
Abstract
Ficus benjamina, the weeping fig, is one of the most widely distributed and cultivated figs, with important ecological functions and landscape value. However, the lack of a reference genome has hindered molecular and functional research on this well-known fig-tree. Here we present a chromosome-scale genome assembly and annotation for F. benjamina, based on a combination of Illumina short-reads, PacBio subreads, and Hi-C sequencing data. The genome consists of 13 pseudochromosomes that contain 362.73 Mb of assembled sequences, with a contig N50 length of 25.76 Mb and a complete BUSCO score of 98.10%. In total, 28,840 protein-coding genes were identified, of which 96.22% were functionally annotated. Our study provides the first chromosome-level genome of F. benjamina, providing an important resource for exploring the genetic basis of its ecological and horticultural characters.
Similar content being viewed by others

Background & Summary
Ficus is a large plant genus with over 800 species and a largely tropical and subtropical distribution1,2,3. This woody genus displays a wide range of growth-forms, including shrubs, trees, hemiepiphytes, and lianas, thriving in various climatic and geographic conditions and playing crucial roles in tropical and subtropical ecosystems3,4,5. Because they can fruit year-round, fig trees play a vital role in sustaining a broad range of frugivorous animal communities6,7,8. The obligate mutualism between figs and their pollinating wasps also serves as an excellent model system for studying coevolutionary relationships9,10,11,12,13,14,15,16,17,18,19.
Because of the vigorous growth and great plasticity, some strangler figs of Ficus subg. Spherosuke ( = subg. Urostigma) have high ornamental value. Among these, Ficus benjamina is the second most widely distributed and cultivated species. This fig tree is notable for its diverse morphology, weeping branches and foliage, and indistinct lateral veins20. Among the many cultivars of F. benjamina, some have variegated leaves, while others have contorted wavy branches or curled leaves21.
There are currently eight published Ficus genomes, ranging in size from 297.27 Mb to 426.56 Mb22,23,24,25,26,27. Recently, researchers have employed whole-genome sequencing data to explore various topics, including sex-determining genes, the development of aerial roots, the mechanisms underlying plant longevity22,23,26,28, and the obligate mutualistic relationships between figs and fig wasps24,29. Comparative genomics analyses involving multiple Ficus species have facilitated a better understanding of their evolutionary history. However, the genomics underlying many of the horticultural properties that are important in ornamental fig trees remain unclear.
Here, we aimed to produce a high-quality, chromosome-scale, de novo genome assembly of Ficus benjamina using Illumina, PacBio, and chromosome conformation capture (Hi-C) sequencing technologies. This high-quality F. benjamina genome will help to elucidate the mechanisms of the ecological and horticultural characters of fig trees.
Methods
Sample collection, library construction, and sequencing
All samples of this research were taken from a living individual of Ficus benjamina cultivated at the South China Botanical Garden, Guangzhou, China (23°10′43.4″N 113°21′07.6″E). Fresh and healthy young leaves were collected for genome sequencing. Tissues, including leaves, stems, and inflorescences were sampled for transcriptome sequencing. All materials were promptly frozen using liquid nitrogen and stored at −80 °C until nucleic acid isolation. High-quality genomic DNA was extracted from sampled leaves using the conventional cetyltrimethylammonium bromide method (CTAB)30. Short-read libraries were constructed using Rapid Plus DNA Lib Prep Kit for Illumina (ABclonal, Cat. RK20208).
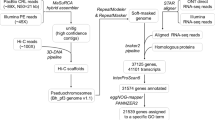
Paired-end reads of 150 bp were generated using an Illumina NovaSeq X Plus platform. For de novo genome assembly, high-molecular-weight DNA was used to construct a 15–20-kb SMRTbell library (SMRTbell Express Template Prep Kit 2.0, Pacific Biosciences). The library was sequenced on the PacBio Sequel II platform using circular consensus sequencing (CCS) mode with a minimum read quality of Q20 (≥99% accuracy). HiFi reads were generated using the CCS algorithm with ≥3 full passes per molecule. A Hi-C library was constructed using DpnII following the standard protocol described previously with modifications for plant samples31. The library was sequenced on an Illumina NovaSeq X Plus platform, generating 150 bp paired-end reads32. Total RNA was isolated using RNAprep Pure Plant Kit (Tiangen, China) and mRNA was purified from total RNA using poly-T oligo-attached magnetic beads. RNA-seq libraries were prepared using Fast RNA-seq Lib Prep Kit V2 (ABclonal, Cat. RK20306) and sequenced on an Illumina NovaSeq X Plus platform using paired-end reads of 150 bp. All Illumina sequencing data were filtered using the fastp v0.23.1 software33 with default parameters. For genome sequencing, we generated: (1) 30.46 Gb of high-quality Illumina short-reads (97.80% Q20, 84.14 × coverage) for genome survey; (2) 32.70 Gb of PacBio HiFi reads (90.33 × coverage) for assembly; (3) 61.27 Gb of Hi-C data (97.60% Q20, 169.25 × coverage) for scaffolding; and (4) 25.21 Gb of RNA-seq data (98.95% Q20, 69.64 × coverage) for annotation (Table 1).
Genome survey
The genome features of Ficus benjamina were surveyed using the k-mer method based on Illumina short-reads. The k-mer count histogram was generated using Jellyfish v2.2.734 with the following parameters: ‘count -G 2 -m 17 -C -o kmercount’. The analysis based on 17-mers estimated the genome size of F. benjamina to be approximately 419.6 Mb, with repeat sequences of highly approximate 52.4% and a heterozygosity of 1.57% (Fig. 1a).

Overview of the Ficus benjamina genome assembly: (a) genome survey based on the K-mer distribution analysis using a k-mer size of 17-mers, (b) Hi-C interaction heat map for the assembled genome.
Genome assembly
High-quality PacBio HiFi long-reads were assembled into contigs using hifiasm v0.15.435 with default parameters, yielding a preliminary assembly of 409.26 Mb. Given the high heterozygosity, we performed deduplication using purge_dups v1.2.536 to remove haplotypic redundancies, followed by assembly polishing with NextPolish237. To anchor the contigs into pseudochromosomes, Hi-C data were aligned to the final assembled contigs by juicer pipeline v1.638 to obtain an interaction matrix. The contigs were then ordered and anchored using the Hi-C scaffolding tool, YaHS v1.239. The diploid chromosome number of F. benjamina (2n = 26) was confirmed using the Chromosome Counts Database (CCDB; https://taux.evolseq.net/CCDB_web), guiding the pseudochromosome construction. The Hi-C contact maps of the final assembly result were examined manually with Juicebox v2.2040. The Hi-C interaction heat map showed a strong intrachromosomal interactive signal along the diagonal (Fig. 1b). Finally, a gap-free Ficus benjamina genome of 362.73 Mb was constructed, with a contig N50 length of 25.76 Mb (Table 2), and 13 large contigs representing 13 pseudochromosomes (Fig. 2a).

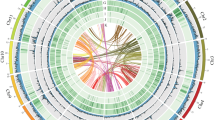
Genomic features of Ficus benjamina showed by concentric circles from outside to inside: (a) chromosomes, (b) gene density, (c) repeat density, (d) Copia density, (e) Gypsy density, (f) GC density, (g) syntenic gene blocks.
Transposable elements and non-coding RNA annotation
Transposable elements (TEs) were identified and classified using Extensive de-novo TE Annotator (EDTA) v2.1.041. To predict non-coding RNA, tRNA genes were identified with tRNAscan-SE v2.0.642. Others, including miRNA, rRNA and snRNA genes, were detected by comparison with the Rfam database43 using CMsearch v1.1.344 under default parameters. The composition of these TEs included 24.20% long terminal repeat (LTR) elements, 8.49% terminal inverted repeat (TIR) elements, and 4.04% Helitrons (Table 3). Among the classified retroelements, the Copia and Gypsy superfamilies accounted for 4.36% and 19.52% of the assembly, respectively (Fig. 2c–e; Table 3). The most abundant DNA transposon superfamily was Mutator, comprising 4.86% of the assembly (Table 3). Genome-wide screening for non-coding RNAs revealed 526 tRNAs, 125 miRNAs, 3,514 rRNAs, and 523 snRNAs (Table 4). In addition, we found most of the LTRs have been accumulated recently over a short time span with the peak of 0.15 million years ago (Ma), suggesting an expansion event (Fig. 3).

The distribution of insertion time (Ma, million years ago) of intact LTRs in Ficus benjamina.
Gene prediction and functional annotation
For protein-coding gene prediction, we used the pipeline MAKER v3.01.0245 with combined homology-based, transcriptome-based, and ab initio prediction methods. First, we used homologies from related species as protein-based evidence for gene sets prediction using GeneWise v2.4.146. The related species include Ficus carica, F. hispida, F. microcarpa, Morus notabilis, Vitis vinifera, and Arabidopsis thaliana. Transcriptome data, including leaf, stem, and inflorescence RNA-seq reads were mapped using HISAT2 v2.1.047. Ab initio gene prediction was carried out using AUGUSTUS v3.4.048, trained by the transcriptome data. To functionally annotate the predicted gene models, several different databases were searched, including NCBI nr49, Swiss-Prot50, eggNOG51, and Pfam52 using BLASTP53. Finally, we annotated 28,840 protein-coding genes with an average exon length of 337.6 bp, and an average intron length of 445 bp (Table 2, Fig. 2b). In total, 26,892 (96.22%) genes were assigned specific functions (Table 5).
Genome synteny analysis
To reveal the syntenic relationships between the protein-coding genes of Ficus benjamina and other four representative figs, collinear blocks between them were identified based on protein sequences using MCScan implemented in jcvi v1.2.754. The syntenic gene blocks and syntenic depth showed 1:1 syntenic patterns between F. benjamina and other four figs (Fig. 4), indicating a conserved genome structure across the genus.

Syntenic blocks and syntenic depth between assembly of Ficus benjamina and other four figs.
Data Records
The raw sequencing data have been deposited in the Genome Sequence Archive (GSA) in National Genomics Data Center (NGDC) database (https://ngdc.cncb.ac.cn/) under the accession number CRA01800655. The final chromosome assembly was deposited in NCBI GenBank under accession number JBFTXC00000000056. The draft genome assembly and genome annotation were deposited in the Figshare database (https://doi.org/10.6084/m9.figshare.27980945)57.
Technical Validation
The quality of the Ficus benjamina genome assembly was evaluated using four approaches. First, the completeness of the genome assembly was assessed using BUSCO v5.4.558 against the embryophyta_odb10 database (containing 1614 orthologs). The results showed 98.10% completeness (1584 complete BUSCOs), comprising 96.30% single-copy (1555) and 1.80% duplicated (29) orthologs (Table 6). Then, the assembly continuity was determined by analyzing the LTR Assembly Index (LAI)59, which had a value of 21.14 (Table 2). Additionally, for the assessment of the assembly’s correctness, we re-aligned Illumina DNA sequencing data and PacBio HiFi long-reads against the genome using BWA v0.7.1560 and minimap2 v2.24-r11226261, respectively. The results indicated high mapping rates of Illumina short-reads (98.05%) and HiFi long-reads (99.86%). Finally, quality value (QV) was estimated using Merqury v1.36562, resulting in a value of 73.33 (Table 2). All these results indicate that the F. benjamina genome assembly presented here is of high quality.
Code availability
All software and pipelines used in this study were implemented according to the manuals and protocols provided by the software developers. Versions of the software have been described in Methods. No custom code was used in this study.
References
Berg, C. C. Classification and distribution of. Ficus. Experientia 45, 605–611, https://doi.org/10.1007/BF01975677 (1989).
Moonlight, P. W. et al. Twenty years of big plant genera. P. Roy. Soc. B-Biol. Sci. 291, 20240702, https://doi.org/10.1098/rspb.2024.0702 (2024).
Harrison, R. D. Figs and the diversity of tropical rainforests. Bioscience 55, 1053–1064, https://doi.org/10.1641/0006-3568(2005)055[1053:FATDOT]2.0.CO;2 (2005).
Berg, C. C. & Corner, E. J. H. in Flora Malesiana Vol. 17 (ed Nooteboom, H. P.) 1–730 (National Herbarium of the Netherlands, 2005).
Beck, H. in Encyclopedia of Ecology (ed Brian Fath) 671–678 (Elsevier, 2019).
Cottee-Jones, H. E. W., Bajpai, O., Chaudhary, L. B. & Whittaker, R. J. The importance of Ficus (Moraceae) trees for tropical forest restoration. Biotropica 48, 413–419, https://doi.org/10.1111/btp.12304 (2016).
Shanahan, M., So, S., Compton, S. G. & Corlett, R. Fig-eating by vertebrate frugivores: a global review. Biol. Rev. 76, 529–572, https://doi.org/10.1017/S1464793101005760 (2001).
Kissling, W. D., Rahbek, C. & Böhning-Gaese, K. Food plant diversity as broad-scale determinant of avian frugivore richness. P. Roy. Soc. B-Biol. Sci. 274, 799–808, https://doi.org/10.1098/rspb.2006.0311 (2007).
Hill, D. S. Figs (Ficus spp.) and fig-wasps (Chalcidoidea). J. Nat. Hist. 1, 413–434, https://doi.org/10.1080/00222936700770401 (1967).
Ramírez, W. Host specificity of fig wasps (Agaonidae). Evolution, 680–691, https://doi.org/10.2307/2406549 (1970).
Janzen, D. H. How to be a fig. Ann. Rev. Ecol. Syst. 10, 13–51, https://doi.org/10.1146/annurev.es.10.110179.000305 (1979).
Machado, C. A., Robbins, N., Gilbert, M. T. P. & Herre, E. A. Critical review of host specificity and its coevolutionary implications in the fig/fig-wasp mutualism. P. Natl. Acad. Sci. USA. 102, 6558–6565, https://doi.org/10.1073/pnas.0501840102 (2005).
Rønsted, N. et al. 60 million years of co-divergence in the fig–wasp symbiosis. P. Roy. Soc. B-Biol. Sci. 272, 2593–2599, https://doi.org/10.1098/rspb.2005.3249 (2005).
Rønsted, N., Weiblen, G. D., Clement, W., Zerega, N. & Savolainen, V. Reconstructing the phylogeny of figs (Ficus, Moraceae) to reveal the history of the fig pollination mutualism. Symbiosis 45, 45–55 (2008).
Cruaud, A. et al. An extreme case of plant–insect codiversification: figs and fig-pollinating wasps. Syst. Biol. 61, 1029–1047, https://doi.org/10.1093/sysbio/sys068 (2012).
Yu, H. et al. Multiple parapatric pollinators have radiated across a continental fig tree displaying clinal genetic variation. Mol. Ecol. 28, 2391–2405, https://doi.org/10.1111/mec.15046 (2019).
Yu, H., Liao, Y. L., Cheng, Y. F., Jia, Y. J. & Compton, S. G. More examples of breakdown the 1:1 partner specificity between figs and fig wasps. Bot. Stud. 62, 1–12, https://doi.org/10.1186/s40529-021-00323-8 (2021).
Su, Z. H. et al. Pollinator sharing, copollination, and speciation by host shifting among six closely related dioecious fig species. Commun. Biol. 5, 1–15, https://doi.org/10.1038/s42003-022-03223-0 (2022).
Zhang, Q., Onstein, R. E., Little, S. A. & Sauquet, H. Estimating divergence times and ancestral breeding systems in Ficus and Moraceae. Ann. Bot. 123, 191–204, https://doi.org/10.1093/aob/mcy159 (2019).
Liao, S. A systematic study of Ficus subsect. Conosycea Doctor thesis, East China Normal University, (2022).
Meislik, J. The world of Ficus Bonsai. (FriesenPress, 2019).
Chakraborty, A., Mahajan, S., Bisht, M. S. & Sharma, V. K. Genome sequencing and comparative analysis of Ficus benghalensis and Ficus religiosa species reveal evolutionary mechanisms of longevity. iscience 25, https://doi.org/10.1016/j.isci.2022.105100 (2022).
Zhang, X. T. et al. Genomes of the banyan tree and pollinator wasp provide insights into fig-wasp coevolution. Cell 183, 875–889, https://doi.org/10.1016/j.cell.2020.09.043 (2020).
Wang, R. et al. Molecular mechanisms of mutualistic and antagonistic interactions in a plant–pollinator association. Nat. Ecol. Evol. 5, 974–986, https://doi.org/10.1038/s41559-021-01469-1 (2021).
Huang, W. C. et al. A high-quality chromosome-level genome assembly of Ficus hirta. Sci. Data 11, 526, https://doi.org/10.1038/s41597-024-03376-z (2024).
Liao, Z. Y. et al. A telomere-to-telomere reference genome of ficus (Ficus hispida) provides new insights into sex determination. Hortic. Res. 11, uhad257, https://doi.org/10.1093/hr/uhad257 (2024).
Shirasawa, K. et al. The Ficus erecta genome aids Ceratocystis canker resistance breeding in common fig (F. carica). Plant J. 102, 1313–1322, https://doi.org/10.1111/tpj.14703 (2020).
Mori, K. et al. Identification of RAN1 orthologue associated with sex determination through whole genome sequencing analysis in fig (Ficus carica L.). Sci. Rep. 7, 41124 (2017).
Wang, G. et al. Genomic evidence of prevalent hybridization throughout the evolutionary history of the fig-wasp pollination mutualism. Nat. Commun. 12, 1–14, https://doi.org/10.1038/s41467-021-20957-3 (2021).
Doyle, J. J. & Doyle, J. L. A rapid DNA isolation procedure for small quantities of fresh leaf tissue. Phytochem. Bull. 19, 11–15 (1987).
Belton, J.-M. et al. Hi–C: A comprehensive technique to capture the conformation of genomes. Methods 58, 268–276, https://doi.org/10.1016/j.ymeth.2012.05.001 (2012).
Crémazy, F. G. et al. in Bacterial Chromatin: Methods and Protocols (ed Dame, R. T.) 3–18 (Springer New York, 2018).
Chen, S. F., Zhou, Y. Q., Chen, Y. R. & Gu, J. fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 34, i884–i890, https://doi.org/10.1093/bioinformatics/bty560 (2018).
Marçais, G. & Kingsford, C. A fast, lock-free approach for efficient parallel counting of occurrences of k-mers. Bioinformatics 27, 764–770, https://doi.org/10.1093/bioinformatics/btr011 (2011).
Cheng, H. Y., Concepcion, G. T., Feng, X. W., Zhang, H. W. & Li, H. Haplotype-resolved de novo assembly using phased assembly graphs with hifiasm. Nat. Methods 18, 170–175, https://doi.org/10.1038/s41592-020-01056-5 (2021).
Guan, D. F. et al. Identifying and removing haplotypic duplication in primary genome assemblies. Bioinformatics 36, 2896–2898, https://doi.org/10.1093/bioinformatics/btaa025 (2020).
Hu, J. et al. NextPolish2: A Repeat-aware Polishing Tool for Genomes Assembled Using HiFi Long Reads. Genomics, Proteomics & Bioinformatics 22, https://doi.org/10.1093/gpbjnl/qzad009 (2024).
Durand, N. C. et al. Juicer Provides a One-Click System for Analyzing Loop-Resolution Hi-C Experiments. Cell Syst. 3, 95–98, https://doi.org/10.1016/j.cels.2016.07.002 (2016).
Zhou, C. X., McCarthy, S. A. & Durbin, R. YaHS: yet another Hi-C scaffolding tool. Bioinformatics 39, btac808, https://doi.org/10.1093/bioinformatics/btac808 (2022).
Durand, N. C. et al. Juicebox provides a visualization system for Hi-C contact maps with unlimited zoom. Cell Syst. 3, 99–101, https://doi.org/10.1016/j.cels.2015.07.012 (2016).
Ou, S. et al. Benchmarking transposable element annotation methods for creation of a streamlined, comprehensive pipeline. Genome Biol. 20, 275, https://doi.org/10.1186/s13059-019-1905-y (2019).
Lowe, T. M. & Eddy, S. R. tRNAscan-SE: A Program for Improved Detection of Transfer RNA Genes in Genomic Sequence. Nucleic. Acids Res. 25, 955–964, https://doi.org/10.1093/nar/25.5.955 (1997).
Gardner, P. P. et al. Rfam: updates to the RNA families database. Nucleic. Acids Res. 37, D136–D140, https://doi.org/10.1093/nar/gkn766 (2008).
Cui, X. F., Lu, Z. W., Wang, S., Jing-Yan Wang, J. & Gao, X. CMsearch: simultaneous exploration of protein sequence space and structure space improves not only protein homology detection but also protein structure prediction. Bioinformatics 32, i332–i340, https://doi.org/10.1093/bioinformatics/btw271 (2016).
Cantarel, B. L. et al. MAKER: an easy-to-use annotation pipeline designed for emerging model organism genomes. Genome Res. 18, 188–196, https://doi.org/10.1101/gr.6743907 (2008).
Birney, E., Clamp, M. & Durbin, R. GeneWise and genomewise. Genome Res. 14, 988–995, https://doi.org/10.1101/gr.1865504 (2004).
Kim, D., Langmead, B. & Salzberg, S. L. HISAT: a fast spliced aligner with low memory requirements. Nat. Methods 12, 357–360, https://doi.org/10.1038/nmeth.3317 (2015).
Stanke, M. et al. AUGUSTUS: ab initio prediction of alternative transcripts. Nucleic. Acids Res. 34, W435–W439, https://doi.org/10.1093/nar/gkl200 (2006).
Sayers, E. W. et al. Database resources of the national center for biotechnology information. Nucleic. Acids Res. 50, D20–D26, https://doi.org/10.1093/nar/gkab1112 (2021).
Bairoch, A. & Apweiler, R. The SWISS-PROT protein sequence data bank and its supplement TrEMBL in 1999. Nucleic. Acids Res. 27, 49–54, https://doi.org/10.1093/nar/27.1.49 (1999).
Hernández-Plaza, A. et al. eggNOG 6.0: enabling comparative genomics across 12535 organisms. Nucleic. Acids Res. 51, D389–D394, https://doi.org/10.1093/nar/gkac1022 (2022).
Finn, R. D. et al. Pfam: the protein families database. Nucleic. Acids Res. 42, D222–D230, https://doi.org/10.1093/nar/gkt1223 (2013).
Kent, W. J. BLAT—The BLAST-Like Alignment Tool. Genome Res. 12, 656–664, https://doi.org/10.1101/gr.229202 (2002).
Tang, H. B. et al. Synteny and Collinearity in Plant Genomes. Science 320, 486–488, https://doi.org/10.1126/science.1153917 (2008).
Liao, S. NGDC Genome Sequence Archive (GSA) https://ngdc.cncb.ac.cn/gsa/browse/CRA018006 (2024).
Liao, S. NCBI GenBank https://identifiers.org/ncbi/insdc.gca:GCA_042919405.1 (2024).
Liao, S. A high-quality chromosome-level genome assembly of Ficus benjamina (Moraceae), a fig tree with great ecological and ornamental value https://doi.org/10.6084/m9.figshare.27980945 (2024).
Simão, F. A., Waterhouse, R. M., Ioannidis, P., Kriventseva, E. V. & Zdobnov, E. M. BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 31, 3210–3212, https://doi.org/10.1093/bioinformatics/btv351 (2015).
Ou, S. J., Chen, J. F. & Jiang, N. Assessing genome assembly quality using the LTR Assembly Index (LAI). Nucleic. Acids Res. 46, e126–e126, https://doi.org/10.1093/nar/gky730 (2018).
Li, H. & Durbin, R. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformaticsc 25, 1754–1760, https://doi.org/10.1093/bioinformatics/btp324 (2009).
Li, H. Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics 34, 3094–3100, https://doi.org/10.1093/bioinformatics/bty191 (2018).
Rhie, A., Walenz, B. P., Koren, S. & Phillippy, A. M. Merqury: reference-free quality, completeness, and phasing assessment for genome assemblies. Genome Biol. 21, 245, https://doi.org/10.1186/s13059-020-02134-9 (2020).
Acknowledgements
This research was funded by grants from the National Natural Science Foundation of China (32300178 & 32261123001), Guangdong Flagship Project of Basic and Applied Basic Research (2023B0303050001), a fellowship from the China Postdoctoral Science Foundation (2024M753278) and a grant from Guangzhou Collaborative Innovation Center on Science-Tech of Ecology and Landscape (202206010058) to S.L.
Author information
Authors and Affiliations
Contributions
S.P.D. and Y.F.D. conceived the project and supervised this study. S.L., Y.Q.P. and Y.F.D. provided financial support. S.L. collected samples. S.L., Z.Z. and C.X.Y. performed genome analyses. S.L., Z.Z., C.X.Y., E.M.G., Y.Q.P. and Y.M.X. wrote the manuscript. All authors read, revised and approved the final manuscript for submission.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Liao, S., Zhang, Z., Yang, C. et al. A chromosome-level genome assembly of Ficus benjamina, a fig tree with great ecological and ornamental value. Sci Data 12, 824 (2025). https://doi.org/10.1038/s41597-025-05155-w
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41597-025-05155-w
