
Abstract
Scirpus × mariqueter (Tang & F.T.Wang) Tatanov, which is endemic to eastern estuaries in China, is a tidal zone-engineering species with great promise for managing greenhouse gases and enhancing ecosystem resilience against invasive species. Although S. mariqueter is widely recognized as a hybrid species derived from Bolboschoenus planiculmis (F. Schmidt) T.V. Egorova and Schoenoplectus triqueter (L.) Palla, its speciation remains highly controversial. The lack of a reference genome is the major cause of this ambiguity. We generated the first chromosome-level genome assembly for S. mariqueter combining PacBio long-reads, Illumina short-reads, and the Hi-C method. The genome assembly consisted of 227.75 Mb (contig N50: 3.89 Mb). We also constructed a haploid karyotype comprising 54 pseudochromosomes. The average size of these pseudochromosomes was small (4.05 Mb). Thirty-two pseudochromosomes were assembled to a telomere to telomere level. Repetitive elements represented approximately 54.12% of the genome. We predicted and annotated 25,239 protein-coding genes. The overall BUSCO score was 95.10%, with notably few duplicated genes (1.70%). This high-quality genome provides critical data for future studies.
Similar content being viewed by others


Background & Summary
Scirpus × mariqueter (Tang & F.T.Wang) Tatanov, which is endemic to China1, belongs to the species-rich Cyperaceae family. This species makes critical contributions to the integration and health of coastal ecosystems. Because of its thriving and resilient underground rhizome system, S. mariqueter is often a pioneer species in inter-tidal zones and forms single-species patches covering vast areas (Fig. 1), thereby prompting the development of coastal lines and off-shore islands2,3.

Scirpus × mariqueter in the field. (a) S. mariqueter inflorescence bearing only one spikelet; (b) Seedling and rhizome at the stem base; (c) a large mono-species patch of S. mariqueter in a coastal area.
Many migratory birds routinely rely on its corms and achenes as a source of food4. It also has a crucial effect on the carbon budget of coastal wetlands. A previous study has shown that S. mariqueter emits substantial amounts of methane (CH4) and responds significantly to tidal variation5. Following an invasion by Spartina alterniflora Loisel. (Poaceae), S. mariqueter can enhance plant–soil feedback and mitigate the negative effects of biological invasions6.
Although S. mariqueter is a key coastal ecosystem species, there are considerable controversies regarding its evolutionary trajectory. It is considered to be a hybrid species derived from Bolboschoenus planiculmis (F. Schmidt) T.V. Egorova and Schoenoplectus triqueter (L.) Palla7,8. However, previous research have shown that S. mariqueter is much more closely related to B. planiculmis than to S. triqueter, with no intermediate individuals confirmed in the field7,9. These results raise questions about the validity of hybrid speciation as the sole mechanism underlying the origin of S. mariqueter. Nevertheless, the absence of an S. mariqueter genome assembly has greatly limited our understanding of the biological mechanisms and evolutionary significance of this species.
In this data descriptor, we report a chromosome-level Scirpus × mariqueter genome assembly. The current formal nomenclature for this species is × Bolboschoenoplectus mariqueter (https://powo.science.kew.org). However, we herein mainly use S. mariqueter in accordance with how this species is most commonly referred to in the published literature.
In general, the final genome assembly comprises 227.75 Mb. The contig N50 value is 3.89 Mb. The scaffold N50 value is 4.07 Mb. The overall BUSCO score is 95.10. Most of the complete BUSCO units are single-copy (93.40%), with few duplicated BUSCOs (1.70%) (Table 1). By integrating valid Hi-C data (Table 2), we determine the association between most contigs. We congregate these contigs into pseudochromosomes (i.e. approximations of the actual chromosomes, especially in the orders and orientations of the compositional bases) and further improve our genome assembly to chromosome level (Fig. 2, Supplementary Table S1).

Heatmap of Hi-C interactions of S. mariqueter pseudochromosomes. The resolution is 300 kb. Color gradient from yellow to red indicates the frequencies of Hi-C interactions (low to high, respectively).
The constructed pseudochromosomes reveal a haploid karyotype (n = 54). According to critical research on Cyperid evolution by Elliot et al.10, the genome size of our sample approximate the average value in the genus Bolboschoenus (223.82 Mb ± 13.54 Mb). Analyses of random reads indicate that B. planiculmis is the most frequently matched species (41.15% of all reads) (Table 3). The average pseudochromosome size in our genome (4.05 Mb) also approaches the lowest mean chromosome size in cyperid species (3.7 Mb from Bolboschoenus robustus10).
Annotation results show that repetitive elements constitute 54.12% (~123.25 Mb) of the S. mariqueter genome. Approximately 35.27% of the genome are comprised of Transposable Elements (TEs), including Long-Terminal Repeat (LTR) retrotransposons (15.87%) and DNA transposons (13.91%) (Supplementary Table S2). Tandem repeats make up 18.85% of the genome (Table 4). By analyzing the repeat-masked genome, 25,239 protein-coding genes are identified in the S. mariqueter genome. Approximately 94.66% of the predicted genes get annotated using canonical databases (Table 5). We also establish a non-coding RNA (ncRNA) library consisting of 3,039 rRNAs, 1,090 tRNAs, 163 miRNAs, and 243 snRNAs (Table 6). A Circos graph (Fig. 3) is provided to intuitively display key information, including GC contents, gene density, intra-genome collinearity, and TEs.

Circos plot of the distribution of S. mariquter genomic features. Four circular tiers represent (a) chromosome ideograms, (b) gene density, (c) transposable element density, and (d) GC content. Central lines indicate putative homology among linked sections. Colors were arbitrarily selected.
Methods
Sampling and pretreatment
We chose a healthy S. mariqueter individual on Yonglongsha island (31.709°N, 121.618°E). We transported this individual to a plantation at the Chinese Academy of Forestry, where it was maintained for long-term research purposes. We carefully collected and cleaned sampled tissues to prevent exogenous contamination. All samples were swiftly transferred to the laboratory and stored at −80 °C.
Genome sequencing
We followed the conventional CTAB (cetyltrimethylammonium bromide) approach to extract genomic DNA, after which DNA quality was assessed via agarose gel electrophoresis. For PacBio long-reads sequencing, SMRTbell (15 kb in length) DNA libraries were generated using the standard protocol for SMRTbell Express Template Prep Kit 2.0 (PacBio, CA, USA). After filtering and quantification, library modules were sequenced using a PacBio Revio platform. Raw data were treated using SMRTLink v.8.0 (https://www.pacb.com/support/documentation/). For Illumina short-reads sequencing, we chose the Next era DNA Flex Library Prep Kit (Illumina, CA, USA) to create pair-end libraries (insertion size 250 bp). Sequencing was performed using a NovaSeq 6000 platform. Raw reads were filtered using SOAPnuke v.2.1.411 (-n 0.01 -l 20 -q 0.1 -i -Q 2 -G -M 2 -A 0.5 -d).
Transcriptome sequencing
For gene prediction, samples were taken from five different parts (root, stem, leaf, bract, and spikelet). All the samples were mixed together to form a pooled sample. We then extracted the total RNA from this pooled sample following the conventional procedure for RNA prep Pure Plant Kit (Tiangen Biotech, Beijing, China). Sequencing was carried out using an Illumina NovaSeq 6000 platform, with pair-end libraries constructed following a standard Illumina protocol (San Diego, CA, USA). The insertion size was 250 bp.
K-mer analysis and genome assembly
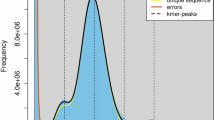
A K-mer analysis was realized using Genome Scope v.2.012 and Jellyfish v. 2.2.11313 (count -m 19 -C -c 7 -t 96 -s 1 G -f 2). The results showed that, according to a 19-mer model, the S. mariqueter genome manifested a genome size of 202.19 Mb, with a heterozygosity of 0.73% (Fig. 4).

K-mer analysis results and preliminary estimation of S. mariqueter genome parameters. Results were based on a 19-mer model. Estimated heterozygosity was 0.73%.
Following this estimation, we obtained a primary assembly using the NextDenovo pipeline14 (read_cutoff = 1k, genome_size = 0.5 g, sort_options = -m 128 g -t 96, nextgraph_options = -a 1 -q 10) (https://github.com/Nextomics/NextDenovo). The primary genome had a length of 227.75 Mb, which approximated the estimated size from K-mer modeling. Double rounds of error correction were performed for the primary assembly using Pilon v.1.2315 (https://github.com/broadinstitute/pilon), after which heterozygous sequences were removed using Purge_haplotigs pipeline v.1.0.416 to decrease ambiguities. The HindIII enzymatic digestion method detailed by Xie et al.17 was selected to guide our Hi-C library construction. Clean Hi-C data were aligned to the primary assembly according to the Burrows-Wheeler-Aligner (BWA) v.0.7.1718 algorithm. Valid-interaction reads which got unique alignment were then filtered using HiCUP v.0.8.019. ALLHiC v.0.9.820 was utilized to group the contigs of the draft assembly into pseudochromosomes with reference to valid interaction information. By applying this Hi-C methodology, we were able to detect the association between most contigs and cluster them into pseudomolecules reflecting real chromosomes. Thus, the primary assembly was improved to produce a chromosome-level genome assembly. Additionally, we used 3D-DNA v.18092221 and Juciebox v.1.11.0822 to further improve contig orientation and order.
Detection of repetitive elements
We built a de novo repeat library using RepeatModeler v.2.0.123. We refined this library using a combination of RepeatMasker v.4.15 (http://www.repeatmasker.org) and RepBase v.2018102624. We performed further predictions for two major repeat components bearing evolutionary significance: LTR and tandem repeats. By integrating LTR_finder, LTR_harvester, and LTR_retriever, we acquired high-quality LTRs following the instructions of Ou & Jiang25. Tandem repeats were predicted using TRF v.4.1.026 and MISA v.2.127. We also identified the potential sites for centromeres and telomeres, as they are vital factors affecting speciation in Cyperid species which commonly have holocentric chromosomes10. We executed the standard Python scripts of quarTeT28 to detect centromeres, telomeres, and gaps. Based on the results from quarTeT, we plotted a comprehensive karyotype ideogram using RIdeogram v.0.2.229 to represent the patterns intuitively.
Gene prediction and annotation
After masking the repeat content, we used Augustus v.3.5.0 to produce de novo gene models30. We inferred homology on the basis of high-quality genome assemblies for the following five species: Rhynchospora breviuscula (GCA_027562975.1)31, Bolboschoenus planiculmis (GCA_031770325.1)32, Oryza sativa (GCA_034140825.1)33, Schoenoplectus tabernaemontani (GCA_037127355.1)34, and Arabidopsis thaliana (GCA_000001735.2)35. We decoded the transcripts using TransDecoder v.5.7.1 (https://github.com/TransDecoder/TransDecoder). Finally, we reconciled these results using Maker v.3.0136 pipeline to get the ultimate gene sets (https://github.com/Yandell-Lab/maker?tab). Gene functions were annotated using the NR, InterPro, UniProt, GO, KEGG, and Pfam databases with an e-value of 1e-5. For ncRNAs, tRNAs were predicted using tRNAscan-SE v.1.3.137. rRNAs were identified using RNAmmer v.1.238 (https://services.healthtech.dtu.dk/services/RNAmmer-1.2/). We further determined miRNAs, snoRNAs and snRNAs using Infernal 1.139 with reference to Rfam (v.14.9) database. Detailed procedures and parameters could be found in the manual of ncRNA analysis using Rfam database40.
Data Records
The genome assembly and all sequence data are deposited in the NCBI database. The genome assembly number is GCA_037678475.141. The Bioproject ID is PRJNA1079027. The Biosample ID is SAMN40029249. Raw reads used to generate the genome assembly are stored in the Sequence Read Archive (SRP491792)42. The complete genome annotation files in gff3 format, including coding sequences, ncRNA sequences, and repeat sequences, are shared in the Figshare database (https://doi.org/10.6084/m9.figshare.25479922.v1)43.
Technical Verification
Data volume and quality
Sequence data volume and quality were sufficient for constructing a high-quality genome assembly. We produced 21.12 Gb PacBio long-reads data for the primary assembly. Among the long-reads, 60.27% got a Phred quality score (Q score) better than Q30. Details regarding PacBio long-read quality are provided in Fig. 5.

Distribution of quality scores and lengths of PacBio reads. The x-axis presents the read length (bp), whereas the y-axis presents the predicted base calling accuracy. Dot colors reflect count abundance (scale bar on the right). The plot presents a high volume of reads with a Phred quality score (Q score) of 28–41 and a length of 13,500–25,000 bp.
We also generated a total of 132.85 Gb Illumina short-reads for genome profiling, gene prediction, pseudochromosome construction, and back-mapping check. The average Q30 value for short-reads data was 93.74. (Table 7). A total of 50,000 random short-reads were blasted into Nucleotide Sequence Database (NT) using BLASTN v. 2.11.0 (-evalue 1e-5 -max_target_seqs1). Eventually, 7,510 reads got matches. The top 20 matched species were listed in Table 3. The results showed that all the matched records belong to Viridiplantae, with approximately 67% of the matched reads belonging to Cyperaceae, indicating that our sample was safeguarded against extraneous contamination. Thus, our genome assembly had a robust foundation in data volume, data quality, and data source.
Continuity and completeness of the genome assembly
We assembled a high-quality genome with the following characteristics: (1) There were relatively few gaps. The total length of the detected gaps was 500Ns. Specifically, there was one gap(100Ns) in Chr3, Chr19, Chr21, Chr23, and Chr44 respectively (Fig. 6).

Chromosome ideograms of S. mariqueter. Ideograms present a haploid karyotype (n = 54). Ideogram length is proportional to chromosome size. Background color scales represent gene densities (100 kb window). Five gaps (orange box) in the genome assembly are shown. Putative telomeres are indicated by green triangles, whereas centromeres are indicated by purple circles.
Gaps were not detected in the other 49 pseudochromosomes (i.e., 90.74% of the total number); (2) In our final genome, 36 pseudochromosomes showed telomeres at both ends, whereas 15 pseudochromosomes had a telomere at only one end. The rest three pseudochromosomes currently manifested no trace of telomeres. Considering the observed gaps, we assembled 32 telomere-to-telomere pseudochromosomes, accounting for 59.25% of the total number; (3) The overall BUSCO score was 95.1% (Table 1), similar to some recently published Cyperid genomes44. In terms of the back-mapping rate, 99.03% of the sequenced reads were aligned in our final genome. Overall, 99.88% of the final genome was covered through back-mapping. Specifically, 99.61% of the final genome was covered at least 4×, 99.25% of the final genome was covered at least 10×, and 98.84% of the final genome was covered at least 20×.
Code availability
Novel codes were not generated in this work. The related bioinformatic suites and R packages were implemented as described in the published literature. All the parameters followed their default values unless specified otherwise in the Method part.
References
Huang, J., Ma, K. & Huang, J. Species Diversity Distribution Patterns of Chinese Endemic Seed Plants Based on Geographical Regions. PLoS ONE 12, e0170276, https://doi.org/10.1371/journal.pone.0170276 (2017).
Yang, S. L. The Role of Scirpus Marsh in Attenuation of Hydrodynamics and Retention of Fine Sediment in the Yangtze Estuary. Estuarine Coastal Shelf Sci. 47, 227–233, https://doi.org/10.1006/ecss.1998.0348 (1998).
Yang, M. et al. Landscape genetic structure of Scirpus mariqueter reveals a putatively adaptive differentiation under strong gene flow in estuaries. Ecol Evol 9, 3059–3074, https://doi.org/10.1002/ece3.4793 (2019).
Ma, Z. et al. Effects of tidewater on the feeding ecology of hooded crane (Grus monacha) and conservation of their wintering habitats at Chongming Dongtan, China. Ecol. Res. 18, 321–329, https://doi.org/10.1046/j.1440-1703.2003.00557.x (2003).
Peng, Y. et al. Tidal variation and litter decomposition co-affect carbon emissions in estuarine wetlands. Sci. Total. Environ. 839, 156357, https://doi.org/10.1016/j.scitotenv.2022.156357 (2022).
Cheng, C. et al. Genotype diversity enhances invasion resistance of native plants via soil biotic feedbacks. Ecol Lett 27, https://doi.org/10.1111/ele.14384 (2024).
Yang, M. et al. AFLP markers in the detection of Scirpus × mariqueter (Cyperaceae) hybrid in China. Aquat. Bot. 91, 298–302, https://doi.org/10.1016/j.aquabot.2009.08.005 (2009).
Deng, H. K. et al. Genome size determination of Scirpus mariqueter and its related species. Guihaia 43(10), 1838–1848, https://doi.org/10.11931/guihaia.gxzw202209007 (2023).
Yang, M. et al. Interspecific hybridization between Scirpus mariqueter Ts. Tang & F.T. Wang and S. planiculmis F. Schmidt and their selfing. Aquat. Bot. 110, 92–96, https://doi.org/10.1016/j.aquabot.2013.05.002 (2013).
Elliott, T. L. et al. Chromosome size matters: genome evolution in the cyperid clade. Ann. Bot. 130, 999–1014, https://doi.org/10.1093/aob/mcac136 (2022).
Chen, Y. et al. SOAPnuke: a MapReduce acceleration-supported software for integrated quality control and preprocessing of high-throughput sequencing data. Gigascience 7, https://doi.org/10.1093/gigascience/gix120 (2017).
Ranallo-Benavidez, T. R., Jaron, K. S. & Schatz, M. C. GenomeScope 2.0 and Smudgeplot for reference-free profiling of polyploid genomes. Nat. Commun. 11, 1–10, https://doi.org/10.1038/s41467-020-14998-3 (2020).
Marcais, G. & Kingsford, C. A fast, lock-free approach for efficient parallel counting of occurrences of k-mers. Comput. Appl. Biosci. 27, 764–770, https://doi.org/10.1093/bioinformatics/btr011 (2011).
Hu, J. et al. NextDenovo: an efficient error correction and accurate assembly tool for noisy long reads. Genome Biol. 25, https://doi.org/10.1186/s13059-024-03252-4 (2024).
Walker, B. J. et al. Pilon: An integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLos ONE 9(11), e112963, https://doi.org/10.1371/journal.pone.0112963 (2014).
Roach, M. J., Schmidt, S. A. & Borneman, A. R. Purge Haplotigs: allelic contig reassignment for third-gen diploid genome assemblies. BMC Bioinform. 19, https://doi.org/10.1186/s12859-018-2485-7 (2018).
Xie, T. et al. De Novo Plant Genome Assembly Based on Chromatin Interactions: A Case Study of Arabidopsis thaliana. Mol. Plant 8, 489–492, https://doi.org/10.1016/j.molp.2014.12.015 (2015).
Li, H. & Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Comput. Appl. Biosci. 25, 1754–1760, https://doi.org/10.1093/bioinformatics/btp324 (2009).
Wingett, S. W. et al. HiCUP: pipeline for mapping and processing Hi-C data. F1000Res 4, 1310, https://doi.org/10.12688/f1000research.7334.1 (2015).
Wang, Y.-B. & Zhang, X. Chromosome Scaffolding of Diploid Genomes Using ALLHiC. Bio. Protoc. 12, https://doi.org/10.21769/bioprotoc.4503 (2022).
Dudchenko, O. et al. De novo assembly of the Aedes aegypti genome using Hi-C yields chromosome-length scaffolds. Science 356, 92–95, https://doi.org/10.1126/science.aal3327 (2017).
Durand, N. C. et al. Juicebox Provides a Visualization System for Hi-C Contact Maps with Unlimited Zoom. Cell Syst. 3, 99–101, https://doi.org/10.1016/j.cels.2015.07.012 (2016).
Flynn, J. M. et al. RepeatModeler2 for automated genomic discovery of transposable element families. Proc. Natl. Acad Sci USA 117, 9451–9457, https://doi.org/10.1073/pnas.1921046117 (2020).
Ou, S. & Jiang, N. LTR_retriever: A Highly Accurate and Sensitive Program for Identification of Long Terminal Repeat Retrotransposons. Plant Physiol. 176, 1410–1422, https://doi.org/10.1104/pp.17.01310 (2017).
Bao, W., Kojima, K. K. & Kohany, O. Repbase Update, a database of repetitive elements in eukaryotic genomes. Mob. DNA 6, https://doi.org/10.1186/s13100-015-0041-9 (2015).
Benson, G. Tandem repeats finder: a program to analyze DNA sequences. Nucl. Acids Res. 27, 573–580, https://doi.org/10.1093/nar/27.2.573 (1999).
Beier, S., Thiel, T., Munch, T., Scholz, U. & Mascher, M. MISA-web: a web server for microsatellite prediction. Comput. Appl. Biosci. 33, 2583–2585, https://doi.org/10.1093/bioinformatics/btx198 (2017).
Lin, Y. et al. quarTeT: a telomere-to-telomere toolkit for gap-free genome assembly and centromeric repeat identification. Hortic Res https://doi.org/10.1093/hr/uhad127 (2023).
Hao, Z. et al. RIdeogram: drawing SVG graphics to visualize and map genome-wide data on the idiograms. PeerJ Comput. Sci. 6, e251, https://doi.org/10.7717/peerj-cs.251 (2020).
Stanke, M., Diekhans, M., Baertsch, R. & Haussler, D. Using native and syntenically mapped cDNA alignments to improve de novo gene finding. Bioinformatics 24, 637–644, https://doi.org/10.1093/bioinformatics/btn013 (2008).
Hofstatter, P. G. et al. Genbank https://identifiers.org/insdc.gca:GCA_027562975.1 (2022).
Ning, Y., Hu, Y. Q. & Xia, X. F. Genbank https://identifiers.org/insdc.gca:GCA_031770325.1 (2023).
Shang, L. et al. Genbank https://identifiers.org/insdc.gca:GCA_034140825.1 (2023).
Li, Y. et al. Genbank https://identifiers.org/insdc.gca:GCA_037127355.1 (2024).
Swarbreck, D. et al. Genbank https://identifiers.org/insdc.gca:GCA_000001735.2 (2018).
Cantarel, B. L. et al. MAKER: An easy-to-use annotation pipeline designed for emerging model organism genomes. Genome Res. 18, 188–196, https://doi.org/10.1101/gr.6743907 (2007).
Lowe, T. & Eddy, S. tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucl Acids Res 25, 955–964, https://doi.org/10.1093/NAR/25.5.0955 (1997).
Lagesen, K. et al. RNAmmer: consistent and rapid annotation of ribosomal RNA genes. Nucl Acids Res 35, 3100–3108, https://doi.org/10.1093/nar/gkm160 (2007).
Nawrocki, E. P. & Eddy, S. R. Infernal 1.1: 100-fold faster RNA homology searches. Comput. Appl. Biosci. 29, 2933–2935, https://doi.org/10.1093/bioinformatics/btt509 (2013).
Kalvari, I. et al. Non-Coding RNA Analysis Using the Rfam Database. CP in Bioinformatics 62, https://doi.org/10.1002/cpbi.51 (2018).
NCBI GenBank https://identifiers.org/insdc.gca:GCA_037678475.1 (2024).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRP491792 (2024).
Ning, Y. Annotation files for the chromosome-level genome assembly of Scirpus × mariqueter (× Bolboschoenoplectus mariqueter). figshare https://doi.org/10.6084/m9.figshare.25479922.v1 (2025).
Planta, J. et al. Chromosome-scale genome assemblies and annotations for Poales species Carex cristatella, Carex scoparia, Juncus effusus, and Juncus inflexus. G3 12, https://doi.org/10.1093/g3journal/jkac211 (2022).
Acknowledgements
We thank Mr. LIN H Y for assisting our field work. We are also grateful to anonymous reviewers whose comments help improve our manuscript. The funding sources for this research are the Fundamental Research Funds of the Chinese Academy of Forestry (CAFYBB2024MA023, CAFYBB2020SZ006) and the National Natural Science Foundation of China (NSFC31800348, NSFC31972948).
Author information
Authors and Affiliations
Contributions
NING, Y. and LI, C.Y. conceived and designed the research; LI, C.Y. issued the funds and organized the related resources; LI, Y., ZHENG, Y.C., WANG, J.Z. and WANG, T.S. participated in the field work and curated the raw data; ZHAN Y.Y. contributed in data visualization; NING, Y., WANG Y.F. and LI, C.Y. drafted the manuscript; XU, S.J. and DONG, S.B. improved the logic and writing. The revision and approval of this final manuscript were established by all the authors.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Ning, Y., Li, Y., Li, C.Y. et al. Chromosome-level genome assembly for clubrush (Scirpus × mariqueter) endemic to China. Sci Data 12, 839 (2025). https://doi.org/10.1038/s41597-025-05204-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41597-025-05204-4
