
Abstract
Heteroaryl substituents are important building blocks of functionalized organic compounds in drug discovery, materials science, and catalysis. Quantitative descriptions of steric and electronic properties of heteroaryl substituents are essential in establishing structure-activity relationships and predicting reactivity and properties of heteroaromatic compounds. We introduce HArD, the HeteroAryl Descriptors database, comprising DFT-computed steric and electronic descriptors of >31,500 heteroaryl substituents. The database features heteroaryl substituents comprising 5- and 6-membered rings as well as 5,6- and 6,6-fused ring systems. Different regioisomers and additional substituents on the heteroaromatic ring were included to describe the diverse chemical space of heteroaryl substituents. The database includes 65 descriptors such as buried volume and Sterimol parameters to describe steric effects, atomic charges and HOMO/LUMO coefficients and energies to describe electronic effects, and Harmonic Oscillator Model of Aromaticity (HOMA) values describing aromaticity. In addition, we developed Hammett-type heteroaryl substituent constants (σHet) based on computed heteroaryl carboxylic acid pKa values, which aim to extend the broadly used Hammett constants (σp and σm) from substituted phenyl groups to substituted heteroaryl groups.
Similar content being viewed by others

Background & Summary
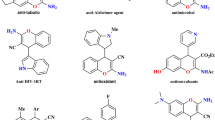
Heteroaryl groups are functional groups in organic molecules that contain a heteroaromatic ring with at least one heteroatom, such as nitrogen, oxygen, or sulphur. They are naturally abundant and widely utilized in functionalized organic molecules. For example, pyridine, one of the most prevalent nitrogen-containing heteroarenes, is present in 54 small molecule drugs approved by the FDA between 2013 and 20231. Heteroaryls are also common structural motifs in both coupling partners and catalysts in transition metal catalysis and organocatalysis2,3,4,5. The prevalence of heteroaromatic compounds is attributed to several factors. First, different heteroarene cores have distinct electronic and steric properties that effectively alter the target compound’s chemical reactivity and biological function. One area where these properties have been leveraged is in the design of covalent modifier drugs6,7,8 with warhead reactivity modulated by heteroaryl groups; examples of this include FDA-approved drugs afatinib and selinexor, as well as other covalent modifiers such as roblitinib, nitrofuran derivative C-176, and TC9-305 (Fig. 1a)7. Second, heteroarene cores could be further functionalized with electron-donating or electron-withdrawing substituents at different sites9,10, leading to a large number of regioisomers with a substantially expanded property space of heteroaromatic compounds. Third, structurally diverse functionalized heteroarenes could be synthesized from readily available starting materials via a number of established synthetic methods, including recently developed site-selective functionalization4,9,10,11 and skeletal editing12 strategies.

Overview and background of this work. (a) Selected covalent modifiers possessing warheads modulated by heteroaryl groups7. (b) Descriptors for aryl and heteroaryl substituents. (c) This work: HArD (HeteroAryl Descriptors) database.
Quantitative description of the intrinsic steric and electronic properties of heteroaryl substituents is essential for establishing structure-activity relationships (SAR) and machine learning models for heteroaromatic compounds used in drug discovery and reaction design. DFT-computed descriptors are widely used in catalyst design13,14,15, materials science16,17, and reactivity and selectivity predictions18,19. Descriptors for heteroaromatic compounds such as HOMO/LUMO orbital coefficients/energies20,21 and atomic charges22,23 have been applied to various reaction types, including electrophilic20,22,23,24,25 and nucleophilic21,26 aromatic substitution, C–N cross-coupling27, and radical C–H functionalization28. Despite these advances, a systematic and comprehensive database that integrates various physical-organic descriptors for heteroaryl substituents is still lacking, which has hindered the development of reactivity and selectivity prediction models. In contrast to the broadly used Hammett substituent constants (σp and σm)29 to describe electronic properties of aryl substituents, similar universal electronic descriptors for heteroaryl substituents have not been developed (Fig. 1b). This is in part due to the lack of experimental data (i.e., pKa values of corresponding heteroaryl carboxylic acids)30 as well as the inherent complexity of heteroaryl groups with different ring types, heteroatom substitutions, and regioisomers. We expect that a database of steric and electronic descriptors of heteroaryl substituents could serve as a foundation for developing robust predictive models that expand to the entire chemical space of heteroaromatic compounds. These could streamline reaction and catalyst developments by enabling the rational selection of heteroaryl substituents based on their electronic and steric features, rather than relying on trial-and-error approaches. In addition, this database for intrinsic chemical reactivity factors would complement existing cheminformatics databases for heteroarene synthetic feasibility31 and ADMET properties32, which have been broadly used in drug discovery.
Here, we present HArD, the HeteroAryl Descriptors database of >31,500 heteroaryl substituents based on 238 commercially available parent heteroarene cores (Fig. 1c). To capture the structural diversity of heteroaryl groups, we included both 5- and 6-membered heteroaromatic rings as well as 5,6- and 6,6-fused ring systems with carbon, nitrogen, oxygen, and sulphur as possible heavy atoms in the ring scaffold (Fig. 2a). Each parent heteroarene was functionalized with commonly used electron-withdrawing and electron-donating substituents to give monosubstituted heteroaryl groups (Fig. 2b). For each heteroaryl substituent, 49 DFT-computed electronic, steric, and geometrical descriptors and 16 fingerprint-type descriptors were included (Fig. 2c,d). This database includes computed Hammett-type substituent constants for heteroaryls (σHet), which would allow straightforward extensions of existing SAR and ML models of aryl compounds based on Hammett substituent constants (σp and σm) into previously unexplored space of heteroaryl-containing compounds. These newly developed σHet electronic parameters were computed based on pKa values of corresponding heteroaryl carboxylic acids (Fig. 3a), in analogy to the original definition of Hammett constants for aryl substituents to enable backward compatibility. In addition, other previously used descriptors, such as HOMO/LUMO coefficients, HOMO/LUMO energies, and partial atomic charges have also been computed for all heteroaryl groups in the database. Overall, HArD not only bridges a critical gap in the quantitative characterization of heteroaryl substituents but also provides a practical tool to design and predict the properties of these key building blocks in drug discovery, catalysis, and materials science.

Workflow to generate the database. The HArD database was created by collecting heteroaryl cores, enumerating possible substituents, and performing high-throughput DFT calculations to provide a set of steric and electronic descriptors. (a) 238 commercially available N-, O-, and S-containing heteroarenes from Reaxys®. (b) SMILES enumeration via RDKit to form approximately 31,500 monosubstituted groups. (c) High-throughput DFT calculations for various descriptors. (d) Descriptors included in the HArD database.

Overview of selected electronic and steric descriptors. (a) Definition of Hammett-type substituent constant (σHet) for heteroaryl groups as an electronic descriptor. (b) Distribution of σHet in the database. (c) σHet of selected heteroaryl groups. (d) Examples demonstrating different steric and electronic properties between two heteroaryl regioisomers.
Methods
Establishing the heteroaryl library
Parent heteroarene cores were selected based on commercially available unsubstituted heteroaromatic compounds with 5- and 6-membered rings, as well as 5,6- and 6,6-fused ring systems from the Reaxys® database (reaxys.com) (Fig. 2a). Only compounds with C, N, O, and S atoms in the heteroaromatic rings were included. A total of 238 unsubstituted parent heteroarenes were selected, including 23 five-membered heteroarenes, 9 six-membered heteroarenes, 157 5,6-fused rings, 47 6,6-fused rings, plus benzene and naphthalene. This resulted in 812 regioisomers of unsubstituted heteroaryl groups. Next, each unsubstituted heteroaryl group was functionalized using the RDKit33 “ReactionFromSmarts” function to substitute a C–H bond on the heteroaromatic ring with a substituent to generate monosubstituted heteroaryl groups. The substituents used include 12 common electron-donating and electron-withdrawing groups—NMe2, NH2, OH, OMe, Me, TMS, F, Cl, Br, Ac, CN, and NO2. This resulted in approximately 31,500 unique heteroaryl groups (Fig. 2b). To calculate the steric and electronic properties of each heteroaryl group (ArHet), SMILES strings of three compounds were used, including ArHet–H, ArHet–CO2H, and ArHet–\({{\rm{CO}}}_{2}^{-}\). The RDKit Experimental-Torsion Distance Geometry (ETDG) method34 was used to generate 3D structures as Gaussian 1635 input files for subsequent DFT calculations.
Density functional theory (DFT) calculations
Geometries of all structures were optimized using the dispersion-corrected36,37 B3LYP-D3(BJ) functional38,39 with the 6–31 + G(d) basis set using the Gaussian 16 program35 (Fig. 2c). Vibrational frequency calculations were performed at the same level of theory as the geometry optimization to confirm that each structure is a local minimum (i.e., with no imaginary frequencies). Single-point energy calculations were carried out using the M06-2X functional40 with the 6–31 + G(d) basis set. Solvation energy corrections were calculated using the SMD solvation model41 in single-point energy calculations with water as the solvent. Carboxylic acids (ArHet–CO2H) and carboxylate anions (ArHet–\({{\rm{CO}}}_{2}^{-}\)) may have several conformers depending on whether the carboxylic acid or carboxylate group is coplanar with the heteroaromatic ring. The “SetDihedralDeg” function in RDKit was used to generate conformers of carboxylic acids and carboxylate anions by rotating about the Cipso − Ccarbonyl bond. Only the lowest energy conformer of each structure was used to compute the reported properties. The Automated Quantum Mechanical Environments (AQME) software42 was used in post-processing to check for self-consistent field (SCF) and geometry optimization convergence errors and imaginary frequencies. Calculations with convergence errors were resubmitted by using the intermediate structure during the previous geometry optimization with the lowest root-mean-square gradient as the input geometry. In cases where imaginary frequencies were present, the calculations were adjusted by slightly perturbing the geometry and resubmitted with the keyword “opt = (calcfc,maxstep = 5)”. This automated process was repeated twice, and any calculations still showing errors after the attempted recalculations were not included in the final database.
Descriptor acquisition
Hammett-type substituent constants for heteroaryl groups (σHet)
Hammett-type substituent constants for heteroaryls were calculated from the difference between the aqueous pKa values of the corresponding heteroaryl carboxylic acid, pKa(Het), and benzoic acid, pKa(Ph), as a reference (Fig. 3a).
The pKa values for benzoic acid and each heteroaryl carboxylic acid were calculated from
where R is the gas constant and T is 298.15 K. The value −270.29 kcal/mol was used for the Gibbs free energy of a proton in aqueous solution (\(\triangle {G}_{{H}^{+}}\)). This is calculated from the sum of the gas-phase free energy of proton (−6.28 kcal/mol), its hydration free energy43 (−265.9 kcal/mol), and a +1.89 kcal/mol correction for standard state conversion. The Gibbs free energies (∆G) of the carboxylate anion and the carboxylic acid were calculated using DFT at the M06-2X/6-31+G(d)/SMD(sSAS,H2O)//B3LYP-D3(BJ)/6-31+G(d) level of theory under standard conditions (i.e., 298.15 K, 1 mol/L). A modified scaled solvent-accessible surface (sSAS) approach was used in the SMD solvation model because it has been demonstrated to improve the accuracy of pKa calculations of carboxylic acids44. The computed σ values of substituted aryl groups have a good linear correlation with the experimentally derived σ values in the literature (R2 = 0.87; MAE = 0.11). Nonetheless, due to the error of the pKa calculations, we note that σHet values should be compared with the computed σ values of substituted aryl groups, which are also included in our database, rather than the experimental σ values when directly comparing the electronic properties of heteroaryl and aryl substituents.
Similar to Hammett constants for substituted aryl groups, a negative σHet value indicates a more electron-donating heteroaryl compared to phenyl, whereas a positive σHet indicates a more electron-withdrawing heteroaryl. While traditional aryl Hammett substituent constants often fall in a range of approximately −1 to +1, σHet values exhibit a broader range (Fig. 3b,c), showcasing the electronic diversity of heteroaryl groups. Regioisomers of the same parent heteroarene could offer significant variance in their σHet values (e.g., 0.91, 0.71, and 1.33 for 2-pyridyl, 3-pyridyl, and 4-pyridyl, respectively, Fig. 3c). Further, while 5-membered rings such as pyrrole are known to often be more electron-donating than 6-membered rings such as pyridine, these rings can be tuned using electron-withdrawing groups to alter their electronic properties (Fig. 3c).
Other electronic descriptors
Electronic properties such as HOMO and LUMO energies, total dipole moments, and quadrupole moments were extracted from Gaussian 16 single-point energy output files using the Python-based cclib library. From these values, the chemical potential, HOMO-LUMO gap, global electrophilicity, and global nucleophilicity were derived. The chemical potential was calculated from the average of the HOMO and LUMO energies45, while the HOMO-LUMO gap is the energy difference between these orbitals. Global electrophilicity (electrophilicity index) was determined using the formula \(\omega =\frac{{\mu }^{2}}{2\eta }\) where μ is the chemical potential and η is the HOMO-LUMO gap46. Global nucleophilicity (nucleophilicity index) was computed as the inverse of global electrophilicity (\(N=\frac{1}{\omega }\)). The HOMO and LUMO coefficients of the ipso carbon were extracted directly from the Gaussian 16 outputs (keyword: pop = (orbitals = 10, ThreshOrbitals = 5)). Partial atomic charges were computed using Natural Population Analysis (NPA)47, Hirshfeld48, and Charge Model 5 (CM5)49 charge schemes for the ipso carbon and the sum of the atomic charges of the heteroaryl group in the ArHet–H, ArHet–CO2H, and ArHet–\({{\rm{CO}}}_{2}^{-}\) compounds.
Steric and geometrical descriptors
All steric descriptors were computed using the MORFEUS program50. These include Sterimol parameters, buried volume, distal volume, Cipso–H bond length in ArHet–H, and Cipso–Ccarbonyl bond lengths in ArHet–CO2H and ArHet–\({{\rm{CO}}}_{2}^{-}\). Sterimol parameters, developed by Verloop, describe substituent size51,52. The Sterimol length (L) is the vector length from the hydrogen on the ipso carbon of ArHet–H through the carbon to the tangent of the van der Waals (vdW) surface. B1 and B5 are the minimum and maximum widths, defined by the shortest and longest vectors from the ipso carbon to the vdW surface and perpendicular to L. Buried volume was originally developed to quantify the steric hindrance caused by ligands in transition metal complexes53,54. Here, the fraction buried volume (VBur) was calculated from the percentage of space occupied by the ArHet substituent within a sphere of 3.5 Å radius centred on the ipso carbon (Bondi radii, 0.10 Å mesh spacing, and excluding hydrogen atoms) (Fig. 3d). The distal volume describes the volume occupied by ArHet outside of this sphere. Using this approach, the computed steric descriptors could distinguish between different regioisomers (e.g., 3-bromo-2-pyridyl vs. 3-bromo-4-pyridyl, Fig. 3d). The universal quantitative dispersion descriptor (Pint) was derived by constructing a molecular vdW surface and calculating dispersion coefficients using Grimme’s D3 dispersion correction method36,55. The solvent-accessible surface area (SASA) of the ipso carbon of ArHet was determined using the double cubic lattice method (DCLM) algorithm, which applies a constant surface density of points using a 1.4 Å probe56. The volume enclosed by this solvent-accessible surface is also computed57.
HOMA aromaticity descriptor
The Harmonic Oscillator Model of Aromaticity (HOMA) descriptor58,59 was calculated using the bond lengths of the heteroaromatic rings (\({R}_{i}\)) from the DFT-optimized geometries:
where Ropt is the optimal bond length for a reference aromatic bond60, n the total number of bonds within the ring, and α serves as a normalization factor60 to scale the result to a value between 1 and 0, where 1 indicates perfectly aromatic benzene and 0 represents the hypothetical Kekulé structure of a nonaromatic 1,3,5-cyclohexatriene ring. The Ropt and α values for CC, CN, CO, CS, NN, and NO bonds were taken from the literature60. For the NS bond, the optimal bond length Ropt = 1.61 Å was also taken from the literature61, whereas the \(\alpha \) (71.875 Å−2) was calculated according to standard procedures58.
Fingerprint descriptors
In addition to DFT-computed descriptors, we included fingerprint-type descriptors relevant to drug discovery applications using RDKit33. These include the Wildman-Crippen partition coefficient (Log P)62, topological polar surface area (TPSA)63, number of hydrogen bond donors/acceptors, molecular weight, number of heavy atoms, and fraction of sp2- and sp3-hybridized carbons.
Data Records
The HeteroAryl Descriptors (HArD) database, along with a Python script for data processing, is available on a publicly accessible FigShare repository64 and GitHub repository (github.com/turkiAlturaifi/HArD). The repository includes the scripts used to generate the database, as well as an Excel file (hard.xlsx) and a database file (hard.db) listing the SMILES representations of monosubstituted heteroaryls along with their associated descriptors. The Excel file (hard.xlsx) contains two sheets. The “database” sheet includes 72 columns, 65 of which correspond to molecular descriptors: 38 electronic, 11 geometric/steric, and 16 fingerprint-type descriptors. The remaining columns provide structural identity information, including the molecule ID, the name of the parent heteroarene, and SMILES strings for the heteroaryl group (ArHet), the unsubstituted heteroarene (ArHet–H), as well as ArHet–CO2H and ArHet–\({{\rm{CO}}}_{2}^{-}\) species used to compute the σHet descriptors. The “descriptors” sheet provides detailed descriptions and units for each of the 65 descriptors. The repository is organized into two main folders: (i) the database processing folder, which contains a Python script (hard.py) for end-users to perform SMILES-based searches of the database and extract descriptor data from the database file (hard.db); and (ii) the database generation folder, which includes scripts and files for developers to further extend the existing data points, including scripts to filter SMILES strings from Reaxys® query search, generate substituted heteroaryls, perform high-throughput calculations, analyse the results, and extract descriptors. Additionally, the Cartesian coordinates of all optimized geometries (provided as XYZ files) are available on FigShare64. Finally, we provide a simple website (hard.pengliugroup.com) to search for the descriptors by SMILES strings or via a graphical interface (see Usage notes).
Technical Validation
In our automated DFT-calculation/descriptor extraction workflow, several validation checks were performed, including post-processing using the AQME software to address convergence issues (vide supra) and connectivity validation to exclude intrinsically unstable compounds. Similar to traditional Hammett substituent constants, which only included meta- and para-substituted benzenes to avoid influences of steric effects of ortho substituents, we excluded σHet values for all heteroaryl groups with another substituent at an ortho position and those with A1,3-type interactions (e.g., 4-benzothiophenyl with another substituent at the 3-position). These procedures ensure that the reported σHet values describe electronic properties only.
Next, we analysed all DFT-computed descriptors in the database using both linear and non-linear dimensionality reduction techniques, specifically Principal Component Analysis (PCA) and Uniform Manifold Approximation and Projection (UMAP)65 (Fig. 4a,b). In the PCA analysis, the principal components with the greatest variances consist of atomic charge descriptors (PC1, explaining 28% of variance) and quadrupole moments, dispersion, and steric (PC2, 19% of variance). The PCA plots of PC1 versus PC2 show broader distributions of properties across each heteroaryl class (purple, red, yellow, and green colours for 5-membered, 6-membered, and 5,6- and 6,6-fused rings, respectively) than those of aryl groups (black colour) (Fig. 4a). Similarly, the UMAP projection (Fig. 4b) revealed that many heteroaryl groups occupy distinct property space not accessible by aryl substituents. Next, we used z-scores (standard scores) to illustrate the differences among subsets of the database, including aryls, 5- and 6-membered rings, 5,6- and 6,6-fused rings, unsubstituted heteroaryls, and monosubstituted heteroaryls with either electron-donating groups (EDGs) or electron-withdrawing groups (EWGs). For each steric and electronic descriptor shown in Fig. 4c, the z-score for each subset was calculated from the average descriptor value of the subset (x), the average descriptor value of the entire dataset (μ), and the standard deviation of the entire dataset (σ). The z-score analysis indicates that, in many cases, different subsets of the database have distinct steric and electronic properties. For example, 5-membered rings are less sterically hindered than other subsets as indicated by their lower z-scores for fraction buried volume (−0.94) and Sterimol length (−1.20). Additionally, these different electronic descriptors do not strongly correlate with each other and thus could provide complementary descriptions of different types of electronic effects on reactivity.

Statistical analysis of the heteroaryl database. (a) Principal component analysis (PCA). (b) Uniform manifold approximation and projection (UMAP) of DFT-computed descriptors showing broader distributions of heteroaryl properties than those of aryl groups. (c) z-score analysis to indicate the normalized average deviation of subsets of data points from the average values of the entire dataset.
Heteroaromatic compounds are known to have distinct electronic and steric properties affected by their heteroarene cores66. We selected a subset of common heteroaryl groups and plotted their average steric and electronic properties based on the heteroarene core (Fig. 5). We chose a commonly-used steric descriptor, fraction buried volume (VBur), and our newly developed electronic descriptor, the Hammett-type substituent constant (σHet), to illustrate the properties of the heteroaryl groups. Each point represents the average value of all heteroaryls with the same heteroarene core shown in the top panel of Fig. 5. This plot revealed several general trends that could be qualitatively validated by previous experimental observations. In terms of steric effects, five-membered heteroaryls are generally less hindered due to the smaller size of the ring67, and sulphur-containing heteroaryls often have a larger fraction buried volume due to the longer C–S bond (e.g., compare thiophenyl 10 with other five-membered heteroaryls). As expected, substituting a C–H moiety in phenyl or naphthyl with a nitrogen atom decreases the fraction buried volume (e.g., benzene 14 > pyridine 15 > pyridazine 16 > triazines 18 and 19). In terms of electronic properties, five-membered heteroaryls exhibited more diverse electronic effects than other ring sizes. Based on σHet values, pyrrole (1) is more electron-donating than benzene (14), consistent with its higher reactivity in electrophilic aromatic substitution reactions68. On the other hand, 1,3,4-thiadiazole (13) and 1,2,4-oxadiazole (9) are among the most electron-withdrawing heteroaryls, which is consistent with previous experimental observations69,70.

Steric and electronic properties of heteroaryls based on the heteroarene core. Visualization of fraction buried volume (a steric descriptor) versus the Hammett-type substituent constant (an electronic descriptor) describes the intrinsic properties of each type of heteroaryl group.
With a wide array of DFT-computed electronic, steric, and geometrical descriptors for diverse heteroaryl groups in the HArD database, we set out to explore whether the computed descriptors correlate with previously reported experimental reactivity and selectivity data. In 2022, the Leitch group published an extensive set of experimentally determined free energies of activation (ΔG‡SNAr) of nucleophilic aromatic substitution (SNAr) of benzyl alcohol reacting with 2-chloropyridines, 4-chloropyridines, and other chloro-substituted (hetero)aryl compounds (Fig. 6a)21. In another recent work, the Baud group performed systematic kinetic studies of 2-sulphonylpyrimidines as thiol-reactive covalent warheads that can undergo SNAr reactions with biological thiols to achieve selective protein arylation (Fig. 6b)71. The authors achieved a wide range of reactivity for these warheads with the glutathione (GSH) nucleophile by altering the substituents at the 4- and 5-positions of the pyrimidinyl group and exchanging the pyrimidine ring for different parent heteroaryl groups. Both sets of experimental reactivity data strongly correlate with the DFT-computed CM5 charge of the heteroaryl group, q(Het)-CM5 (Fig. 6a,b), illustrating the capability of the q(Het)-CM5 descriptor for quantitative SNAr reactivity prediction. Next, we examined whether the computed electronic descriptors correlate with experimental site-selectivity trends in radical-mediated heteroaryl C–H functionalization from Baran et al. (Fig. 6c)10. The computed ipso carbon LUMO coefficients in the HArD database could qualitatively predict the preferred sites for alkyl radical addition in the majority of the examples studied. Taken together, these preliminary examinations suggest that the computed descriptors in the HArD database could potentially be applied in various types of reactivity and selectivity prediction models.

Utilizing heteroaryl descriptors to predict experimental reactivity and selectivity. (a) Reactivity prediction of nucleophilic aromatic substitution with chloro-substituted heteroaryl electrophiles with experimental reactivity data from Leitch et al.21 q(Het)-CM5: Charge Model 5 (CM5) charge of the heteroaryl group. (b) Reactivity prediction of 2-sulphonylpyrimidine warheads with experimental thiol reactivity data from Baud et al.71. (c) Site-selectivity prediction for radical-mediated C–H functionalization of electron-deficient heteroarenes with selectivity reported by Baran et al.10.
Usage Notes
The database can be accessed in three ways: (i) via the interactive interface provided on the website (hard.pengliugroup.com) to search descriptors by entering a SMILES string or drawing the molecule in the JSME editor72 (Fig. 7), (ii) by downloading the Excel file (hard.xlsx) from the repository, which contains descriptors that can be utilized with various data analysis software libraries, and (iii) by using the Python script (hard.py) to perform searches by SMILES and by similarity, which is recommended when searching for a large list of SMILES to study correlation with reactivity/selectivity or building machine learning models. README files are provided to provide guidance for reproducing and expanding this database. Example bash and Slurm scripts for high-throughput calculations on HPC systems are also included.

Annotated screenshots of hard.pengliugroup.com. A structure search can be performed by either using a SMILES string for the heteroarene or drawing the molecule directly in the editor. The site then returns all regioisomers of the specified heteroaryl group, along with their computed descriptors.
Code availability
The code for generating and processing this database is available on FigShare64 and GitHub (github.com/turkiAlturaifi/HArD) under the MIT license, and the database files are licensed under CC-BY.
References
Marshall, C. M., Federice, J. G., Bell, C. N., Cox, P. B. & Njardarson, J. T. An update on the nitrogen heterocycle compositions and properties of U.S. FDA-approved pharmaceuticals (2013–2023). J. Med. Chem. 67, 11622–11655, https://doi.org/10.1021/acs.jmedchem.4c01122 (2024).
Shen, Q., Shekhar, S., Stambuli, J. P. & Hartwig, J. F. Highly reactive, general, and long-lived catalysts for coupling heteroaryl and aryl chlorides with primary nitrogen nucleophiles. Angew. Chem. Int. Ed. 44, 1371–1375, https://doi.org/10.1002/anie.200462629 (2005).
Billingsley, K. L., Anderson, K. W. & Buchwald, S. L. A highly active catalyst for Suzuki–Miyaura cross-coupling reactions of heteroaryl compounds. Angew. Chem. Int. Ed. 45, 3484–3488, https://doi.org/10.1002/anie.200600493 (2006).
Holmberg-Douglas, N. & Nicewicz, D. A. Photoredox-catalyzed C–H functionalization reactions. Chem. Rev. 122, 1925–2016, https://doi.org/10.1021/acs.chemrev.1c00311 (2022).
Yang, Y. et al. Discovery of organic optoelectronic materials powered by oxidative Ar–H/Ar–H coupling. J. Am. Chem. Soc. 146, 1224–1243, https://doi.org/10.1021/jacs.3c12234 (2024).
Singh, J., Petter, R. C., Baillie, T. A. & Whitty, A. The resurgence of covalent drugs. Nat. Rev. Drug Discov. 10, 307–317, https://doi.org/10.1038/nrd3410 (2011).
Hillebrand, L., Liang, X. J., Serafim, R. A. M. & Gehringer, M. Emerging and re-emerging warheads for targeted covalent inhibitors: an update. J. Med. Chem. 67, 7668–7758, https://doi.org/10.1021/acs.jmedchem.3c01825 (2024).
Boike, L., Henning, N. J. & Nomura, D. K. Advances in covalent drug discovery. Nat. Rev. Drug Discov. 21, 881–898, https://doi.org/10.1038/s41573-022-00542-z (2022).
Ma̧kosza, M. & Wojciechowski, K. Nucleophilic substitution of hydrogen in heterocyclic chemistry. Chem. Rev. 104, 2631–2666, https://doi.org/10.1021/cr020086 (2004).
O’Hara, F., Blackmond, D. G. & Baran, P. S. Radical-based regioselective C–H functionalization of electron-deficient heteroarenes: scope, tunability, and predictability. J. Am. Chem. Soc. 135, 12122–12134, https://doi.org/10.1021/ja406223k (2013).
Dixneuf, P. H., Doucet, H. C–H Bond Activation and Catalytic Functionalization I. https://doi.org/10.1007/978-3-319-24630-7 (Springer, Cham, 2016).
Jurczyk, J. et al. Single-atom logic for heterocycle editing. Nat. Synth. 1, 352–364, https://doi.org/10.1038/s44160-022-00052-1 (2022).
Fey, N. et al. Development of a ligand knowledge base, part 1: computational descriptors for phosphorus donor ligands. Chem. Eur. J. 12, 291–302, https://doi.org/10.1002/chem.200500891 (2006).
Gensch, T. et al. A comprehensive discovery platform for organophosphorus ligands for catalysis. J. Am. Chem. Soc. 144, 1205–1217, https://doi.org/10.1021/jacs.1c09718 (2022).
Durand, D. J. & Fey, N. Computational ligand descriptors for catalyst design. Chem. Rev. 119, 6561–6594, https://doi.org/10.1021/acs.chemrev.8b00588 (2019).
Mayo Yanes, E., Chakraborty, S. & Gershoni-Poranne, R. COMPAS-2: a dataset of cata-condensed hetero-polycyclic aromatic systems. Sci. Data 11, 97, https://doi.org/10.1038/s41597-024-02927-8 (2024).
Ai, Q. et al. OCELOT: An infrastructure for data-driven research to discover and design crystalline organic semiconductors. J. Chem. Phys. 154, 174705, https://doi.org/10.1063/5.0048714 (2021).
St. John, P. C. et al. Quantum chemical calculations for over 200,000 organic radical species and 40,000 associated closed-shell molecules. Sci. Data 7, 244, https://doi.org/10.1038/s41597-020-00588-x (2020).
Garwood, J. J. A., Chen, A. D. & Nagib, D. A. Radical polarity. J. Am. Chem. Soc. 146, 28034–28059, https://doi.org/10.1021/jacs.4c06774 (2024).
Kruszyk, M., Jessing, M., Kristensen, J. L. & Jørgensen, M. Computational methods to predict the regioselectivity of electrophilic aromatic substitution reactions of heteroaromatic systems. J. Org. Chem. 81, 5128–5134, https://doi.org/10.1021/acs.joc.6b00584 (2016).
Lu, J., Paci, I. & Leitch, D. C. A broadly applicable quantitative relative reactivity model for nucleophilic aromatic substitution (SNAr) using simple descriptors. Chem. Sci. 13, 12681–12695, https://doi.org/10.1039/D2SC04041G (2022).
Tomberg, A., Johansson, M. J. & Norrby, P.-O. A predictive tool for electrophilic aromatic substitutions using machine learning. J. Org. Chem. 84, 4695–4703, https://doi.org/10.1021/acs.joc.8b02270 (2019).
Ree, N., Göller, A. H. & Jensen, J. H. RegioML: predicting the regioselectivity of electrophilic aromatic substitution reactions using machine learning. Digit. Discov. 1, 108–114, https://doi.org/10.1039/D1DD00032B (2022).
Kromann, J. C., Jensen, J. H., Kruszyk, M., Jessing, M. & Jørgensen, M. Fast and accurate prediction of the regioselectivity of electrophilic aromatic substitution reactions. Chem. Sci. 9, 660–665, https://doi.org/10.1039/C7SC04156J (2018).
Ree, N., Göller, A. H. & Jensen, J. H. RegioSQM20: improved prediction of the regioselectivity of electrophilic aromatic substitutions. J. Cheminformatics 13, 10, https://doi.org/10.1186/s13321-021-00490-7 (2021).
Guan, Y., Lee, T., Wang, K., Yu, S. & McWilliams, J. C. SNAr regioselectivity predictions: machine learning triggering DFT reaction modeling through statistical threshold. J. Chem. Inf. Model. 63, 3751–3760, https://doi.org/10.1021/acs.jcim.3c00580 (2023).
Feng, K. et al. Development of a deactivation-resistant dialkylbiarylphosphine ligand for Pd-catalyzed arylation of secondary amines. J. Am. Chem. Soc. 146, 26609–26615, https://doi.org/10.1021/jacs.4c09667 (2024).
Li, X., Zhang, S.-Q., Xu, L.-C. & Hong, X. Predicting regioselectivity in radical C−H functionalization of heterocycles through machine learning. Angew. Chem. Int. Ed. 59, 13253–13259, https://doi.org/10.1002/anie.202000959 (2020).
Hansch, C., Leo, A. & Taft, R. W. A survey of Hammett substituent constants and resonance and field parameters. Chem. Rev. 91, 165–195, https://doi.org/10.1021/cr00002a004 (1991).
Butler, A. Dissociation constants of thiophencarboxylic acids: calculation of σ constants for the thiophen ring. J. Chem. Soc. B, 867–870 (1970).
Pitt, W. R., Parry, D. M., Perry, B. G. & Groom, C. R. Heteroaromatic rings of the future. J. Med. Chem. 52, 2952–2963, https://doi.org/10.1021/jm801513z (2009).
Gaulton, A. et al. ChEMBL: a large-scale bioactivity database for drug discovery. Nucleic Acids Res. 40, D1100–D1107, https://doi.org/10.1093/nar/gkr777 (2012).
Landrum, G. A. RDKit: open-source cheminformatics, http://www.rdkit.org.
Riniker, S. & Landrum, G. A. Better informed distance geometry: using what we know to improve conformation generation. J. Chem. Inf. Model. 55, 2562–2574, https://doi.org/10.1021/acs.jcim.5b00654 (2015).
Frisch, M. J., et al. Gaussian 16 Rev. C.01. Gaussian 16 (2016).
Grimme, S., Antony, J., Ehrlich, S. & Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 132, 154104, https://doi.org/10.1063/1.3382344 (2010).
Grimme, S., Ehrlich, S. & Goerigk, L. Effect of the damping function in dispersion corrected density functional theory. J. Comput. Chem. 32, 1456–1465, https://doi.org/10.1002/jcc.21759 (2011).
Becke, A. D. Density‐functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 98, 5648–5652, https://doi.org/10.1063/1.464913 (1993).
Lee, C., Yang, W. & Parr, R. G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 37, 785–789, https://doi.org/10.1103/PhysRevB.37.785 (1988).
Zhao, Y. & Truhlar, D. G. The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: two new functionals and systematic testing of four M06-class functionals and 12 other functionals. Theor. Chem. Acc. 120, 215–241, https://doi.org/10.1007/s00214-007-0310-x (2008).
Marenich, A. V., Cramer, C. J. & Truhlar, D. G. Universal solvation model based on solute electron density and on a continuum model of the solvent defined by the bulk dielectric constant and atomic surface tensions. J. Phys. Chem. B 113, 6378–6396, https://doi.org/10.1021/jp810292n (2009).
Alegre-Requena, J. V., Sowndarya, S. V., Pérez-Soto, R., Alturaifi, T. M. & Paton, R. S. AQME: Automated quantum mechanical environments for researchers and educators. WIREs Comput. Mol. Sci. 13, e1663, https://doi.org/10.1002/wcms.1663 (2023).
Kelly, C. P., Cramer, C. J. & Truhlar, D. G. Aqueous solvation free energies of ions and ion−water clusters based on an accurate value for the absolute aqueous solvation free energy of the proton. J. Phys. Chem. B 110, 16066–16081, https://doi.org/10.1021/jp063552y (2006).
Lian, P., Johnston, R. C., Parks, J. M. & Smith, J. C. Quantum chemical calculation of pKas of environmentally relevant functional groups: carboxylic acids, amines, and thiols in aqueous solution. J. Phys. Chem. A 122, 4366–4374, https://doi.org/10.1021/acs.jpca.8b01751 (2018).
Parr, R. G. & Pearson, R. G. Absolute hardness: companion parameter to absolute electronegativity. J. Am. Chem. Soc. 105, 7512–7516, https://doi.org/10.1021/ja00364a005 (1983).
Parr, R. G., Szentpály, L. V. & Liu, S. Electrophilicity index. J. Am. Chem. Soc. 121, 1922–1924, https://doi.org/10.1021/ja983494x (1999).
Weinhold, F. & Landis, C. R. Valency and Bonding: a Natural Bond Orbital Donor-Acceptor Perspective. https://doi.org/10.1017/CBO9780511614569 (Cambridge University Press, 2005).
Hirshfeld, F. L. Bonded-atom fragments for describing molecular charge densities. Theor. Chim. Acta 44, 129–138, https://doi.org/10.1007/BF00549096 (1977).
Marenich, A. V., Jerome, S. V., Cramer, C. J. & Truhlar, D. G. Charge Model 5: an extension of hirshfeld population analysis for the accurate description of molecular interactions in gaseous and condensed phases. J. Chem. Theory Comput. 8, 527–541, https://doi.org/10.1021/ct200866d (2012).
Kjell, J. Source code for: Molecular features for machine learning (MORFEUS). GitHub https://github.com/digital-chemistry-laboratory/morfeus, https://doi.org/10.5281/zenodo.6685218 (2022).
Verloop, A., Hoogenstraaten, W. & Tipker, J. Chapter 4 - development and application of new steric substituent parameters in drug design. in Drug Design (ed. Ariëns, E. J.) vol. 11, 165–207 https://doi.org/10.1016/B978-0-12-060307-7.50010-9 (Academic Press, Amsterdam, 1976).
Verloop, A. The sterimol approach: further development of the method and new applications. in Pesticide Chemistry: Human Welfare and Environment (eds. Doyle, P. & Fujita, T.) 339–344. https://doi.org/10.1016/B978-0-08-029222-9.50051-2 (Pergamon, 1983).
Poater, A. et al. SambVca: a web application for the calculation of the buried volume of N-heterocyclic carbene ligands. Eur. J. Inorg. Chem. 2009, 1759–1766, https://doi.org/10.1002/ejic.200801160 (2009).
Falivene, L. et al. SambVca 2. a web tool for analyzing catalytic pockets with topographic steric maps. Organometallics 35, 2286–2293, https://doi.org/10.1021/acs.organomet.6b00371 (2016).
Pollice, R. & Chen, P. A Universal quantitative descriptor of the dispersion interaction potential. Angew. Chem. Int. Ed. 58, 9758–9769, https://doi.org/10.1002/anie.201905439 (2019).
Eisenhaber, F., Lijnzaad, P., Argos, P., Sander, C. & Scharf, M. The double cubic lattice method: efficient approaches to numerical integration of surface area and volume and to dot surface contouring of molecular assemblies. J. Comput. Chem. 16, 273–284, https://doi.org/10.1002/jcc.540160303 (1995).
Shrake, A. & Rupley, J. A. Environment and exposure to solvent of protein atoms. Lysozyme and insulin. J. Mol. Biol. 79, 351–371, https://doi.org/10.1016/0022-2836(73)90011-9 (1973).
Kruszewski, J. & Krygowski, T. M. Definition of aromaticity basing on the harmonic oscillator model. Tetrahedron Lett. 13, 3839–3842, https://doi.org/10.1016/S0040-4039(01)94175-9 (1972).
Krygowski, T. M., Szatylowicz, H., Stasyuk, O. A., Dominikowska, J. & Palusiak, M. Aromaticity from the viewpoint of molecular geometry: application to planar systems. Chem. Rev. 114, 6383–6422, https://doi.org/10.1021/cr400252h (2014).
Krygowski, T. M. Crystallographic studies of inter- and intramolecular interactions reflected in aromatic character of π-electron systems. J. Chem. Inf. Comput. Sci. 33, 70–78, https://doi.org/10.1021/ci00011a011 (1993).
Frizzo, C. P. & Martins, M. A. P. Aromaticity in heterocycles: new HOMA index parametrization. Struct. Chem. 23, 375–380, https://doi.org/10.1007/s11224-011-9883-z (2012).
Wildman, S. A. & Crippen, G. M. Prediction of physicochemical parameters by atomic contributions. J. Chem. Inf. Comput. Sci. 39, 868–873, https://doi.org/10.1021/ci990307l (1999).
Ertl, P., Rohde, B. & Selzer, P. Fast calculation of molecular polar surface area as a sum of fragment-based contributions and its application to the prediction of drug transport properties. J. Med. Chem. 43, 3714–3717, https://doi.org/10.1021/jm000942e (2000).
Alturaifi, T. M., Scofield, G. E., Shengchun, W. & Liu, P. A Database of Steric and Electronic Properties of Heteroaryl Substituents. Figshare https://doi.org/10.6084/m9.figshare.28385759 (2025).
McInnes, L., Healy, J., Saul, N. & Großberger, L. UMAP: uniform manifold approximation and projection. J. Open Source Softw. 3, 861, https://doi.org/10.21105/joss.00861 (2018).
Katritzky, A. R., Ramsden, C. A., Joule, J. A. & Zhdankin, V. V. Handbook of Heterocyclic Chemistry. https://doi.org/10.1016/C2009-0-05547-0 (Elsevier, Chantilly, United Kingdom, 2010).
Ji Ram, V., Sethi, A., Nath, M. & Pratap, R. Chapter 5 – five‐membered heterocycles. In The Chemistry of Heterocycles, pp. 149–478. https://doi.org/10.1016/B978-0-08-101033-4.00005-X (Elsevier, 2019).
Jolicoeur, B., Chapman, E. E., Thompson, A. & Lubell, W. D. Pyrrole protection. Tetrahedron 62, 11531–11563, https://doi.org/10.1016/j.tet.2006.08.071 (2006).
Hu, Y., Li, C.-Y., Wang, X.-M., Yang, Y.-H. & Zhu, H.-L. 1,3,4-thiadiazole: synthesis, reactions, and applications in medicinal, agricultural, and materials chemistry. Chem. Rev. 114, 5572–5610, https://doi.org/10.1021/cr400131u (2014).
Piccionello, A. P., Pace, A. & Buscemi, S. Rearrangements of 1,2,4-oxadiazole: “one ring to rule them all”. Chem. Heterocycl. Compd. 53, 936–947, https://doi.org/10.1007/s10593-017-2154-1 (2017).
Pichon, M. M. et al. Structure–reactivity studies of 2-sulfonylpyrimidines allow selective protein arylation. Bioconjug. Chem. 34, 1679–1687, https://doi.org/10.1021/acs.bioconjchem.3c00322 (2023).
Bienfait, B. & Ertl, P. JSME: a free molecule editor in JavaScript. J Cheminform 5, 24, https://doi.org/10.1186/1758-2946-5-24 (2013).
Acknowledgements
We thank Prof. Geoffrey Hutchison (Pitt) for helpful conversations about RDKit, Prof. Kjell Jorner (ETH Zürich) for useful suggestions with HOMA calculations, Prof. Juan V. Alegre-Requena (CSIC-University of Zaragoza) for general guidance, and Joel Muyskens for assistance with the web design. This work was supported by the NIH (R35 GM128779). DFT calculations were carried out at the University of Pittsburgh Center for Research Computing and Data (RRID: SCR_022735), and the Advanced Cyberinfrastructure Coordination Ecosystem: Services & Support (ACCESS) program, supported by NSF award numbers OAC-2117681, OAC-1928147, and OAC-1928224. G.E.S. was supported by a US Department of Education GAANN grant, award number: P200A240158.
Author information
Authors and Affiliations
Contributions
P.L., T.M.A. and G.E.S. conceived the project and designed the study. T.M.A. designed and implemented the database generation–high-throughput calculation pipeline, collected the data, and developed the website with input from P.L. and G.E.S. G.E.S. performed benchmark studies on the Hammett-type substituent constants for heteroaryl groups and validated the dataset using case studies from the literature. S.W. contributed to the visualization of the manuscript. P.L. directed the project. All authors contributed to writing the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Alturaifi, T.M., Scofield, G.E., Wang, S. et al. A database of steric and electronic properties of heteroaryl substituents. Sci Data 12, 1319 (2025). https://doi.org/10.1038/s41597-025-05198-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41597-025-05198-z
