
Abstract
Verpa, commonly known as “early morel” or “false morel”, plays an important ecological role and offers considerable economic and medicinal potential. Despite their significance, research on Verpa species, particularly V. bohemica and V. conica, remains limited. In this study, we assembled high-quality sub-chromosomal genomes of six Verpa strains using Nanopore and Illumina sequencing, with average sizes of 44.38 Mb for V. bohemica and 45.40 Mb for V. conica. Specifically, the assemblies of V. bohemica strain 21108 and V. conica strain 21120 were anchored to 26 and 25 chromosomes with Hi-C technologies, respectively. The consensus quality value (QV) of both V. bohemica and V. conica exceeded 40. In addition, an average of 11,024 and 11,052 protein-coding genes were identified for V. bohemica and V. conica, respectively, with BUSCO completeness scores ranging from 98.71% to 99.24%. Overall, these reported genomes will provide valuable genomic resources for the evolution and ecological roles research of Verpa.
Similar content being viewed by others


Background & Summary
Mushrooms are not only a nutrient-rich food but also offer a variety of health benefits, medicinal value, and significant ecological importance1,2. Among the numerous mushroom species, Morchella, referred to as “true morel”, is highly prized for its distinctive honeycomb appearance and exceptional taste3. However, beyond Morchella, Verpa is another closely related and equally intriguing genus. The genus Verpa belongs to the phylum Ascomycota, order Pezizales, and family Morchellaceae4,5. Due to its fruiting season being slightly earlier than that of Morchella species and its morphological similarities to morels, it is often referred to as “early morels” or “false morels”6. In the wild, Verpa species are often confused with some species of the genus Morchella, such as M. diminutiva7 and M. semilibera8. Despite their many similarities in appearance, there are notable differences in their morphological structures. Specifically, in mature Morchella species, the stem is attached to the base of the cap, whereas in Verpa, the cap is attached to the top of the stem, without any attachment at the base of the stipe7,9,10. The Verpa species are widely distributed, with their presence in various regions across Asia, America, and Europe. However, compared to Morchella, research on the Verpa genus is relatively limited, with studies mainly focusing on V. bohemica and V. conica. For example, isotope analysis has found that both species are typical saprophytic fungi11, acquiring nutrients by biodegrading the substratum, thus playing an important ecological role.
Both V. bohemica and V. conica also have potential economic value in food and medicinal applications. In countries such as Italy and Turkey, V. bohemica has a long history of being collected and consumed6. Although there are still reports of poisoning from consuming morels12, and V. bohemica was previously described as a toxic look-alike species by the Food and Drug Administration (FDA)10, no studies have identified common fungal toxins like gyromitrin or coprine in V. bohemica6,10. Instead, research on V. bohemica indicates that it shares a similar amino acid profile with true morels, and like them, it is rich in proteins, vitamins, fibers, and minerals, while having a low fat content13. This suggests that V. bohemica has considerable potential as an edible mushroom, though further research is needed to investigate the mechanisms behind gastrointestinal discomfort or other adverse effects in sensitive individuals, in order to ensure its safety as food. In addition, species of Verpa have been found to contain bioactive compounds that exhibit excellent antibacterial, antifungal, antioxidant, and DNA protective properties14,15. These bioactive substances not only demonstrate significant potential for use but also have promising applications in biopharmaceutical fields. Although V. bohemica and V. conica exhibit significant potential in the food and medicinal fields, more research is needed to fully understand and utilize these species. One of the main challenges is the lack of high-quality genome sequences, which limits our understanding of their evolutionary history and ecological roles.
In this study, we constructed sub-chromosomal genome assemblies for six strains of Verpa by combining Nanopore and Illumina sequencing technologies. Hi-C data were also used to conduct chromosome-level scaffolding for V. bohemica strain 21108 and V. conica strain 21120. For each genome assembly, nearly 11,000 protein-coding genes were predicted based on an integrated approach. These high-quality genome assemblies and gene annotations offer a robust and comprehensive resource for exploring the evolutionary, ecological, and applied aspects of V. bohemica and V. conica.
Methods
Sample preparation and genome sequencing
Briefly, six Verpa strains used for sequencing were collected from six distinct regions across China (Table 1). For each strain, tissue blocks were taken from the stipe parts of the ascomata and subjected to tissue isolation to obtain pure cultures. The mycelia were then grown using liquid culture techniques. These strains were identified through a combination of ITS phylogenetic analysis and morphological characteristics16. High-quality genomic DNA was extracted from the fresh mycelia of each Verpa strain using the CTAB method17. Total RNA was extracted from each strain using TRIzol® Reagent (Invitrogen) extraction kit and purified using Plant RNA Purification Reagent (Invitrogen) purification kit.
For long-read sequencing, Nanopore sequencing libraries of all samples were constructed and sequenced using the Nanopore PromethION platform (Oxford Nanopore Technologies, UK). For short-read sequencing and RNA sequencing, 150 bp paired-end libraries were prepared and sequenced on the Illumina HiSeq4000 platform (San Diego, CA, USA). Two of the samples, V. bohemica strain 21108 and V. conica strain 21120, were prepared to construct high-throughput chromatin capture (Hi-C) sequencing libraries using the approach published previously18. The extracted DNA of V. bohemica strain 21108 and V. conica strain 21120 was digested with the DpnII and Sau3AI restriction enzymes, respectively. Both libraries were sequenced with 2 × 150 bp chemistry on the Illumina HiSeq4000 platform.
The Nanopore reads were filtered and trimmed using the NanoFilt (v2.8.0) tool of NanoPack19. Meanwhile, the raw reads generated by Illumina sequencing were trimmed and filtered to remove adapter sequences and low-quality reads using Trimmomatic (v0.39)20. A total of 35. 67 Gb of Nanopore long reads, 29.23 Gb of short reads, 26.10 Gb of transcriptome data and 78.92 Gb of Hi-C sequencing data were finally obtained (Table 2).
Genome size estimation and genome assembly
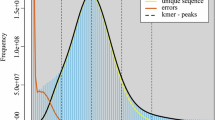
Before genome assembly, the Illumina paired-end sequencing reads were used to determine the overall characteristics of the genomes of the six Verpa strains. The 17-mer count distribution from Jellyfish (v2.3.0)21 served as input of GenomeScope (v1.0.0)22 to predict genomic features. The genome was determined to range from 47 Mb to 49 Mb in size, with heterozygosity rates ranging from 0.02% to 0.44% and the repetitive elements accounting for approximately 15% of the total length of the genome (Fig. 1b).

Genome assemblies of V. bohemica and V. conica. Images of V. bohemica strain 21108 and V. conica strain 21120 in the natural habitat (a). k-mer (17-mer) frequency distribution plot for genomes of Verpa (b). Abbreviations: He (heterozygosity). Hi-C interaction heatmaps for genomes of V. bohemica strain 21108 (c) and V. conica strain 21120 (d). Interaction frequency distribution of Hi-C links among chromosomes shows in color key of heatmap ranging from white to dark red indicating the frequency of Hi-C interaction links from low to high.
Six draft genome assemblies from filtered Nanopore reads were generated using NextDenovo (v2.4.0)23, with the parameter “-g” set according to the results of GenomeScope. The contigs of the six draft assemblies were subsequently polished over three rounds of Illumina reads using NextPolish (v1.3.1)24. For chromosome construction, Hi-C reads were aligned to the assemblies of V. bohemica strain 21108 and V. conica strain 21120, respectively, using Juicer (v1.6)25. Then the 3D-DNA pipeline (v180922)26 was used to eliminate mis-joins, anchor, order, and orient the assembled contigs. The Hi-C heatmap generated by 3D-DNA was loaded into Juicebox (v1.9.8)27 to manually correct potential errors and help determine chromosome boundaries. Finally, combining coverage from an average of 132.49 × Nanopore long reads, 101.7 × short reads, and 880.21 × Hi-C reads, we obtained six genome assemblies, of which four were sub-chromosomal assemblies and the other two were chromosome-level assemblies. The genome assembly sizes, averaged 44.38 Mb in V. bohemica and 45.40 Mb in V. conica (Table 3), were slightly smaller compared to the k-mer estimates. For the chromosome-level assemblies, all 27 contigs of V. bohemica strain 21108 were anchored and assembled consistently into 26 chromosomes, collectively spanning 44.44 Mb (Table 3, Fig. 1c), with a scaffold N50 of 1.65 Mb. While all contigs of V. conica strain 21120 were anchored and assembled into 25 chromosomes, resulting in a total assembly size of 45.03 Mb (Table 3, Fig. 1d), with a scaffold N50 of 1.61 Mb.
Different methods were employed to assess the obtained genome assemblies. BUSCO (v5.2.226)28 analysis, using Ascomycota (odb10) gene set, showed an average score of 95.53% (single-copy and duplicated). Consensus quality value (QV) of our assemblies, assessed by Merqury (v1.3)29, ranged from 40.60 to 54.13 (average of 46.80), indicating high base accuracy (Table 3). The high contiguity and completeness of the assemblies were also supported by the average mapping rates of Nanopore (95.2%), Illumina (98.7%), and RNA-seq (92.5%) read mapping.
Genome annotation
Prior to gene structure prediction and annotation, repetitive sequences in the genome assemblies were identified and masked. RepeatModeler (v2.0.2)30 was used to build the de novo repeat libraries of each genome assembly. Then, sequences from the assembly were aligned to the de novo repeat libraries and known repeat databases to identify and mask repetitive elements, using RepeatMasker (v4.1.2)31. Consequently, we identified an average length of 4.07 Mb (average ratio: 9.17%) and 4.27 Mb (average ratio: 9.40%) repetitive sequences in V. bohemica and V. conica (Table 4), respectively. Notably, major repetitive sequence classes differed between the two species. In V. bohemica, long interspersed nuclear elements (LINEs) and long terminal repeat elements (LTRs) accounted for the majority of repetitive content across strains, with strain 20020 containing 1.04 Mb of LINEs and 1.17 Mb of LTR elements (Table 4). In contrast, V. conica featured rolling-circle transposons in strains 21110 and 21120, which were undetected in V. bohemica (Table 4). Unclassified repeats also represented a significant portion in V. conica, with strain 21120 harboring 2.98 Mb (Table 4). These compositional differences contributed to overall variation in repetitive sequence profiles between the species. Additionally, both species showed fewer repetitive sequences than predicted by GenomeScope, which may explain why the final genome assemblies were smaller than estimated, yet still maintained high completeness in BUSCO assessments.
Protein-coding genes were predicted using GETA (v2.6.1) pipeline (https://github.com/chenlianfu/geta), integrating evidence from ab initio, homology-based, and RNA-seq-based prediction methods. The ab initio prediction was conducted using Augustus (v3.4.0)32. The GeneWise (v2.4.1)33 was applied for homology-based prediction using whole-genome homologous protein sequences of Tuber melanosporum34, M. importuna35, Pyronema confluens36 and Gyromitra esculenta37. RNA-seq reads obtained from each strain were aligned to the corresponding assembly using HISAT2 (v2.1.0)38, and protein-coding regions were predicted using TransDecoder (v5.5.0) (https://github.com/TransDecoder/TransDecoder). The predictions from the three methods were then integrated and filtered against the PFAM database. Finally, we annotated an average of 11,024 and 11,052 protein-coding genes for V. bohemica and V. conica (Table 5), respectively. To validate the protein annotations and assess the consistency among the genomes of different V. bohemica and V. conica strains, we conducted a genome-wide synteny analysis using JCVI (v1.4.21)39, based on protein sequences to identify corresponding syntenic blocks between all pairs of strains. The results demonstrated that, despite the limited utilization of Hi-C data, the sub-chromosome level assemblies of V. bohemica (strain 20020, 20124) and strain 21108 exhibited high collinearity (Fig. 2a). Similarly, the sub-chromosome level assemblies of V. conica (strain 21110, 21117) and strain 21120 also showed a high degree of collinearity (Fig. 2a). Additionally, BUSCO analysis was performed using the Ascomycota (odb10) protein dataset to confirm the completeness of the annotations. The results showed that 98.71% to 99.24% (average 99.04%) of single-copy and duplicated conserved genes were identified across the six Verpa strains (Fig. 2b). Specifically, V. bohemica and V. conica contained an average of 97.73% and 97.95% complete single-copy genes, respectively, with low proportions of duplicated (1.23% and 1.17%) genes (Fig. 2b). These results highlight the exceptional quality of the genome assemblies and annotations.

Genome assemblies and BUSCO assessments of Verpa Species. (a) Synteny relationships. Left column numbers represent strains of V. bohemica (20020, 20124, 21108) and V. conica (21110, 21117, 21120). Other numbers correspond to scaffold or chromosome identifiers within each genome assembly. Blocks colored in gray highlight conserved syntenic regions. (b) BUSCO assessment results.
To annotate the predicted protein-coding genes, we performed alignments against several databases, including NR, Swiss-Prot, PFAM, InterPro, KEGG, and GO. For NR and Swiss-Prot annotations, DIAMOND (v2.0.2)40 was used with BLASTp, filtering results by e-value (1e-5) and identity threshold (20%). HMMER41 was employed to identify PFAM domains. InterPro annotations were obtained online42. KEGG annotations were assigned using BlastKOALA43 platform for pathway mapping, and GO terms were assigned using the PANNZER244 web server. In total, at least 96.50% (average 96.81%) of gene models in each Verpa strain genome were annotated in at least one database (Table 5), highlighting the robustness of the gene prediction results and providing a robust foundation for further genomic studies of these species.
Data Records
The raw sequencing data have been deposited in the National Center for Biotechnology Information (NCBI) under the BioProject accession number of PRJNA904943 (SRR22519296, SRR22519297, SRR22519298, SRR22519299, SRR22519300, SRR22519301, SRR22519302, SRR22519303, SRR22519304, SRR22519305, SRR22519306, SRR22519307, SRR22519308, SRR22519309, SRR22519310, SRR22519311, SRR22519312, SRR22519313, SRR31828003, and SRR31828004)45,46,47,48,49,50,51,52,53,54,55,56,57,58,59,60,61,62,63,64. The Sequence Read Archive (SRA) accession numbers SRR22519296-SRR22519313 correspond to genome and RNA sequencing data, while SRR31828003-SRR31828004 represent Hi-C sequencing data. The sub-chromosomal genome assemblies are available in NCBI GenBank under accession numbers GCA_033030385.165, GCA_033030375.166, GCA_033030345.167, GCA_033030205.168, GCA_033030305.169, and GCA_033030425.170. Additionally, all genome assemblies and gene annotation results have been archived in the figshare database and can be accessed at https://doi.org/10.6084/m9.figshare.28141691.v171.
Technical Validation
The quality of the genome assemblies was assessed across six Verpa samples in the following aspects: (1) Genome completeness was evaluated using BUSCO v5.2.2 with the ascomycota_odb10 orthologous gene set. (2) Mapping rates for Nanopore, Illumina, and RNA-seq reads were used to evaluate assembly accuracy. (3) Merqury was employed to estimate the consensus quality value of the genome assemblies using Illumina k-mers. The quality of the predicted gene set was supported by BUSCO cores and functional annotation results.
Code availability
No specific code or script was used in this work. All parameters and software versions were described in the Methods.
References
Manzi, P., Aguzzi, A. & Pizzoferrato, L. Nutritional value of mushrooms widely consumed in Italy. Food Chem 73(3), 321–325, https://doi.org/10.1016/S0308-8146(00)00304-6 (2001).
Bhambri, A., Srivastava, M., Mahale, V. G., Mahale, S. & Karn, S. K. Mushrooms as potential sources of active metabolites and medicines. Front Microbiol 13, 837266, https://doi.org/10.3389/fmicb.2022.837266 (2022).
Liu, W. et al. Large-scale field cultivation of Morchella and relevance of basic knowledge for its steady production. J Fungi (Basel) 9(8), 855, https://doi.org/10.3390/jof9080855 (2023).
Bunyard, B. A., Nicholson, M. S. & Royse, D. J. Phylogenetic resolution of Morchella, Verpa, and Disciotis [Pezizales: Morchellaceae] based on restriction enzyme analysis of the 28S ribosomal RNA gene. Exp Mycol 19(3), 223–33, https://doi.org/10.1006/emyc.1995.1027 (1995).
Trappe, M. J., Trappe, J. M. & Bonito, G. M. Kalapuya brunnea gen. & sp. nov. and its relationship to the other sequestrate genera in Morchellaceae. Mycologia 102(5), 1058–65, https://doi.org/10.3852/09-232 (2010).
Davoli, P. & Sitta, N. Early morels and little friars, or a short essay on the edibility of Verpa bohemica. Fungi 8(1), 5 (2015).
Wei, C. et al. Identification and mycelial growth characteristics of two wild strains of Verpa. J. Fungal Res. 19, 263–270, https://doi.org/10.13341/j.jfr.2021.1448 (2021).
Wu, D. et al. Study on species diversity of wild Morchella in Xinjiang. Sci Technol Food Ind 36(2), 167–172, https://doi.org/10.13386/j.issn1002-0306.2015.02.027 (2015).
Lagrange, E. & Vernoux, J. P. Warning on false or true morels and button mushrooms with potential toxicity linked to hydrazinic toxins: an update. Toxins (Basel) 12(8), 345, https://doi.org/10.3390/toxins12080482 (2020).
Gecan, J. S. & Cichowicz, S. M. Toxic mushroom contamination of wild mushrooms in commercial distribution. J Food Prot 56(8), 730–734, https://doi.org/10.4315/0362-028x-56.8.730 (1993).
Hobbie, E. A., Weber, N. S. & Trappe, J. M. Mycorrhizal vs saprotrophic status of fungi: the isotopic evidence. New Phytol 150(3), 601–610, https://doi.org/10.1046/j.1469-8137.2001.00134.x (2001).
Demorest, H. et al. Outbreak linked to morel mushroom exposure - Montana, 2023. MMWR Morb Mortal Wkly Rep 73(10), 219–224, https://doi.org/10.15585/mmwr.mm7310a1 (2024).
McKellar, R. L. & Kohrman, R. E. Amino acid composition of the morel mushroom. J Agric Food Chem 23(3), 464–7, https://doi.org/10.1021/jf60199a047 (1975).
Shameem, N., Kamili, A. N., Ahmad, M., Masoodi, F. A. & Parray, J. A. Antimicrobial activity of crude fractions and morel compounds from wild edible mushrooms of north western Himalaya. Microb Pathog 105, 356–360, https://doi.org/10.1016/j.micpath.2017.03.005 (2017).
Elmastas, M., Isildak, O., Turkekul, I. & Temur, N. Determination of antioxidant activity and antioxidant compounds in wild edible mushrooms. J Food Compos Anal 20(3-4), 337–345, https://doi.org/10.1016/j.jfca.2006.07.003 (2007).
Sun, W. et al. Structure of the mating-type genes and mating systems of Verpa bohemica and Verpa conica (Ascomycota, Pezizomycotina). J Fungi (Basel) 9(12), 1202, https://doi.org/10.3390/jof9121202 (2023).
Porebski, S., Bailey, L. G. & Baum, B. R. Modification of a CTAB DNA extraction protocol for plants containing high polysaccharide and polyphenol components. Plant Mol Biol Rep 15(1), 8–15, https://doi.org/10.1007/BF02772108 (1997).
Belton, J. M. et al. Hi-C: a comprehensive technique to capture the conformation of genomes. Methods 58(3), 268–76, https://doi.org/10.1016/j.ymeth.2012.05.001 (2012).
De Coster, W., D’Hert, S., Schultz, D. T., Cruts, M. & Van Broeckhoven, C. NanoPack: visualizing and processing long-read sequencing data. Bioinformatics 34(15), 2666–2669, https://doi.org/10.1093/bioinformatics/bty149 (2018).
Bolger, A. M., Lohse, M. & Usadel, B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30(15), 2114–20, https://doi.org/10.1093/bioinformatics/btu170 (2014).
Marçais, G. & Kingsford, C. A fast, lock-free approach for efficient parallel counting of occurrences of k-mers. Bioinformatics 27(6), 764–70, https://doi.org/10.1093/bioinformatics/btr011 (2011).
Vurture, G. W. et al. GenomeScope: fast reference-free genome profiling from short reads. Bioinformatics 33(14), 2202–2204, https://doi.org/10.1093/bioinformatics/btx153 (2017).
Hu, J. et al. NextDenovo: an efficient error correction and accurate assembly tool for noisy long reads. Genome Biol 25(1), 107, https://doi.org/10.1186/s13059-024-03252-4 (2024).
Hu, J., Fan, J., Sun, Z. & Liu, S. NextPolish: a fast and efficient genome polishing tool for long-read assembly. Bioinformatics 36(7), 2253–2255, https://doi.org/10.1093/bioinformatics/btz891 (2020).
Durand, N. C. et al. Juicer provides a one-click system for analyzing loop-resolution Hi-C experiments. Cell Syst 3(1), 95–8, https://doi.org/10.1016/j.cels.2016.07.002 (2016).
Dudchenko, O. et al. De novo assembly of the Aedes aegypti genome using Hi-C yields chromosome-length scaffolds. Science 356(6333), 92–95, https://doi.org/10.1126/science.aal3327 (2017).
Durand, N. C. et al. Juicebox provides a visualization system for Hi-C contact maps with unlimited zoom. Cell Syst 3(1), 99–101, https://doi.org/10.1016/j.cels.2015.07.012 (2016).
Simão, F. A., Waterhouse, R. M., Ioannidis, P., Kriventseva, E. V. & Zdobnov, E. M. BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 31(19), 3210–2, https://doi.org/10.1093/bioinformatics/btv351 (2015).
Rhie, A., Walenz, B. P., Koren, S. & Phillippy, A. M. Merqury: reference-free quality, completeness, and phasing assessment for genome assemblies. Genome Biol 21(1), 245, https://doi.org/10.1186/s13059-020-02134-9 (2020).
Flynn, J. M. et al. RepeatModeler2 for automated genomic discovery of transposable element families. Proc Natl Acad Sci USA 117(17), 9451–9457, https://doi.org/10.1073/pnas.1921046117 (2020).
Chen, N. Using RepeatMasker to identify repetitive elements in genomic sequences. Curr Protoc Bioinformatics Chapter 4, Unit 4.10, https://doi.org/10.1002/0471250953.bi0410s05 (2004).
Stanke, M. et al. AUGUSTUS: ab initio prediction of alternative transcripts. Nucleic Acids Res 34, 435–9, https://doi.org/10.1093/nar/gkl200 (2006).
Birney, E., Clamp, M. & Durbin, R. GeneWise and Genomewise. Genome Res 14(5), 988–95, https://doi.org/10.1101/gr.1865504 (2004).
Martin, F. et al. Périgord black truffle genome uncovers evolutionary origins and mechanisms of symbiosis. Nature 464(7291), 1033–8, https://doi.org/10.1038/nature08867 (2010).
Liu, W., Chen, L., Cai, Y., Zhang, Q. & Bian, Y. Opposite polarity monospore genome de novo sequencing and comparative analysis reveal the possible heterothallic life cycle of Morchella importuna. Int J Mol Sci 19(9), 2525, https://doi.org/10.3390/ijms19092525 (2018).
Traeger, S. et al. The genome and development-dependent transcriptomes of Pyronema confluens: a window into fungal evolution. PLoS Genet 9(9), e1003820, https://doi.org/10.1371/journal.pgen.1003820 (2013).
Liu, W. et al. Two near-chromosomal-level genomes of globally-distributed Macroascomycete based on single-molecule fluorescence and Hi-C methods. Sci Data 11(1), 964, https://doi.org/10.1038/s41597-024-03794-z (2024).
Kim, D., Langmead, B. & Salzberg, S. L. HISAT: a fast spliced aligner with low memory requirements. Nat Methods 12(4), 357–60, https://doi.org/10.1038/nmeth.3317 (2015).
Tang, H. et al. JCVI: a versatile toolkit for comparative genomics analysis. Imeta 3(4), e211, https://doi.org/10.1002/imt2.211 (2024).
Buchfink, B., Xie, C. & Huson, D. H. Fast and sensitive protein alignment using DIAMOND. Nat Methods 12(1), 59–60, https://doi.org/10.1038/nmeth.3176 (2015).
Söding, J. Protein homology detection by HMM-HMM comparison. Bioinformatics 21(7), 951–60, https://doi.org/10.1093/bioinformatics/bti125 (2005).
Mitchell, A. L. et al. InterPro in 2019: improving coverage, classification and access to protein sequence annotations. Nucleic Acids Res 47(D1), D351–d360, https://doi.org/10.1093/nar/gky1100 (2019).
Kanehisa, M., Sato, Y. & Morishima, K. BlastKOALA and GhostKOALA: KEGG tools for functional characterization of genome and metagenome sequences. J Mol Biol 428(4), 726–731, https://doi.org/10.1016/j.jmb.2015.11.006 (2016).
Törönen, P., Medlar, A. & Holm, L. PANNZER2: a rapid functional annotation web server. Nucleic Acids Res 46(W1), W84–W88, https://doi.org/10.1093/nar/gky350 (2018).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR22519296 (2025).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR22519297 (2025).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR22519298 (2025).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR22519299 (2025).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR22519300 (2025).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR22519301 (2025).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR22519302 (2025).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR22519303 (2025).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR22519304 (2025).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR22519305 (2025).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR22519306 (2025).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR22519307 (2025).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR22519308 (2025).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR22519309 (2025).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR22519310 (2025).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR22519311 (2025).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR22519312 (2025).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR22519313 (2025).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR31828003 (2025).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR31828004 (2025).
NCBI GenBank http://identifiers.org/ncbi/insdc.gca:GCA_033030385.1 (2023).
NCBI GenBank http://identifiers.org/ncbi/insdc.gca:GCA_033030375.1 (2023).
NCBI GenBank http://identifiers.org/ncbi/insdc.gca:GCA_033030345.1 (2023).
NCBI GenBank http://identifiers.org/ncbi/insdc.gca:GCA_033030205.1 (2023).
NCBI GenBank http://identifiers.org/ncbi/insdc.gca:GCA_033030305.1 (2023).
NCBI GenBank http://identifiers.org/ncbi/insdc.gca:GCA_033030425.1 (2023).
Yan, Z. Genome assemblies and annotation of Verpa bohemica and Verpa conica. Figshare https://doi.org/10.6084/m9.figshare.28141691.v1 (2025).
Acknowledgements
This work was supported by the Yunnan Key Project of Science and Technology (202402AE090030), the Basic Research Project of Yunnan Provincial Department of Science and Technology (202101AT070541), Yunnan Revitalization Talent Support Program to Jesús Pérez-Moreno and Xinhua He, and the Yunnan Technology Innovation Program (202205AD160036) to Fuqiang Yu.
Author information
Authors and Affiliations
Contributions
F.Q.Y. and W.L. conceived and supervised the project; X.F.S., L.Y.W., J.Z., X.G. and B.W. collected and isolated the samples; X.F.S., Y.L.C. and L.Y.W. conducted cultivation, molecular identification and sequencing experiments; Z.Y.Y., W.L., and W.H.S. assembled and annotated the genomes; Z.Y.Y. and W.L. drafted the manuscript, with contribution from all authors. P.X.H., F.Q. Y. and W.L. oversaw the writing, revised the manuscript, and ensured the overall progress of the project.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Yan, Z., Shi, X., Cai, Y. et al. Chromosome-level genome assemblies of Verpa bohemica and Verpa conica. Sci Data 12, 880 (2025). https://doi.org/10.1038/s41597-025-05224-0
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41597-025-05224-0
