose gel electrophoresis. The RIN (RNA Integrity Number) was counted using Agilent2100.
Raw and clean sequence base quality
During sequencing, along with the consumption of chemical reagent and reduction of enzymatic activity, the quality of sequence will decline. Sequence machine itself and quality of the samples would affect the sequence quality as well. Using SeqPrep and Sickle software, adaptors on reads and self-linked reads with adaptors were removed. Low-quality 3′-end sequences (<Q20) were trimmed and then reads with low quality (<Q10) still were removed, reads with N base (unclear information base) rate >10% were removed, reads base less than 30 bp after trimmed were removed. Using the software fastx_toolkit35, the base quality, base error rate and the base content were assessed as well as the statistic of raw and clean sequence reads (Table 2).
Assembly quality
After assembling the high-seq reads with Trinity, usual errors from de novo assembly output were assessed and the quality score was calculated using TransRate and then the comprehensive score of the assembly was obtained after integration. After that, redundant transcripts were removed by CD-HIT24. A total of 283,246 non-redundant transcripts with an average length of 815.32 bp were obtained as well as 174,758 non-redundant unigenes with an average length of 709.28 bp (Table 3). Unigenes and transcripts length distribution were pictured (Fig. 2). The sequence lengths ranging from 200 to 500 account for most (61%; 106,740) of the total unigenes. Similarly, sequence lengths of 200 to 500 account for most (53%; 149,424) of all transcripts. The ExN50 plots showed that the N50 > 1000 bp when a fraction of the most highly expressed transcripts >42% and reach the highest at E86N50 = 1947 bp, while the N50 > 1000 bp when a fraction of the most highly expressed unigenes >32% and reach the highest at E85N50 = 2569 bp, which indicate a good quality of the assembly (Fig. 3).

Sequence length distribution of unigenes and transcripts. Two bar plots depict the count of sequence length ranges for unigenes (left panel) and transcripts (right panel). Most sequences for both unigenes and transcripts fall within the 200–500 nucleotide (nt) range, followed by the 501–1,000 nt range. The smallest number of sequences is found in the length category exceeding 4,500 nt.

Expression-dependent N50 (ExN50) against a fraction of the total expressed data (Ex). The ExN50 at the point of assembly saturation (85%) are highlighted for Transcripts (E86N50 = 1947 bp) and Unigenes (E85N50 = 2569 bp).
The completeness of transcripts and unigenes was assessed by BUSCO25. The complete BUSCO is 92.8% for the non-redundant transcripts, with which single-copy BUSCO account for 29.7%, duplicated BUSCOs is 63.1%, fragmented BUSCO is 2.5%, while missing BUSCO is 4.7%. Meanwhile, the complete BUSCO is 85.1% for non-redundant unigenes, with which single-copy BUSCO account for 82.8%, duplicated BUSCOs is 2.2%, fragmented BUSCO is 6.7%, while missing BUSCO is 8.2% (Table 3). Additionally, the clean reads from each sample were mapped onto the final assembly showed the 68.21–75.57% mapping rates (Table 4).
Annotation to database
To obtain comprehensive information on gene function in the B. glabra de novo transcriptome assembly, the transcripts and unigenes were finally mapped onto several databases (Fig. 4, Table 5). Among them, 127,894(45.15%) transcripts and 65,361(37.4%) unigenes were mapped onto GO (Fig. 5), KEGG (Fig. 6), eggNOG, NR (Fig. 7), Swiss-Prot and Pfam databases for annotation.

Functional annotation of unigenes and transcripts. Annotation of unigenes and transcripts using six databases (GO, KEGG, eggNOG, NR, Swiss-Prot, and Pfam), with 84,065 (29.68%) transcripts and 41,644 (23.83%) unigenes were annotated, respectively.

GO annotation classification of unigenes. The bar plot showing the counts of unigenes in each annotation terms within three categories, including: molecular function, cellular component, and biological process.

KEGG annotation classification of unigenes. The counts of unigenes within each corresponding KEGG pathway terms are shown. The pathways are separated into five categories, as indicated by different colors.

NR annotation of unigenes. Each pie chart segment represents the number and proportion of unigenes that match the homologous species in the NR database.
Gene structure statistics
A total of 100,115 CDSs with different lengths was obtained (Fig. 8). Among them, CDS in length 201–400 are the most. SNPs in each accession were detected and counted according to functional area type (CDS or non-CDS) and gene type (Homo or Hete) (Table 6). Four types of transition variations and eight types of transversion variations were shown in Tables 7, 8. On average, each variety have 69,990 Unigenes containing SNPs, with 35,682 annotated per accession. Over 70% of SNPs are on depth ≤30.

Length distribution of CDS. Bar plot displays the count of CDS within each group of ten categories of CDS lengths.
Total 35,498 unigenes were found to contain 49,349 SSRs (Simple Sequence Repeats) and 7,066 unigenes had more than 1 SSRs. The SSRs contained 34,801 (70.52%) mononucleotide, 7,802 (15.81%) dinucleotide, 5,817 (11.79%) trinucleotide, 527 (1.07%) tetranucleotide, 164 (0.33%) pentanucleotide, and 238 (0.48%) hexanucleotide motifs (Fig. 9).

Distribution of different type of SSRs. The stacked plot shows the sequence counts in each of six types of SSRs. The SSRs exhibit variations in repeat numbers, as indicated by different colors.
Code availability
No custom code was generated.
References
Saleem, H., Usman, A., Mahomoodally, M. F. & Ahemad, N. Bougainvillea glabra (choisy): A comprehensive review on botany, traditional uses, phytochemistry, pharmacology and toxicity. J Ethnopharmacol 266, 113356, https://doi.org/10.1016/j.jep.2020.113356 (2021).
Roy, R., Singh, S. & Rastogi, R. Bougainvillea: Identification, gardening and landscape use. (CSIR-National Botanical Research Institute, 2015).
El-Deeb, A.-n. E., Abdel-Aleem, I. M., El-Amin, S. M., Refahy, L. A. & El-Shazly, M. A. Active cytotoxic compounds and essential oil from Bougainvillea alba. J Nat Remedies 15, https://doi.org/10.18311/jnr/2015/486 (2015).
Enciso-Díaz, O. J. et al. Antibacterial activity of Bougainvillea glabra, Eucalyptus globulus, Gnaphalium attenuatum, and propolis collected in Mexico. Pharmacol Pharm 03, 433–438, https://doi.org/10.4236/pp.2012.34058 (2012).
Ashmawy, N. A., Behiry, S. I., Al-Huqail, A. A., Ali, H. M. & Salem, M. Z. M. Bioactivity of selected phenolic acids and hexane extracts from Bougainvilla spectabilis and Citharexylum spinosum on the growth of Pectobacterium carotovorum and Dickeya solani bacteria: An opportunity to save the environment. Processes 8, https://doi.org/10.3390/pr8040482 (2020).
Ahmar Rauf, M., Oves, M., Ur Rehman, F., Rauf Khan, A. & Husain, N. Bougainvillea flower extract mediated zinc oxide’s nanomaterials for antimicrobial and anticancer activity. Biomed & Pharmacother 116, 108983, https://doi.org/10.1016/j.biopha.2019.108983 (2019).
Abdel-Salam, O. M. E. et al. Bougainvillea spectabilis flowers extract protects against the rotenone-induced toxicity. Asian Pac J Trop Med 10, 478–490, https://doi.org/10.1016/j.apjtm.2017.05.013 (2017).
Abarca-Vargas, R., Pena Malacara, C. F. & Petricevich, V. L. Characterization of chemical compounds with antioxidant and cytotoxic activities in Bougainvillea x buttiana Holttum and Standl, (var. Rose) extracts. Antioxidants (Basel) 5, https://doi.org/10.3390/antiox5040045 (2016).
Saleem, H. et al. HPLC–PDA polyphenolic quantification, UHPLC–MS secondary metabolite composition, and In Vitro enzyme inhibition potential of Bougainvillea glabra. Plants 9, https://doi.org/10.3390/plants9030388 (2020).
Hernandez-Martinez, A. R., Estevez, M., Vargas, S., Quintanilla, F. & Rodriguez, R. New dye-sensitized solar cells obtained from extracted bracts of Bougainvillea glabra and spectabilis betalain pigments by different purification processes. Int J Mol Sci 12, 5565–5576, https://doi.org/10.3390/ijms12095565 (2011).
Nores, M. J., López, H. A., Rudall, P. J., Anton, A. M. & Galetto, L. Four o’clock pollination biology: nectaries, nectar and flower visitors in Nyctaginaceae from southern South America. Bot J Linn Soc 171, 551–567, https://doi.org/10.1111/boj.12009 (2013).
Nimbolkar, P. K., Gupta, Y. C., Bhargav, V. & Salam, P. Evolution in Bougainvillea (Bougainvillea Commers.) - A review. J App Nat Sci 9, 1489–1494, https://doi.org/10.31018/jans.v9i3.1389 (2017).
Lan, L. et al. A high-quality Bougainvillea genome provides new insights into evolutionary history and pigment biosynthetic pathways in the Caryophyllales. Hortic Res 10, uhad124, https://doi.org/10.1093/hr/uhad124 (2023).
Zhang, W. et al. Transcriptome analyses shed light on floral organ morphogenesis and bract color formation in Bougainvillea. BMC Plant Biol 22, 97, https://doi.org/10.1186/s12870-022-03478-z (2022).
Ohno, S., Makishima, R. & Doi, M. Post-transcriptional gene silencing of CYP76AD controls betalain biosynthesis in bracts of Bougainvillea. J Exp Bot 72, 6949–6962, https://doi.org/10.1093/jxb/erab340 (2021).
Masotti, A. & Preckel, T. Analysis of small RNAs with the Agilent 2100 Bioanalyzer. Nature Methods 3, 658–658, https://doi.org/10.1038/nmeth908 (2006).
Green, M. R. & Sambrook, J. Isolation of Poly(A)(+) messenger RNA using Magnetic Oligo(dT) Beads. Cold Spring Harb Protoc 2019, https://doi.org/10.1101/pdb.prot101733 (2019).
Modi, A., Vai, S., Caramelli, D. & Lari, M. In Bacterial Pangenomics: Methods and Protocols (eds Mengoni, A., Bacci, G. & Fondi, M.) 15–42 (Springer US, 2021).
John, J. S., Allison, Stajich, J., Reynolds, A. & Thompson, R. C. Tool for stripping adaptors and/or merging paired reads with overlap into single reads., https://github.com/jstjohn/SeqPrep (2016).
Joshi, N. A., & Fass, J. Sickle: A sliding-window, adaptive, quality-based trimming tool for FastQ files (Version 1.33), https://github.com/najoshi/sickle (2011).
Grabherr, M. G. et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat Biotechnol 29, 644–652, https://doi.org/10.1038/nbt.1883 (2011).
Haas, B. J. et al. De novo transcript sequence reconstruction from RNA-seq using the Trinity platform for reference generation and analysis. Nat Protoc 8, 1494–1512, https://doi.org/10.1038/nprot.2013.084 (2013).
Smith-Unna, R., Boursnell, C., Patro, R., Hibberd, J. M. & Kelly, S. TransRate: reference-free quality assessment of de novo transcriptome assemblies. Genome Res 26, 1134–1144, https://doi.org/10.1101/gr.196469.115 (2016).
Li, W. & Godzik, A. Cd-hit: a fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics 22, 1658–1659, https://doi.org/10.1093/bioinformatics/btl158 (2006).
Simao, F. A., Waterhouse, R. M., Ioannidis, P., Kriventseva, E. V. & Zdobnov, E. M. BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 31, 3210–3212, https://doi.org/10.1093/bioinformatics/btv351 (2015).
Haas, B., Zimmermann, B., Crusoe, M. R., Plessy, C. & MacManes, M. TransDecoder: Find coding regions within Transcripts, https://github.com/TransDecoder/TransDecoder/ (2018).
Langmead, B. & Salzberg, S. L. Fast gapped-read alignment with Bowtie 2. Nature Methods. 9, 357–359 (2012).
Thiel, T. MISA: MIcroSAtellite Identification Tool, http://pgrc.ipk-gatersleben.de/misa/ (2001).
NGDC GSA https://ngdc.cncb.ac.cn/gsa/browse/CRA008624 (2022).
NGDC OMIX https://ngdc.cncb.ac.cn/omix/release/OMIX002070 (2025).
NCBI TSA https://identifiers.org/ncbi/insdc:GLCL00000000.1 (2025).
Huang, H. et al. De novo transcriptomes of floral bracts for 22 Bougainvillea accessions. Figshare https://doi.org/10.6084/m9.figshare.26489128 (2025).
NGDC GVM https://ngdc.cncb.ac.cn/gvm/getProjectDetail?project=GVM000984 (2025).
EMBL-EBI EVA https://identifiers.org/ena.embl:PRJEB87743 (2025).
Hannon, G. J. FASTX-Toolkit:FASTQ/A short-reads pre-processing tools http://hannonlab.cshl.edu/fastx_toolkit (2010).
Acknowledgements
This work has been supported by the Earmarked Fund of Science and Technology Innovation for Fujian Agriculture and Forestry University (Project No. KFb22112XA), as well as the grant from the Yuanshan Institute of Bougainvillea (Project No. KH200285A), and the grants from Fujian Province Forestry Science and Technology Project (Project No. 2023FKJ30).
Author information
Authors and Affiliations
Contributions
X.M. and J.Y. conceived the project. X.M., J.Y. and D.Q. designed the experiments. J.Y., H.H., S.J., H.J. collected the flower bract samples. H.H., H.W. and B.R. extracted the RNA samples. S.J., H.W. and Y.Y. inspected the RNA quality. S.J., H.W., B.R., Z.M., W.L. and Y.Y. constructed the RNA library. H.H., Y.S., S.J., H.W., B.R., Z.M. and L.L. analyzed the RNA-seq data. H.H., Y.S., S.J., H.W., B.R., Z.M., W.L. and Y.W. visualized the transcriptome results. Y.S., X.M., H.H. and H.W. processed and uploaded the final datasets to the data repositories. X.M., H.H., Y.S. and S.J. wrote the manuscript. Y.S., X.M., J.Y. and D.Q. revised the manuscript. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Huang, H., Sun, Y., Ju, S. et al. De novo transcriptomes of floral bracts for 22 Bougainvillea accessions. Sci Data 12, 645 (2025). https://doi.org/10.1038/s41597-025-04968-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41597-025-04968-z
