
Abstract
Apple and stone fruit trees are vital components of Ontario’s agricultural landscape. However, since 2016, these trees have been facing alarming mortality rates, exhibiting symptoms collectively referred to as Fruit Tree Decline (FTD) and Rapid Apple Decline (RAD). Despite its widespread occurrence, the exact cause of FTD and RAD remains elusive, with various pathogenic fungi and viruses implicated, along with abiotic stressors such as drought, winter injury and nutrient deficiency. In this study, we sequenced, assembled and annotated the genomes of 12 fungi associated with FTD and RAD syndromes in Ontario, Canada. We present the first and only publicly available assemblies for three ascomycete species including Diplodia intermedia, Diatrype stigma, and Nothophoma quercina. Additionally, we present high-quality reference genome sequences for Diplodia seriata, Didymella pomorum and Neofusicoccum ribis. These genomic resources are valuable for understanding the molecular mechanisms behind FTD and RAD, and for developing strategies for disease prevention and management in fruit trees.
Similar content being viewed by others


Background & Summary
Apple and stone fruit trees are integral to Ontario’s agriculture economy. The primary areas for apple production in Ontario are located along the shores of Lake Ontario, Lake Erie, Lake Huron and the Georgian Bay. Recent years have seen a concerning downturn, with apple cultivation decreasing to 6,414 hectares in 2023, a 28% drop from the 8,903 hectares recorded in 20021. Similarly, the Niagara region, responsible for over 90% of Ontario’s stone fruit production, has seen a decrease in cultivation area, down 37% from 5,080 hectares in 2007 to 3,192 hectares in 20231.
Since 2016, both apple and stone fruit trees have faced alarming levels of mortality in Ontario, with reports of up to 42% mortality in apples and up to 72% in stone fruits, particularly affecting trees aged between 2 to 10 years2,3,4,5,6,7,8. Symptoms of decline vary, from leaf discoloration and canker development in apples to progressive dieback in stone fruits. These symptoms collectively define Fruit Tree Decline (FTD), with Rapid Apple Decline (RAD) specifically identified in apples9. The decline observed in apple and stone fruit trees extends beyond Ontario, echoing in other regions across North America10. While the exact cause of FTD remains elusive, the emergence of new pathogenic fungi2,3,4,5,6,7,8,11 and viruses12,13,14, as well as the re-emergence of known pathogens15,16,17, compounded by unexpected abiotic stressors such as drought stress, heat waves, floods, winter injury, and nutrient deficiency, are thought to play a significant role in its development9,18,19.
Fungal pathogenicity in a plant depends on multiple factors, including the specific traits of the pathogen, the plant’s defenses, and the conditions within their shared environment20. While weak pathogens may individually induce negligible FTD symptoms, the severity of the disease can escalate under the influence of other biotic or abiotic stresses. Understanding these interactions and the underlying genetic mechanisms is key for effective disease management.
Sequencing fungal genomes lays the foundation for exploring their ecological niche, evolutionary patterns, and disease-causing capabilities21. By deciphering the genetic makeup of these fungi, we aim to improve disease diagnosis, elucidate pathogenic mechanisms, and support the development of targeted strategies for disease prevention and management22. In the present study, we sequenced, de novo assembled and annotated the genomes of 12 fungi associated with FTD and RAD syndromes in Ontario, Canada, spanning a spectrum from pathogens to weak pathogens and non-pathogens/endophytes. By identifying genetic differences between pathogens and non-pathogens, we can gain a stronger understanding into the genetic determinants driving pathogenic evolution23. This knowledge is required for developing predictive models for fungal pathogen emergence and designing effective control measures.
This study provides the first assembled and annotated genomes for the Ascomycetes species Diplodia intermedia, Diatrype stigma, and Nothophoma quercina. These represent the only genome assemblies currently available for the Diatrype and Nothophoma genera. We also present high-quality and well-annotated reference genome for Diplodia seriata, Didymella pomorum and Neofusicoccum ribis. These new genomic resources significantly enhance our ability to study fungal pathogenicity in agricultural systems and inform the development of targeted control measures against pathogens. Moreover, the availability of these assemblies enables downstream functional analyses, including the identification of effector proteins, which are key virulence factors used by pathogens to facilitate host colonization24. Since effector characterization can inform resistance breeding and integrated disease management strategies, this work supports future discovery efforts and comparative genomic studies aimed at improving the resilience and sustainability of fruit production systems.
Methods
Sample collection, fungal isolation and identification
Between 2018 and 2021, wood samples were collected from 25 apple, 30 apricot, six peach, and six nectarine trees showing extensive symptoms of tree fruit decline across 15 commercial orchards in Ontario. Tree mortality rates reached up to 42% in apples and up to 72% in stone fruits, with the highest impact observed in trees between 2 and 10 years of age across the sampled orchards. Small sections of diseased wood (1 cm long) underwent surface sterilization with 70% ethanol for 30 seconds, followed by treatment with 1% NaClO for 20 minutes. After rinsing thrice in sterile water, the wood samples were placed on 2% potato dextrose agar (PDA, Difco™, Franklin Lakes, NJ, USA) with kanamycin (50 mg/L) and incubated at 22 °C for 5 days in darkness. Fungal colonies consistently isolated underwent hyphal-tip transfer to individual PDA plates, then incubated at 22 °C for 7 days in darkness. Purified isolates were morphologically classified into morphotypes before molecular identification.
Genomic DNA was extracted from the mycelium grown on PDA of 7-day-old cultures of one representative isolate from each morphotype group using the Plant/Fungi DNA Isolation Kit (Norgen Biotech, ON, Canada, cat. no. 26200), following the manufacturer’s instructions with slight modifications. Specifically, fungal tissue was vortexed for 15 minutes with 1 mm glass beads in 500 μL of lysis buffer supplemented with 1 μL of RNase A, followed by incubation at 65 °C for 10 minutes. Subsequently, 100 μL of Binding Buffer I was added, the mixture was thoroughly mixed, and incubated on ice for 5 minutes before centrifugation at 10,000 rpm to separate the lysate from the beads and biomass. Resin drying during the column wash was achieved by spinning for 10 minutes at 14,000 rpm. Finally, DNA was eluted at 10,000 rpm for 2 minutes.
Polymerase chain reaction (PCR) was performed in a VWR PCR Thermal Cycler XT96 Gradient (VWR, USA) under the following conditions: 98 °C for 30 sec; 35 × (98 °C, 10 sec; 60 °C, 30 sec; 72 °C, 30 sec); 72 °C, 2 min. The internal transcribed spacer (ITS), translation elongation factor 1-α (EF1-α) and β-tubulin (TUB2) gene regions were each amplified in separate PCR reactions using the primers ITS1/ITS425, EF1-728 F/EF1-986R26 and Bt2a/Bt2b27, respectively. Each 30 μL reaction contained 9 μL of nuclease-free Water, 15 μL of Q5 Hot Start High-Fidelity 2X Master Mix (New England Biolabs, ON, Canada, cat. no. M0494S), 3 μL of extracted genomic DNA and 1.5 μL (10 μM stock) of the appropriate fungal-specific primers. Reactions were performed with negative controls containing no DNA.
The quality of the PCR products was examined using electrophoresis in 1% agarose gel. Sanger sequencing was carried out at Genome Quebec’s Sequencing Facility (Montreal, Canada). Sequences were analyzed with the Basic Local Alignment Search Tool (BLAST)28 against the nucleotide database for an early approximation of species classification.
DNA isolation and genome sequencing
Genomic DNA was extracted from fungal mycelium obtained from 12 pure cultures of fungi identified as previously described. These fungi include strains such as Botryosphaeria dothidea, Cytospora paraplurivora, Diaporthe eres, Diatrype stigma, Didymella pomorum, Diplodia intermedia, Diplodia seriata, Neofusicoccum ribis, Nothophoma quercina, and Paraconiothyrium brasiliense, all isolated from symptomatic apple, apricot, and nectarine trees (Table 1). Eleven of the 12 strains were deposited at the Canadian Collection of Fungal Cultures (DAOMC), each assigned a unique identification number as detailed in Table 1. The mycelium was cultured in potato dextrose broth (PDB, Difco™, Franklin Lakes, NJ, USA) for 5 days at room temperature on an orbital shaker (3 g), followed by filtration through sterilized WhatmanTM Qualitative filter paper grade 1 (Cytiva, Marlborough, MA, USA), freeze-drying for 48 hours using a Benchtop Freeze Dry System (Labconco FreeZone® 4.5 Liter, Kansas City, MO, USA), and bead beating using 1 mm glass beads until a powdered form was obtained. Subsequently, the modified DNA extraction protocol, as previously described (Norgen Biotech, Thorold, ON, Canada), was employed.
Sequencing strategies were designed to species abundance and data requirements. Diplodia seriata FDS-637, one of the most abundant species in the study, was sequenced using both PacBio and Illumina platforms to generate high-quality long-read data for comprehensive genome assembly. Two additional abundant species, Cytospora paraplurivora FDS-564 and Diaporthe eres M63-4 were sequenced using PacBio only, while the remaining nine isolates were sequenced using Illumina, which provided sufficient resolution.
Genomic DNA (gDNA) libraries for Cytospora paraplurivora FDS-564, Diaporthe eres M63-4, and Diplodia seriata FDS-637 were constructed and sequenced at the SickKids sequencing facility (Toronto, ON, Canada). Combinations of barcoded primers and SMRTbell adapters were used to prepare the samples, which were afterward pooled. Long reads for genome assembly were generated from a library prepared with 5 µg of unsheared gDNA using the Express Template Prep Kit (v2.0) (Pacific Biosciences, Menlo Park, CA). A post-library size-selection step targeting fragments > 14 kb was performed using the Agilent 4200 TapeStation System (Agilent, Waldbronn, Germany). The final size-selected library was sequenced on a PacBio Sequel® II sequencer using a single 8 M SMRT Cell in continuous long-read (CLR) mode with a 15-hour movie acquisition time. Raw reads were processed using PacBio’s P-filter to remove low-quality reads and adapter sequences.
Illumina gDNA libraries for Diplodia seriata FDS-637 and the remaining nine fungal isolates were prepared and sequenced at the Centre d’Expertise et de Services, Génome Québec, McGill University (Montréal, QC, Canada). Genomic DNA was quantified using the Quant-iT™ PicoGreen® dsDNA Assay Kit (Life Technologies™, Grand Island, NY, USA), and its integrity was assessed on a TapeStation 2200 (Agilent Technologies, Inc. Santa Clara, CA, USA). Libraries were prepared using the NEBNext Ultra II DNA Library Prep Kit for Illumina (New England Biolabs, Whitby, ON, Canada) according to the manufacturer’s instructions. Adapters and PCR primers were purchased from Integrated DNA Technologies (IDT, Coralville, IA, USA). Size selection to obtain the desired insert size was performed using sparQ beads (Quantabio, Beverly, MA, USA). Library quantification was carried out using the KAPA Library Quantification Universal Complete Kit (Kapa Biosystems; Wilmington, MA, USA), and average fragment size was determined using a LabChip GX II instrument (PerkinElmer, Waltham, MA, USA). Libraries were normalized, pooled, denatured with 0.02 N NaOH, and neutralized using HT1 buffer. The pooled libraries were loaded at 225 pM on an Illumina NovaSeq 6000 S4 flow cell using the Xp protocol, following the manufacturer’s guidelines. Sequencing was conducted in paired-end mode (2 × 150 bp cycles). A 1% PhiX control library was spiked into the run. Base calling was performed using Illumina Real-Time Analysis (RTA) software (v3), and demultiplexing and FASTQ file generation were done using bcl2fastq2 Conversion Software (v2.20) (Illumina, San Diego, CA, USA).
Genome de novo assembly and annotation
Illumina NovaSeq reads (shown in Table 1) were trimmed with Trimmomatic (v0.38.1)29. The ILLUMINACLIP setting was used to remove adapter sequences, and a sliding window approach (4 bp window with an average quality threshold of Q = 20) was applied to trim low quality base calls. The quality of the reads were analyzed before and after Trimmomatic by FastQC (v0.72). Trimmed reads were assembled and gaps were filled using SPAdes (v3.12.0)30 with the K-mer values 21, 33, 45, 69, 81, 93, 105, and 117.
PacBio reads for Cytospora paraplurivora FDS-564, Diaporthe eres M63-4 and Diplodia seriata FDS-637 were trimmed and assembled using Canu (v2.1.1)29 with default settings, as described by Ilyukhin et al.8. For Diplodia seriata FDS-637, Illumina paired-end reads were aligned to the Canu-assembled PacBio genome using Burrow Wheeler Alignment (BWA (v0.7.17))31 with default settings to generate a SAM file. This file was then converted to a sorted BAM file using SAMtools (v1.14)32, which served as input for Pilon (v1.23)33 to correct base-level errors and enhance the overall accuracy of the Canu assembly. Quast (v5.0.2)34 was used to determine assembly statistics and genome coverage (Table 1).
The Funannotate (v1.8.18)35 pipeline was used for genome annotation following the recommended instructions (https://funannotate.readthedocs.io) for gene prediction. All assemblies were cleaned and masked using Funannotate default settings, which utilize Tantan (v40)36 (https://gitlab.com/mcfrith/tantan) for repeat masking. Subsequently, Benchmarking Universal Copy Orthologs (BUSCO (v2.0.0))37 was run on all genomes using the dikarya_odb9 dataset and its results were used to train GlimmerHMM (v3.0.4)38, SNAP (2006-07-28)39, and Augustus v3.3.340 for ab initio gene prediction. Gene predictions were generated using the aforementioned trained gene models and GeneMark-ES (v4.59)41. Weights were assigned to each prediction, based on the source and the strength of each prediction, and passed into EVidenceModeler42, which was used to generate a consensus gene model. Finally, tRNAscan-SE43 was used to generate tRNA predictions.
Functional Annotation was performed using the funannotate wrapper through which the results of numerous analyses were combined. Secondary metabolic gene clusters were predicted using antiSMASH (v6.0)44. The number of each category of biosynthetic gene clusters, found by antiSMASH are listed in Table 2. Interproscan (v5)45, eggnog-mapper (v2)46 (utilizing eggNOG (v5.0)47), HMMer3 searches against Pfam (v32.0)48, dbCAN (v8.0)49; and diamond searches against MEROPS (v12.0)50, uniprot and CAZYmes51 databases were used to annotate protein functional domains. Phobius (v1.01)52 was used to predict protein secretion and transmembrane domains, while signal peptides were predicted using SignalP (v6.0)53. The number of annotations added by these steps is summarized in Table 3.
Genomes were visualized using Circos diagrams to assess quality (Fig. 1). GC skew and GC content were calculated over a sliding window of size 100 kbp. Counts of the canonical telomeric sequences”TTAGGG”, and its reverse complement “CCCTAA” were found over a 50 kbp window. Spikes at the ends of scaffolds likely indicate the presence of telomeres. Scaffolds with telomere count spikes on either side can be found in Paraconiothyrium Brasiliense strain M42-189, Nothophoma quercina strain M97-236 and Diplodia seriata strain FDS-637, indicating that some of the scaffolds in these assemblies may be chromosomes sequence telomere-to-telomere. Functional annotations was assessed by evaluating the number of Gene Ontology (GO) terms (Fig. 2 and Tables S1–S3) and the Clusters of Orthologous Groups of proteins (COGs) present in each annotation, as shown in Fig. 3 and Table S4. Relatively similar percents of different COG groups and GO annotation terms were found between genomes of the same species.

Circos plots for all fungal assemblies in this study, each with 7 rings lettered A to G. Only scaffolds greater than 100 kbp are shown. Ring A illustrates scaffolds in the assembly, with smaller ticks are placed at intervals of 100 kbp, and larger ticks are placed at intervals of 1Mbp. Ring B highlights the coding sequences (CDSs) on the forward strand, while Ring C does the same for the reverse strand and Ring D depicts the tRNAs. Ring E presents the direction and magnitude of GC skew, with black fill signifying an increase in GC skew relative to the average of the entire genome, while gray represents a decrease. Similarly, Ring F presents the GC content, with olive fill representing an increase in GC content over the average of the assembly, while magenta represents a decrease. Ring G plots the counts of telomeric sequence.

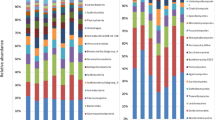
A series of plots representing the number of GO ontology terms in each annotation. In order of top to bottom, the first plot shows a line graph for the number of instances of the three different categories of GO terms: GO process, referring to the biological processes in which a particular gene may play a part; GO function, the activity the product of the gene may possess; and GO component, the location where the gene product is active. The next three plots (in order: GO function, GO component, and GO process) represent the relative percentages of each of the top 15 most common GO terms from their respective category, while terms not in the top 15 are summarized in the “other” group.

Plot of COGs in each annotation. Each bar represents the percent number of each of the COGS (the corresponding y-axis is on the left). The line graph shows the total number of COG annotations in each assembly (with the corresponding y-axis on the right).
Phylogenomic analyses
Genomes of the 12 fungal strains sequenced, assembled, and annotated in this study, along with 90 fungal genomes downloaded from NCBI using the get-assemblies tool (GitHub - davised/get_assemblies), were included in the phylogenetic analysis (Fig. 4). All genomes were retrieved by using all genera names of the sequenced organisms in this study as queries, such as “Didymella”, “Paraconiothyrium”, and more. Sixty-one core genes, listed in Table 4, were extracted, aligned and concatenated using the Universal Fungal Core Genes (UFCG) pipeline version 1.0.554. Maximum Likelihood-based phylogenetic analysis of the concatenated sequences was performed using IQ-Tree (v2.2.6)55, with 1,000 bootstrap replicates processed to determine the best-scoring Maximum Likelihood tree. The phylogenetic tree was rooted with Venturia oleaginea Yun35.

Maximum Likelihood phylogenetic tree generated using a concatenated alignment of 61 core genes extracted from the genomes of Dothideomycetes and Sordariomycetes species sourced from NCBI. The 12 fungal genomes obtained in this study are highlighted in bold. The tree was rooted to Venturia oleaginea Yun35. The numbers at each node represent bootstrap support, expressed as percentages.
Data Records
The datasets have been deposited in the Sequenced Read Archive (SRA) under accession numbers: SRX14291869, SRX14155424, SRX14046716, SRX14003295, SRX13989866, SRX13979984, SRX13961038, SRX13940527, SRX13921100, SRX13894123, SRX13883208, SRX13877378, SRX1344580056. All assemblies and annotations can be found on NCBI GenBank under BioProject PRJNA79001357, with the following accession numbers: JAJVDB00000000058, JAJSPL00000000059, JAKOOP00000000060, JAKNSF00000000061, JAKJXP00000000062, JAKJXN00000000063, JAKEKT00000000064, JAJVDA00000000065, JAJVCZ00000000066, JAJVDC00000000067, JAKIXB00000000068, JAKJXO00000000069.
Technical Validation
BUSCO (v5.8.2) employing the Ascomycota_odb12 dataset was used to assess whether core genes were adequately captured in the genomes sequenced. All genomes were of high quality (Table 5) and had greater than 97% of BUSCOs complete.
Code availability
Funannotate-based annotations were automated with the scripts available at the following gitub: https://github.com/Ellouzlab/Fun_pipeline. Bioinformatics programs were used as per their manual or protocol. If settings were omitted, default parameters were used.
References
Statistics Canada. Area, production and farm gate value of marketed fruits: Table 32-10-0364-01, https://doi.org/10.25318/3210036401-eng (2024).
Ellouze, W. & Ilyukhin, E. First report of Diplodia seriata associated with canker and dieback diseases of apricot and nectarine trees in Ontario, Canada. Journal of Plant Pathology 106, 759–760, https://doi.org/10.1007/s42161-023-01579-8 (2024).
Ilyukhin, E. & Ellouze, W. First Report of Neofusicoccum ribis causing cankers and dieback diseases on apricot trees in Canada and worldwide. Plant Disease 108, 222, https://doi.org/10.1094/PDIS-08-23-1588-PDN (2024).
Ellouze, W., Ilyukhin, E., Sulman, M. & Ali, S. First Report of Diplodia intermedia Causing Canker and Dieback Diseases on Apple Trees in Canada. Plant Disease 108, 217, https://doi.org/10.1094/PDIS-07-23-1361-PDN (2024).
Villanueva, O. & Ellouze, W. First report of a Canadian isolate of Phytopythium vexans causing root rot disease on apple and peach under laboratory conditions. New Disease Reports 48, e12195, https://doi.org/10.1002/ndr2.12195 (2023).
Ilyukhin, E., Schneider, K. & Ellouze, W. First Report of Botryosphaeria dothidea Causing Stem Canker and Dieback of Apple Trees in Ontario, Canada. Plant Disease 106, 2994, https://doi.org/10.1094/PDIS-12-21-2838-PDN (2022).
Ilyukhin, E. & Ellouze, W. First report of Phaeobotryon negundinis associated with twig and branch dieback of Malus domestica trees in southern Ontario, Canada and worldwide. Journal of Plant Pathology 105, 355–356, https://doi.org/10.1007/s42161-022-01272-2 (2023).
Ilyukhin, E., Nguyen, H. D. T., Castle, A. J. & Ellouze, W. Cytospora paraplurivora sp. nov. isolated from orchards with fruit tree decline syndrome in Ontario, Canada. PLOS ONE 18, e0279490, https://doi.org/10.1371/journal.pone.0279490 (2023).
Singh, J., Silva, K. J. P., Fuchs, M. & Khan, A. Potential role of weather, soil and plant microbial communities in rapid decline of apple trees. PLOS ONE 14, e0213293, https://doi.org/10.1371/journal.pone.0213293 (2019).
Stokstad, E. Rapid apple decline has researchers stumped. Science 363, 1259–1259, https://doi.org/10.1126/science.363.6433.1259 (2019).
Ali, S., Renderos, W., Bevis, E., Hebb, J. & Abbasi, P. A. Diaporthe eres causes stem cankers and death of young apple rootstocks in Canada. Canadian Journal of Plant Pathology 42, 218–227, https://doi.org/10.1080/07060661.2019.1653377 (2020).
Wright, A. A., Szostek, S. A., Beaver-Kanuya, E. & Harper, S. J. Diversity of three bunya-like viruses infecting apple. Archives of Virology 163, 3339–3343, https://doi.org/10.1007/s00705-018-3999-z (2018).
Liu, H. et al. Characterization of a new apple luteovirus identified by high-throughput sequencing. Virology journal 15, 85, https://doi.org/10.1186/s12985-018-0998-3 (2018).
Xiao, H., Hao, W., Storoschuk, G., MacDonald, J. L. & Sanfaçon, H. Characterizing the virome of apple orchards affected by rapid decline in the Okanagan and Similkameen valleys of British Columbia (Canada). Pathogens 11, 1231, https://doi.org/10.3390/pathogens11111231 (2022).
Gottwald, T. R., Wierenga, E., Luo, W. & Parnell, S. Epidemiology of Plum pox ‘D’ strain in Canada and the USA. Canadian Journal of Plant Pathology 35, 442–457, https://doi.org/10.1080/07060661.2013.844733 (2013).
Gougherty, A. V. & Nutter, F. W. Impact of eradication programs on the temporal and spatial dynamics of Plum pox virus on Prunus spp. in Pennsylvania and Ontario, Canada. Plant Disease 99, 593–603, https://doi.org/10.1094/PDIS-03-14-0224-RE (2015).
Parcey, M. et al. Comparative genomic analysis of Erwinia amylovora reveals novel insights in phylogenetic arrangement, plasmid diversity, and streptomycin resistance. Genomics 112, 3762–3772, https://doi.org/10.1016/j.ygeno.2020.04.001 (2020).
Serrano, A. et al. The comparative root system architecture of declining and non-declining trees in two apple orchards in New York. Plants 12, 2644, https://doi.org/10.3390/plants12142644 (2023).
Xu, H., Hannam, K. D., MacDonald, J. L. & Ediger, D. Field investigation into tree fates from recent apple tree decline: Abrupt hydraulic failure versus gradual hydraulic loss. Stresses 3, 256–269, https://doi.org/10.3390/stresses3010019 (2023).
Termorshuizen, A. J. Ecology of Fungal Plant Pathogens. Microbiol Spectr. 4, 1–11, https://doi.org/10.1128/microbiolspec.funk-0013-2016 (2016).
Aylward, J. et al. A plant pathology perspective of fungal genome sequencing. IMA Fungus 8, 1–15, https://doi.org/10.5598/imafungus.2017.08.01.01 (2017).
Kelman, A. Plant pathology at the crossroads. Annual review of phytopathology 23, 1–12, https://doi.org/10.1146/annurev.py.23.090185.000245 (1985).
Hill, R. et al. Tapping culture collections for fungal endophytes: First genome assemblies for three genera and five species in the Ascomycota. Genome Biology and Evolution 15, https://doi.org/10.1093/gbe/evad038 (2023).
Villanueva, O., Nguyen, H. D. T. & Ellouze, W. Comparative genomic and secretome analysis of Phytophthora capsici strains: Exploring pathogenicity and evolutionary dynamics. Agronomy 14, 2623 https://www.mdpi.com/2073-4395/14/11/2623 (2024).
White, T. J., Bruns, T., Lee, S. & Taylor, J. in PCR Protocols: A Guide to Methods and Applications (eds Innis, M. A., Gelfand, D. H., Sninsky, J. J. & White, T. J.) 315–322 (Academic Press, 1990).
Carbone, I. & Kohn, L. M. A method for designing primer sets for speciation studies in filamentous ascomycetes. Mycologia 91, 553–556, https://doi.org/10.1080/00275514.1999.12061051 (1999).
O’Donnell, K., Kistler, H. C., Cigelnik, E. & Ploetz, R. C. Multiple evolutionary origins of the fungus causing Panama disease of banana: Concordant evidence from nuclear and mitochondrial gene genealogies. Proc. Natl. Acad. Sci. 95, 2044–2049, https://doi.org/10.1073/pnas.95.5.2044 (1998).
Altschul, S. F., Gish, W., Miller, W., Myers, E. W. & Lipman, D. J. Basic local alignment search tool. Journal of Molecular Biology 215, 403–410, https://doi.org/10.1016/S0022-2836(05)80360-2 (1990).
Koren, S. et al. Canu: scalable and accurate long-read assembly via adaptive k-mer weighting and repeat separation. Genome Res 27, 722–736, https://doi.org/10.1101/gr.215087.116 (2017).
Bankevich, A. et al. SPAdes: A new genome assembly algorithm and its applications to single-cell sequencing. Journal of Computational Biology 19, 455–477, https://doi.org/10.1089/cmb.2012.0021 (2012).
Li, H. & Durbin, R. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 25, 1754–1760, https://doi.org/10.1093/bioinformatics/btp324 (2009).
Danecek, P. et al. Twelve years of SAMtools and BCFtools. GigaScience 10, https://doi.org/10.1093/gigascience/giab008 (2021).
Walker, B. J. et al. Pilon: An integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLOS ONE 9, e112963, https://doi.org/10.1371/journal.pone.0112963 (2014).
Mikheenko, A., Prjibelski, A., Saveliev, V., Antipov, D. & Gurevich, A. Versatile genome assembly evaluation with QUAST-LG. Bioinformatics 34, i142–i150, https://doi.org/10.1093/bioinformatics/bty266 (2018).
Palmer, J. M. & Stajich, J. Funannotate v1.8.1: Eukaryotic genome annotation. Zenodo, Zenodo. https://doi.org/10.5281/zenodo.4054262 (2020).
Frith, M. C. A new repeat-masking method enables specific detection of homologous sequences. Nucleic Acids Research 39, e23–e23, https://doi.org/10.1093/nar/gkq1212 (2010).
Waterhouse, R. M. et al. BUSCO Applications from Quality Assessments to Gene Prediction and Phylogenomics. Mol Biol Evol 35, 543–548, https://doi.org/10.1093/molbev/msx319 (2018).
Majoros, W. H., Pertea, M. & Salzberg, S. L. TigrScan and GlimmerHMM: two open source ab initio eukaryotic gene-finders. Bioinformatics 20, 2878–2879, https://doi.org/10.1093/bioinformatics/bth315 (2004).
Korf, I. Gene finding in novel genomes. BMC Bioinformatics 5, 59, https://doi.org/10.1186/1471-2105-5-59 (2004).
Stanke, M., Diekhans, M., Baertsch, R. & Haussler, D. Using native and syntenically mapped cDNA alignments to improve de novo gene finding. Bioinformatics 24, 637–644, https://doi.org/10.1093/bioinformatics/btn013 (2008).
Borodovsky, M. & Lomsadze, A. Eukaryotic gene prediction using GeneMark.hmm-E and GeneMark-ES. Current Protocols in Bioinformatics 35, 4.6.1–4.6.10, https://doi.org/10.1002/0471250953.bi0406s35 (2011).
Haas, B. J. et al. Automated eukaryotic gene structure annotation using EVidenceModeler and the Program to Assemble Spliced Alignments. Genome Biology 9, R7, https://doi.org/10.1186/gb-2008-9-1-r7 (2008).
Chan, P. P. & Lowe, T. M. in Gene Prediction: Methods and Protocols (ed Kollmar, M.) 1–14 (Springer New York, 2019).
Blin, K. et al. antiSMASH 6.0: improving cluster detection and comparison capabilities. Nucleic Acids Research 49, W29–W35, https://doi.org/10.1093/nar/gkab335 (2021).
Quevillon, E. et al. InterProScan: protein domains identifier. Nucleic Acids Res 33, W116–120, https://doi.org/10.1093/nar/gki442 (2005).
Cantalapiedra, C. P., Hernández-Plaza, A., Letunic, I., Bork, P. & Huerta-Cepas, J. eggNOG-mapper v2: Functional annotation, orthology assignments, and domain prediction at the metagenomic scale. Molecular Biology and Evolution 38, 5825–5829, https://doi.org/10.1093/molbev/msab293 (2021).
Huerta-Cepas, J. et al. eggNOG 5.0: a hierarchical, functionally and phylogenetically annotated orthology resource based on 5090 organisms and 2502 viruses. Nucleic Acids Res 47, D309–d314, https://doi.org/10.1093/nar/gky1085 (2019).
El-Gebali, S. et al. The Pfam protein families database in 2019. Nucleic Acids Research 47, D427–D432, https://doi.org/10.1093/nar/gky995 (2019).
Yin, Y. et al. dbCAN: a web resource for automated carbohydrate-active enzyme annotation. Nucleic Acids Res 40, W445–451, https://doi.org/10.1093/nar/gks479 (2012).
Rawlings, N. D., Tolle, D. P. & Barrett, A. J. MEROPS: the peptidase database. Nucleic Acids Res 32, D160–164, https://doi.org/10.1093/nar/gkh071 (2004).
Cantarel, B. L. et al. The Carbohydrate-Active EnZymes database (CAZy): an expert resource for Glycogenomics. Nucleic Acids Research 37, D233–D238, https://doi.org/10.1093/nar/gkn663 (2009).
Käll, L., Krogh, A. & Sonnhammer, E. L. A combined transmembrane topology and signal peptide prediction method. J Mol Biol 338, 1027–1036, https://doi.org/10.1016/j.jmb.2004.03.016 (2004).
Teufel, F. et al. SignalP 6.0 predicts all five types of signal peptides using protein language models. Nat Biotechnol 40, 1023–1025, https://doi.org/10.1038/s41587-021-01156-3 (2022).
Kim, D., Gilchrist, C. L. M., Chun, J. & Steinegger, M. UFCG: database of universal fungal core genes and pipeline for genome-wide phylogenetic analysis of fungi. Nucleic Acids Research 51, D777–D784, https://doi.org/10.1093/nar/gkac894 (2022).
Minh, B. Q. et al. IQ-TREE 2: New models and efficient methods for phylogenetic inference in the genomic era. Molecular Biology and Evolution 37, 1530–1534, https://doi.org/10.1093/molbev/msaa015 (2020).
Sulman, M. et al. Fungi associated with Fruit Tree Decline symptoms in Ontario, Canada. NCBI Sequenced Read Archive https://identifiers.org/ncbi/insdc.sra:SRP351463 (2024).
Sulman, M. et al. Fungi associated with Fruit Tree Decline symptoms in Ontario, Canada. NCBI BioProject https://www.ncbi.nlm.nih.gov/bioproject/PRJNA790013/ (2024).
Sulman, M., Ilyukhin, E., Ali, S. & Ellouze, W. Botryosphaeria dothidea strain M68-17, whole genome shotgun sequencing project. GenBank https://identifiers.org/ncbi/insdc:JAJVDB000000000 (2024).
Sulman, M., Ilyukhin, E., Nguyen, H. D. T., Ali, S. & Ellouze, W. Cytospora paraplurivora strain FDS-564, whole genome shotgun sequencing project. GenBank https://identifiers.org/ncbi/insdc:JAJSPL000000000 (2024).
Sulman, M., Ilyukhin, E., Nguyen, H. D. T., Ali, S. & Ellouze, W. Diaporthe eres strain M63-4, whole genome shotgun sequencing project. GenBank https://identifiers.org/ncbi/insdc:JAKOOP000000000 (2024).
Sulman, M., Ilyukhin, E., Ali, S. & Ellouze, W. Diaporthe eres strain M169, whole genome shotgun sequencing project. GenBank https://identifiers.org/ncbi/insdc:JAKNSF000000000 (2024).
Sulman, M., Ilyukhin, E., Ali, S. & Ellouze, W. Diatrype stigma strain M11/M66-122, whole genome shotgun sequencing project. GenBank https://identifiers.org/ncbi/insdc:JAKJXP000000000 (2024).
Sulman, M., Ilyukhin, E., Ali, S. & Ellouze, W. Didymella pomorum strain M27-16, whole genome shotgun sequencing project. GenBank https://identifiers.org/ncbi/insdc:JAKJXN000000000 (2024).
Sulman, M., Ilyukhin, E., Ali, S. & Ellouze, W. Diplodia intermedia strain M45-28, whole genome shotgun sequencing project. GenBank https://identifiers.org/ncbi/insdc:JAKEKT000000000 (2024).
Sulman, M., Ilyukhin, E., Ali, S. & Ellouze, W. Diplodia seriata strain M28-159, whole genome shotgun sequencing project. GenBank https://identifiers.org/ncbi/insdc:JAJVDA000000000 (2024).
Sulman, M., Ilyukhin, E., Nguyen, H. D. T., Ali, S. & Ellouze, W. Diplodia seriata strain FDS-637, whole genome shotgun sequencing project. https://identifiers.org/ncbi/insdc:JAJVCZ000000000 (2024).
Sulman, M., Ilyukhin, E., Ali, S. & Ellouze, W. Neofusicoccum ribis strain M1-105, whole genome shotgun sequencing project. GenBank https://identifiers.org/ncbi/insdc:JAJVDC000000000 (2024).
Sulman, M., Ilyukhin, E., Ali, S. & Ellouze, W. Nothophoma quercina strain M97-236, whole genome shotgun sequencing project. GenBank https://identifiers.org/ncbi/insdc:JAKIXB000000000 (2024).
Sulman, M., Ilyukhin, E., Ali, S. & Ellouze, W. Paraconiothyrium brasiliense strain M42-189, whole genome shotgun sequencing project. GenBank https://identifiers.org/ncbi/insdc:JAKJXO000000000 (2024).
Acknowledgements
The authors gratefully acknowledge the Ontario Fruit & Vegetable Growers association, the Ontario Apple Growers organization and the growers who generously provided access to their orchards for sample collection. We extend our sincere thanks to Kathryn Carter and Kristy Grigg-McGuffin, Horticulture IPM Specialists with the Ontario Ministry of Agriculture, Food and Rural Affairs (OMAFRA), for their assistance during the sample collection process. We also sincerely thank Karin Schneider for her technical assistance. Illumina genome sequencing was performed by the Centre d’Expertise et de Services, Génome Québec, McGill University (Montréal, QC, Canada), and PacBio genome sequencing was conducted by The Hospital for Sick Children (SickKids, Toronto, ON, Canada). This research was funded by Agriculture and Agri-Food Canada under grant number J-002199.
Author information
Authors and Affiliations
Contributions
M.S.: Conceptualization, methodology, experimentation, investigation, data analysis and visualization, data curation, writing original draft, review and editing. E.I.: Conceptualization, methodology, experimentation, data analysis, data curation, review and editing. O.V.: Experimentation, review and editing. H.N.: Data analysis, review and editing. S.A.: Conceptualization, funding acquisition, review and editing. W.E.: Conceptualization, funding acquisition, resources, project administration, supervision, data curation, writing original draft, review and editing, corresponding author.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Sulman, M., Ilyukhin, E., Villanueva, O. et al. De novo Genome Assembly and Annotation of 12 Fungi Associated with Fruit Tree Decline Syndrome in ON, Canada. Sci Data 12, 1098 (2025). https://doi.org/10.1038/s41597-025-05192-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41597-025-05192-5
