
Abstract
The S. heraclei (Takahashi) (Hemiptera: Aphididae) is a destructive pest of cultivated insectary plant Cnidium monnieri (L.) Cuss. However, to date, no S. heraclei-related genomic information has been reported. Here, we present the first chromosomal-scale genome assembly of S. heraclei approximately 440.3 Mb with contig N50 of 81.7 Mb. Using PacBio long-read sequencing, Illumina sequencing, and Hi-C scaffolding techniques, 94.24% of the assembled sequences were successfully anchored to the four pseudochromosomes. BUSCO assessment showed a completeness score of 95.4%. The S. heraclei genome consisted of 32.02% repetitive elements and 13,983 predicted protein-coding genes. Phylogenetic analysis showed that S. heraclei was closely related to Diuraphis noxia. This high-quality genome assembly of S. heraclei will serve as a genomic resource for aphid evolution and pave the way for deciphering the tri-trophic interaction mechanisms between plants, herbivores, and natural enemies.
Similar content being viewed by others


Background & Summary
The host-alternating aphid S. heraclei is a polyphagous host-alternating aphid that has been reported to use Lonicera spp. as its primary host plant and Apiaceae plants as its secondary host plants1. S. heraclei is predominantly found on umbelliferous and honeysuckle plants2, such as C. monnieri, and Lonicera japonica Thunb. The life cycle of this aphid survives from the winter as diapausing eggs on the honeysuckle plants, which emerge in early spring and reproduce asexually on honeysuckle, winged virginoparae migrate to other host plants by early summer, and in late autumn, winged gynoparae and males return to honeysuckle, and the gynoparae give rise to sexual females, males, and sexual females, then mate and lay eggs1,3. Alternating generations of parthenogenesis and sexual reproduction are common in aphids. Parthenogenetic individuals are all female aphids that are pregnant at birth and parthenogenetic viviparous. Intriguingly, both phenotypes of asexual aphids play important roles in the process of damaging the insectary plant C. monnieri, which coincides with the blooming period of C. monnieri from April to July. C. monnieri conserves natural enemies, such as Coccinellidae, Chrysopidae, and Syrphidae by providing them with food (S. heraclei and pollens) and suitable shelter, enabling them to propagate prolifically to control the wheat aphids into low occurrence in the spring and summer1,4,5. In addition, planting C. monnieri flower strips at the border of wheat-maize rotation fields served as a bridge habitat to conserve ladybeetles in wheat fields during harvest and helped the predator migrate to adjacent maize fields for pest control6,7.
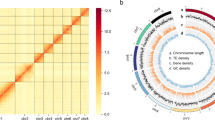
Here, we report the first high-quality draft genome assembly of S. heraclei, generated using PacBio long-read sequencing (~28.11 Gb HiFi reads, with N50 = 15.3 kb) (Table 1). After assembling long reads into contigs, bacterial contamination was removed using BLAST 2.13.0 + (-evalue 1e-5 -outfmt 6 -task megablast -num_threads 5 -max_target_seqs 5), compared the assembly genome with NCBI nucleotide database library of bacterial. There were 75 contigs in the final monoploid genome assembly of S. heraclei with a total of 440.3 Mb (Table 2). The contig N50 reaches 81.7 Mb, and the longest contig was 93.7 Mb (Table 2). 94.24% of the assembled sequences were successfully anchored to the four pseudochromosomes (2n = 8) (Figs. 1C,D). Repetitive components of 140.99 MB were found to make up 32.02% of the S. heraclei assembly (Table 3). The contiguity of the S. heraclei genome assembly, as evidenced by these findings, appears to be on par with that observed in the 10 previously published aphid genomes8,9,10,11,12,13,14,15,16,17. After soft-masking the S. heraclei genome, we predicted 13,983 protein-coding genes with an average length of 10,274 bp (Table 4) using the BRAKER pipeline18,19,20,21,22,23, the methodology incorporated empirical data derived from transcript assemblies, utilizing both short-read sequencing (RNA-seq) and full-length transcript analysis via long-read PacBio sequencing (Iso-seq). Additionally, extrinsic evidence based on homologous sequences from other aphid species was integrated into the analysis (refer to methods for details) Fig. 2.

Heatmap of genome-wide Hi-C data and circular representation of Semiaphis heraclei chromosomes. Pictures show S. heraclei alatae (A) and apterae (B) feeding on Cnidium monnieri (L.), whose genome was sequenced, at the Institute of Plant Protection, Shandong Academy of Agricultural Science (Jinan, China). Photo by Xin Jiang. (C) Heatmap of chromosomal interactions in S. heraclei. The frequency of Hi-C interaction links is represented by colors ranging from yellow (low) to red (high). (D) Circos plot of genomic element distribution in S. heraclei. The tracks indicate (a) length of the chromosome, (b) distribution of transposable element (TE) density ranges from 688 to 2085, (c) gene density ranges from 0 to 74, and (d) GC density ranges from 25 to 53. The densities of TEs, genes, and GC were calculated in 1 Mb sliding windows.

The composition of gene elements in the S. heraclei genome to other closely related species (Apis: Acyrthosiphon pisum, Rpad: Rhopalosiphum padi, Dmel: Drosophila melanogaster) (A) and Venn diagram of number of genes with homology or functional classification by each method (B).
We constructed a maximum likelihood phylogenetic tree based on single-copy orthologs to determine the relationship between S. heraclei and other 14 members of Aphididea. This shows that S. heraclei is closely related to D. noxia (Fig. 3).

Phylogeny and orthology analyses between S. heraclei and other 14 aphid species. The phylogenetic tree was constructed based on 7,168 single-copy orthogroups obtained from the genomes of 14 tested aphids. The estimated species divergence times (million years ago, Mya) are indicated at each branch point. Cinara_cedri was selected as the outgroup. Aphid species are clustered according to their divergent times.
Our investigation encompassed the genetic composition of orthologs, including those with single and multiple copies, as well as the unique orthologous genes specific to each species under examination. There were 857 single-copy and 200 multicopy orthologs in the 14 species, and 42 unique orthologous groups were found in the S. heraclei genome (Fig. 4B). To investigate the rapidly evolving orthologous groups in S. heraclei, we used orthologous group evolution analysis to uncover the changes that occurred in certain orthologous groups over time. We found 833 orthologous groups that had undergone expansions, whereas 9,709 orthologous groups had experienced contractions (Fig. 4A). Of these, 44 orthologous groups (expansions) were identified as rapidly evolving orthogroups. The significantly expanded orthologous groups were primarily associated with heat resistance (heat shock protein), detoxification (carboxylesterase, cytochrome P450), glycometabolism (glycosyl hydrolase), and DNA transposition (DDE superfamily endonuclease, PiggyBac transposable element-derived protein). The rapidly expanded orthologous groups were further confirmed to be involved in metabolic detoxification, digestion, and secondary metabolite synthesis, as shown by the GO and KEGG enrichment analyses (Fig. 4C,D). These results indicate that S. heraclei possesses strong digestion and detoxification abilities, which may enable it to respond effectively to the toxic compounds present in its prey.

Phylogenetic analyses of Semiaphis heraclei and GO, KEGG of rapid evolved genes in expansion. (A) Node values indicate gene families showing expansion (red) or contraction (green). (B) The bar chart indicates the number of genes classified into six groups (single-copy, two-copy, three-copy, four-copy, over four-copy, and unclustered genes). (C) Gene ontology (GO) enrichment of rapidly evolved genes during expansion. (D) KEGG pathway analysis of rapidly evolved genes during expansion.
We conducted a genome synteny analysis between S. heraclei, Acyrthosiphon pisum (clone JIC1), and Myzus. persicae (clone O)14 (Fig. 5A,B). Most chromosomal regions from the S. heraclei genome were aligned with the M. persicae and A. pisum genome assemblies. Assessment of chromosomal rearrangements showed a lack of large-scale rearrangements between the X chromosome and autosomes for any of the aphid species analyzed, whereas aphid autosomes underwent extensive structural changes with many rearrangements between chromosomes. For example, M. persicae scaffold 1 and A. pisum scaffold 1 are homologous to S. heraclei chr 2. In contrast, M. persicae scaffolds 4 and 5 were homologous to S. heraclei chr 1, and A. pisum scaffolds 2 and 3 were homologous to S. heraclei chr 1, with the breakpoint clearly delineated. Comparing the more divergent species pair of M. persicae and A. pisum, which belong to Macrosiphini, revealed highly rearranged autosomes with no clear homology.

Genome synteny between (A) Semiaphis heraclei and Acythosiphon pisum, and (B) Semiaphis heraclei and Myzus persicae. Links indicate the edges of syntenic blocks of gene pairs identified by synteny analysis and are shown in the same color as that of the chromosome ID of S. heraclei.
In summary, this study presents the first chromosome-level reference genome for the aphid of S. heraclei. This work will provides a valuable dataset for understanding genome evolution in aphids and experimental evolution studies, which aims to decipher the adaptive mechanisms of this organism in a changing environment.
Methods
Sample preparation and DNA sequencing
The S. heraclei colony was originally collected in the summer of 2023 from the C. monnieri fields at the Jiyang Experimental Station of the Shandong Academy of Agricultural Sciences and reared on C. monnieri in natural light in a greenhouse maintained at 25 ± 2 °C and relative humidity of 75%. We aimed to create a colony consisting entirely of asexual females; therefore, we carefully selected a single female from the original population to establish a new colony. From this colony, we selected one offspring to generate the next colony, and we repeated this process until we obtained the fifteenth aphid colony, which comprised solely and steadily of asexual females. This pure parthenogenetic colony was used as the sample for all genome-sequencing experiments.
For PacBio sequencing, total RNA was extracted from 200 parthenogenetic female adults. Two 20-kb single-end libraries were built with PacBio SMRT (Single-Molecule Real-Time) sequencing system (Pacific Biosciences, SMRTbell Express Template Prep Kit 2.0). Raw reads were generated from one cell sequence on the PacBio Sequel II/IIe platform at Novogene, Beijing, China. 28.11 Gb (~61.34 × coverage) of SMRT PacBio sequences with a mean read length of 15.1 kb (N50 = 15.3 kb) were retrieved following quality control filtering. Using total RNAs from the entire body of S. heraclei, we created Illumina short-read RNA-seq libraries (5.93 Gb of data with 150 bp paired-end reads) to aid in the prediction of protein-coding genes. Using procedures outlined in earlier research9,16, we created a Hi-C library to further assemble the contigs into chromosomes. Paraformaldehyde was used to crosslink fresh tissues from over 150 distinct samples, including adults and nymphs, in order to produce interacting DNA segments. Following Mbo I digestion of the cross-linked material, the ends of the restriction fragments were labeled with biotinylated nucleotides. The Illumina PE150 platform was used to quantify and sequence the library.
RNA sequencing
TRIzol reagent (Invitrogen, Carlsbad, CA, USA) was used to extract total RNA from 100 parthenogenetic female adults, which was subsequently dissolved in water free of RNase. The integrity of the RNA was evaluated using 2% agarose gel electrophoresis. Using a NanoDrop ND-2000 spectrophotometer (Thermo Fisher Scientific, USA), the concentration and purity of RNA were evaluated. The cDNA libraries were constructed using qualified RNA. An Illumina NovaSeq 6000 platform (Illumina, San Diego, CA, USA) with a 150 bp paired-end approach was used to create the raw sequencing data. 39,031,428 clean readings in all, with a Q30 rate higher than 95%, were produced.
Genome assembly and Hi-C scaffolding
The initial findings of the S. heraclei genome survey revealed a modest degree of heterozygosity (0.24%) in a genome that was 458.27 Mb in size. We assembled the genome using Hifiasm-0.19.6 (default parameters, https://github.com/chhylp123/hifiasm)24,25 with high-quality HiFi reads. Quality control of raw Illumina reads was performed using Fastp v0.23.126. Clean Illumina reads were used to construct a 17-mer frequency distribution map using jellyfish v2.2.727. Using Hifiasm-0.19.6, a contig-level assembly was created with a total length of 440.3 Mb, which is equivalent to the projected genome size, and the contig N50 length was 81.7 Mb (Table 2). FASTP v0.23.1 was used to exclude Low-quality raw reads (quality score < 5 and shorter than 30 bp) and adaptors, then the clean reads were then mapped to the contig assembly using ALLHIC v 0.9.8 (allhic extract group. clean. bam group. fasta--RE GATC allelic partition--pairsfile group.clean.pairs.txt --contigfile group.clean.counts_GATC.txt -K 19--minREs 50--maxlinkdensity 3--Noninformative Rabio 0). The manual changes based on chromosomal interaction were visualized using Juicebox v 1.11.08 with default parameters. Consequently, Hi-C data and contig-level assembly were employed to create a chromosome-level assembly with four sizable scaffolds that matched the species’ previously documented haploid chromosomal number28. The scaffold N50 length was 105.9 Mb, with about 94.24% of the contigs attached to chromosomes (Table 2). The shortest chromosome measured 13.17 Mb, and the longest was 122.47 Mb.
Repeat annotation
In our repeat annotation workflow, we used a combination approach based on homology alignment and a de novo search to find whole-genome repetitions. The RepeatMasker29 (http://www.repeatmasker.org/) software and its proprietary scripts (RepeatProteinMask) with default parameters are used to extract repeat regions from the widely used homolog prediction datebase Repbase30 (http://www.girinst.org/repbase). Using default parameters, RepeatModeler (http://www.repeatmasker.org/RepeatModeler.html) constructed a de novo repeating elements database for ab initio prediction. The raw TE library consisted of all repeat sequences with lengths greater than 100 bp and gaps “N” smaller than 5%. A custom library (a combination of Repbase and our de novo TE library, which was processed using vsearch v1.11.2 (https://github.com/torognes/vsearch) to yield a non-redundant library) was supplied to RepeatMasker for DNA-level repeat identification. According to the findings, repeat sequences made up 32.02% of the genome, with TEs accounting for the majority (31.15%) (Table 3).
Protein coding gene prediction and functional annotation
Ab initio, homology-based, and RNA-seq-assisted gene model prediction are all included in the TE soft-masked S. heraclei genome. Trinity v2.8.5 (--normalize_reads--full_cleanup--min_glue 2--min_kmer_cov 2) was used to produce transcriptome read assemblies for genomic annotation. The RNA-Seq reads from various tissues were aligned to genome fasta using Hisat v2.2.1, with default parameters to detect exon regions and splice points, in order to optimize the genome annotation. The alignment results were then used as input for Stringtie v2.2.1, with the default parameters for genome-based transcript assembly. The non-redundant reference gene set was generated by merging genes predicted by the three methods with EvidenceModeler31 EVM v1.1.1 (-segmentSize 200000-overlapSize 20000-min_intron_length 20) using the Program to Assemble Spliced Alignment (PASA) terminal exon support and including masked transposable elements as input for gene prediction. Our automated gene prediction pipeline employed SNAP (2013-111-29) and Augustus v3.5 (--species = pasa1-uniqueGeneId = TRUE--noInFrameStop = TRUE--GFF3 = on--genemodel = complete--strand = both) to predict genes based on Ab initio. For de novo gene model prediction, the transcript set generated by PASA was utilized in GENEMARK-ST v5.152 for self-training. The training set was applied to AUGUSTUS v3.5 for gene model prediction. Homologous protein sequences were obtained from Ensembl, NCBI, and other sources for the homology-based gene modeling procedure. The software GeneWise32 (v2.4.1) was used to predict the gene structure present in each protein region after protein sequences were aligned to the genome using TblastN v2.2.26 (E-value ≤ 1e−5). The corresponding proteins were then aligned to the homologous genome sequences for precise spliced alignments. Lastly, we used the EVM v1.1.1 to generate a consensus gene model set by combining the outcomes of the three gene prediction methods. This led to the prediction of 13,983 protein-coding gene models were predicted in the S. heraclei genome, with an average coding sequence (CDS) length of 1,487.29 bp and an average transcript length of 10,274.52 bp. On average, each gene comprised 6.98 exons with an average exon length of 213.14 bp and average intron length of 1,469.92 bp (Table 4). The statistical characteristics of gene models, including the lengths of genes, coding sequences (CDS), introns, and exons in S. heraclei, were comparable to those of closely related species. tRNAs were predicted using the tRNAscan-SE33 program (http://lowelab.ucsc.edu/tRNAscan-SE/). We used BLAST to predict rRNA sequences and used relative species rRNA sequences as references due to the high degree of conservation of rRNAs. Additional non-coding RNAs (ncRNAs), including microRNAs (miRNAs) and small nuclear RNAs (snRNAs), were identified through a search against the Rfam34 database using default parameters with the Infernal software (http://infernal.janelia.org/) (Table 5). Protein sequences were aligned to Swiss-Prot using Blastp (with a threshold of E-value ≤ 1e−5), and the best match was used to provide gene functional annotation. The motifs and domains were annotated using InterProScan35 v5.59-91.0 (-cpu 20 -format tsv -appl ProDom, SMART, ProSiteProfiles, PRINTS, Pfam, Panther -iprlookup -dp -goterms) by searching publicly available databases including ProDom, PRINTS, Pfam, SMRT, PANTHER, and PROSITE. Protein function was predicted by transferring annotations from the closest BLAST hit (E-value < 10-5) in the SwissProt36 database and DIAMOND (v0.8.22) / BLAST hit (E-value < 10-5) hit (E-value < 10-5) in the NR37 database. The Gene Ontology (GO)38 IDs for each gene were assigned according to the corresponding InterPro entry. In our analysis, we conducted a mapping of the gene set to a KEGG pathway39, determining the most suitable match for individual genes. The annotation process, utilizing at least one public database, yielded successful results for 13,731 genes, representing 98.2% of the total set Table 6.
Phylogenetic and comparative genomic analyses
The longest predicted protein sequences of 14 aphid genomes, namely Diuraphis noxia3, Sitobion miscanthi10, Aphis gossypii11, Aphis glycines13, Acyrthosiphon pisum14, Myzus perisicae14, Rhopalosiphum nymphaeae16, Therioaphis trifolii17, Melanaphis sacchari (GCF_002803265.2), Myzus cerasi40, Rhopalosiphum padi40, Rhopalosiphum maidis41, Schizaphis graminum (GCA_003264975.1), and the Cinara cedri42 was used as an outgroup, were utilized for identifying orthologous groups among aphids using ORTHOFINDER v2.5.543. Using iqtree v2.3.344 (-B 1000 -seqtype DNA -mset HKY, GTR), we constructed maximum likelihood (ML) trees using 7167 single-copy orthologs that contained 80% of all 15 aphids that were clustered and linked to a supergene. Iqtree chose the model based on modelfinder45 with default parameters. The confidence of the tree node was obtained using ufboot246 (-B 1000) with 1000 iterations. C. cedri was used as the root in order to produce a rooted tree.
Synteny analysis
The chromosome-level genome assemblies of S. heraclei, A. pisum (JIC1 v1), and M. persicae (O. v2)14 were compared using synteny analysis. In order to obtain syntenic blocks, we uploaded the official gene sets to ORTHOVENN2 server47. The following criteria were used to identify the 1:1 single-copy ortholog pairs from each comparison (S. heraclei vs. A. pisum and S. heraclei vs. M. persicae):--no-unlink -k 0 -f 6-e 1e-5--query-cover 50--subject-cover 50--max-target-seqs 10--salltitles--more sensitive). Diamondblastp v2.1.7.16148 (--no-unlink -k 0 -f 6-e 1e-5--query-cover 50--subject-cover 50--max-target-seqs 10--salltitles--more sensitive) was used to select these gene pairings for genome synteny analysis. Jcvi v1.3.949 was used to visualize genome synteny.
Data Records
The raw sequence data reported in this paper have been deposited in the Genome Sequence Archive50 under the accession number CRA01805551 at the National Genomics Data Center (NGDC)52, China National Center for Bioinformation/Beijing Institute of Genomics. All raw data in the NGDC are related to bioproject PRJCA027460. The assembled genome can be found on NCBI under the accession number GCA_046119115.153 The genome assembly and gene annotation of S. heraclei are related to the bioproject PRJNA1184189 and has been deposited in Figshare54.
Technical Validation
Four criteria were used to evaluate the correctness and completeness of the S. heraclei genome assembly. Using BWA v0.7.8, clean Illumina reads were first mapped to the contigs constructed. SAMTOOLS v1.472 was then used to calculate the total number of mapped reads and the mapping rate, which came out to be 98.56%. Second, we compared the k-mers from the final assembly with those in the PacBio HiFi reads using Merqury55 to estimate the base-level correctness and completeness of the S. heraclei assembly. A consensus quality (QV) of 60 was reported by Merqury. Third, the completeness of the genome assembly was evaluated using Benchmarking Universal Single-Copy Orthologs (BUSCO) v5.2.2. The BUSCO analysis revealed that 95.4% of gene orthologs were found in S. heraclei, based on the metazoa_odb10 database. Lastly, contig N50 reached 81.7 Mb, the longest contig reached 93.7 Mb, and 11 scaffolds were positioned on the genome, making up 94.24% of the assembly, in accordance to the genome assembly summary statistics. These data indicate that this assembly is one of the highest contiguous genome assemblies among the 14 aphids compared.
Three validation techniques were used to make sure the annotated gene set was comprehensive. Initially, BUSCO analysis was performed on the annotation using the Metazoa _odb10 database. The findings showed that the annotated gene set contained 93.3% of intact genes, including 89.6% single copies and 1.6% duplicates. Second, whole-body transcriptomes were used to obtain RNA-Seq data for gene expression investigation. Lastly, a number of protein databases (GO, KOG, InterPro, Pfam, KEGG, SWISS-PROT, and others) were compared with the projected gene models. Of the projected gene models, 13,732 (98.2%) had considerable homology to proteins in at least one database, according to the results. These findings collectively validate our conclusion that the genome assembly is of excellent quality.
Phylogenetic analysis revealed that S. heraclei and D. noxia were closely related. The gene sequence and rooted tree were built using an ML tree, and the differentiation time was inferred using the mcmctree2.ctl module of paml56, utilizing the divergence periods of S. heraclei vs. D. noxia as reference (previous time) of 0.078-4.782 million years ago (mya). The divergence time tree was displayed using the ggtree v3.2.057. We also used CAFE v 5.1.058 to analyze the expansion and contraction of gene families in all 15 tested aphid lineages. The results from the phylogenetic tree with divergence times were used as input.
Code availability
No custom code was used for this study. All the software and pipelines used for data processing were executed according to the manuals and protocols of the bioinformatics software cited above. These parameters are described in the methods section. If no detailed parameters were mentioned for the software, the default parameters were used. The software version is described in the methods section.
References
Zhang G-X, Zhong T-S. Economic Insects of China, Vol 25 Homoptera Aphids, 1st ed (Science Press,1983).
Chen J, Ding W-L, Cheng H-Z. Medicinal Plant Protection (Publishing House of Electronics Industry, 2019).
Zhang, Y. et al. Research on population dynamics of Lonicera macranthoides aphid and natural enemy in Xiushan and evolution of pesticides. Chin J Tradit Chin Med. 37, 3219–3222 (2012). (in Chinese).
Li, Z. et al. Functional plant, Cnidium monnieri, facilitates the conservation and the biocontrol performance of natural enemies. The Innovation Geoscience 1, 100045 (2023).
Su, W. et al. Cnidium monnieri (L.) Cusson Flower as a Supplementary Food Promoting the Development and Reproduction of Ladybeetles Harmonia axyridis (Pallas) (Coleoptera: Coccinellidae). Plants 12, 1786 (2023).
Yang, Q.-F. et al. Flower strips as a bridge habitat facilitate the movement of predatory beetles from wheat to maize crops. Pest Manag Sci. 4, 1839–1850 (2021).
Yang, Q.-F. et al. Discovery and utilization of a functional plant, rich in the natural enemies of insect pests, in northern China. Chin J Appl Entomol. 55, 942–947 (2018).
Nicholson, S. J. et al. Te genome of Diuraphis noxia, a global aphid pest of small grains. BMC Genomics. 16, 429 (2015).
Torpe, P. et al. Shared Transcriptional Control and Disparate Gain and Loss of Aphid Parasitism Genes. Genome Biol Evol. 10, 2716–2733 (2018).
Jiang, X. et al. A chromosome-level draft genome of the grain aphid Sitobion miscanthi. Gigascience. 8, giz101 (2019).
Quan, Q.-M. et al. Draft genome of the cotton aphid Aphis gossypii. Insect Biochem Mol Biol. 105, 25–32 (2019).
Mathers, T. C. et al. Genome Sequence of the Banana Aphid, Pentalonia nigronervosa Coquerel (Hemiptera: Aphididae) and Its Symbionts. G3-Genes Genom Genet. 10, 4315–4321 (2020).
Wenger, J. A. et al. Whole genome sequence of the soybean aphid, Aphis glycines. Insect Biochem Mol Biol. 123, 102917 (2020).
Mathers, T. C. et al. Chromosome-Scale Genome Assemblies of Aphids Reveal Extensively Rearranged Autosomes and Long-Term Conservation of the X Chromosome. Mol Biol Evol. 38, 856–875 (2021).
Wei, H.-Y. et al. Chromosome-level genome assembly for the horned-gall aphid provides insights into interactions between gallmaking insect and its host plant. Ecol Evol. 12, e8815 (2022).
Wang, Y. & Xu, S. A high-quality genome assembly of the waterlily aphid Rhopalosiphum nymphaeae. Sci Data 11, 194 (2024).
Huang, T. et al. Chromosome-level genome assembly of the spotted alfalfa aphid Therioaphis trifolii. Sci Data 10, 274 (2023).
Stanke, M. et al. Gene prediction in eukaryotes with a generalized hidden Markov model that uses hints from external sources. BMC Bioinformatics 7, 62 (2006).
Stanke, M. et al. Using native and syntenically mapped cDNA alignments to improve de novo gene finding. Bioinformatics 24, 637–644 (2008).
Hoff, K. J. et al. BRAKER1: Unsupervised RNA-Seq-Based Genome Annotation with GeneMark-ET and AUGUSTUS. Bioinformatics 32, 767–769 (2016).
Hoff, K. J. et al. Whole-Genome Annotation with BRAKER. Methods Mol Biol. 1962, 65–95 (2019).
Bruna, T. et al. BRAKER2: automatic eukaryotic genome annotation with GeneMark-EP plus and AUGUSTUS supported by a protein database. NAR Genom Bioinform. 3, lqaa108 (2021).
Gabriel, L. et al. TSEBRA: transcript selector for BRAKER. BMC Bioinformatics. 22, 566 (2021).
Cheng, H. et al. Haplotype-resolved de novo assembly using phased assembly graphs with hifiasm. Nat Methods. 18, 170–175 (2021).
Cheng, H. et al. Haplotype-resolved assembly of diploid genomes without parental data. Nat Biotechnol. 40, 1332–1335 (2022).
Chen, S. et al. fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics. 34, i884–i890 (2018).
Marçais, G. & Kingsford, C. A fast, lock-free approach for efficient parallel counting of occurrences of k-mers. Bioinformatics. 27, 764–770 (2011).
Chen, X.-S. & Zhang, G.-X. The karyotypes of fifty-one of aphids (homoptera, aphioidea) in. Beijing area. 31, 12–19 (1985).
Smit, A. F. A., Hubley, R, and Green, P. RepeatMasker Open-3.0 (Seattle: The Institute for Systems Biology) (2010).
Jurka, J. et al. Repbase Update, a database of eukaryotic repetitive elements. Cytogenet. Genome Res. 110, 462–467 (2005).
Haas, B. J. et al. Automated eukaryotic gene structure annotation using EVidenceModeler and the Program to Assemble Spliced Alignments. Genome Biol. 9, R7 (2008).
Birney, E., Clamp, M. & Durbin, R. GeneWise and Genomewise. Genome Res. 14, 988–995 (2004).
Lowe, T. M. & Eddy, S. R. tRNAscan-SE: A program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 25, 955–964 (1997).
Griffiths-Jones, S. et al. Rfam: annotating non-codin RNAs in complete genomes. Nucleic Acids Res. 33, D121–4 (2005).
Mulder, N. & Apweiler, R. InterPro and InterProScan: tools for protein sequence classification and comparison. Methods Mol Biol. 396, 59–70 (2007).
Kretschmann, E., Fleischmann, W. & Apweiler, R. Automatic rule generation for protein annotation with the C4.5 data mining algorithm applied on SWISS-PROT. Bioinformatics. 17, 920–926 (2001).
Schoch, C. L. et al. NCBI Taxonomy: a comprehensive update on curation, resources and tools. Database (Oxford). 2020, baaa062 (2020).
Gene Ontology Consortium. Gene Ontology Consortium: going forward. Nucleic Acids Res. 43(Database issue), D1049–D1056 (2015).
Kanehisa, M. et al. KEGG for integration and interpretation of large-scale molecular data sets. Nucleic Acids Res. 40(Database issue), D109–D114 (2012).
Thorpe, P. et al. Shared transcriptional control and disparate gain and loss of aphid parasitism genes. Genome Biol Evol. 110, 2716–2733 (2018).
Chen, W. B. et al. Genome sequence of the corn leaf aphid (Rhopalosiphum maidis Fitch). Gigascience. 8, giz033 (2019).
Julca, I. et al. Phylogenomics identifies an ancestral burst of gene duplications predating the diversification of aphidomorpha. Mol Biol Evol. 37, 730–756 (2020).
Emms, D. M. & Kelly, S. OrthoFinder: phylogenetic orthology inference for comparative genomics. Genome Biol. 20, 238 (2019).
Minh, B. Q. et al. IQ-TREE 2: new models and efficient methods for phylogenetic inference in the genomic era. Mol Biol Evol. 37, 1530–1534 (2020).
Kalyaanamoorthy, S. et al. ModelFinder: fast model selection for accurate phylogenetic estimates. Nat Methods. 14, 587–589 (2017).
Hoang, D. T. et al. UFBoot2: improving the ultrafast bootstrap approximation. Mol Biol Evol. 35, 518–522 (2018).
Xu, L. et al. OrthoVenn2: a web server for whole-genome comparison and annotation of orthologous clusters across multiple species. Nucleic Acids Res. 47, w52–w58 (2019).
Buchfink, B., Reuter, K. & Drost, H. G. Sensitive protein alignments at tree-of-life scale using DIAMOND. Nat Methods. 18, 366–368 (2021).
Tang et al. jcvi: JCVI utility libraries. Zenodo. https://doi.org/10.5281/zenodo.31631 (2015).
Chen, T. et al. The Genome Sequence Archive Family: Toward Explosive Data Growth and Diverse Data Types. Genomics Proteomics Bioinformatics. 19, 578–583 (2021).
National Genomics Data Center, China National Center for Bioinformation https://ngdc.cncb.ac.cn/gsa/browse/CRA018055 (2025).
CNCB-NGDC Members and Partners. Database Resources of the National Genomics Data Center, China National Center for Bioinformation in 2022. Nucleic Acids Res. 50, 27–38 (2022).
Jiang, X. Semiaphis heraclei isolate she-1, whole genome shotgun sequencing project. GenBank https://identifiers.org/ncbi/insdc:GCA_046119115.1 (2024).
jiang, X. et al. Genome assembly and gene annotation of Semiaphis heraclei. figshare. https://doi.org/10.6084/m9.figshare.26779861 (2024).
Rhie, A. et al. Merqury: reference-free quality, completeness, and phasing assessment for genome assemblies. Genome Biol. 21, 245 (2020).
Yang, Z. PAML 4: phylogenetic analysis by maximum likelihood. Mol Biol Evol. 24, 1586–1591 (2007).
Yu, G. et al. ggtree: an R package for visualization and annotation of phylogenetic trees with their covariates and other associated data. Methods Ecol Evol. 8, 28–36 (2017).
Mendes, F. K. et al. CAFE 5 models variation in evolutionary rates among gene families. Bioinformatics. 36, 5516–5518 (2021).
Acknowledgements
This research was supported by Introducing Top Talent Program of Shandong (2023YSYY-006), the National Key R&D Program of China (2023YFD1400800), Agricultural Science and Technology Innovation Project of Shandong Academy of Agricultural Sciences (grant no. CXGC2023F04), and Postdoctoral Innovation Project of Shandong (SDBX2023059).
Author information
Authors and Affiliations
Contributions
X.J. and Z.L. collected the aphid samples in the field and kept the aphid colony in the lab. X.J. and G.F. conceived and supervised the study. X.J., Z.L., J.F., C.C., X.Z., Z.L. and J.F. performed data analyses. X.J. wrote the manuscript. X.J. and G.F. analyzed the data. All authors read, edited, and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Jiang, X., Zhao, L., Fan, J. et al. A chromosome-level genome assembly of the aphid Semiaphis heraclei (Takahashi). Sci Data 12, 770 (2025). https://doi.org/10.1038/s41597-025-04994-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41597-025-04994-x
