
Abstract
The family Scarabaeidae is one of the largest and most ecologically significant groups within the order Coleoptera, comprising over 35,000 described species. However, the limited availability of high-quality genome assemblies has hindered comprehensive studies on their ecology and evolutionary biology. In this study, we present a high-quality, chromosome-level genome assembly of Kibakoganea sinica, generated by integrating PacBio HiFi, Illumina, and Hi-C sequencing data. The final assembly spans 601.44 Mb, comprising 23 scaffolds (scaffold N50: 60.23 Mb) and 70 contigs (contig N50: 24.49 Mb), with 99.57% of the total assembly (598.84 Mb) successfully anchored to 10 chromosomes. BUSCO analysis (n = 1,367) indicates a high level of completeness, with 99.2% of genes detected: 95.2% as single-copy and 4.0% as duplicated. Repetitive elements account for 44.43% (267.24 Mb) of the genome, and a total of 12,940 protein-coding genes were predicted. This chromosome-scale genome assembly provides a foundational resource for future research into the biology and adaptation of Scarabaeidae.
Similar content being viewed by others


Background & Summary
The Scarabaeidae family, commonly known as scarab beetles, is one of the most diverse lineages within the order Coleoptera, comprising approximately 35,000 described species worldwide1. These beetles are distributed worldwide, occurring on all continents except Antarctica and occupying a wide range of ecosystems, including forests, grasslands, deserts, and agricultural landscapes2. Based on their feeding strategies, scarab beetles are broadly classified into two groups: coprophagous (dung-feeding) and phytophagous (plant-feeding) species3,4. The coprophagous group, also known as dung beetles, includes two ecologically and economically significant subfamilies: Scarabaeinae and Aphodiinae5,6. Among them, members of Scarabaeinae are considered the “true dung beetles,” primarily utilizing fresh dung as both a food source and for reproductive purposes7. In contrast, phytophagous scarabs, commonly known as Pleurosticti8, comprise more than 20,000 species, representing nearly 70% of the entire Scarabaeidae family4,9. These beetles feed on a wide array of plant materials, including leaves, roots, decaying wood, and partially decomposed plant litter10. Such a diet requires specialized digestive adaptations to break down complex plant cell wall components, including lignocellulose and various hemicelluloses, which are chemically resistant to degradation11.
High-quality genome assemblies are essential for uncovering the genetic mechanisms underlying feeding adaptations in Scarabaeidae beetles. A recent study generated a chromosome-scale genome assembly and comprehensive intestinal transcriptome for Trypoxylus dichotomus (Coleoptera: Scarabaeidae), offering valuable insights into its ability to digest lignocellulose-rich plant material12. As of April 2025, a total of 62 Scarabaeidae genomes have been deposited in the NCBI database. However, only 13 species have been reported at the chromosome level. The majority of the remaining genomes, assembled from short-read sequencing data, are of limited quality, with scaffold N50 values typically below 100 kb. This shortage of high-quality genomic resources continues to hinder in-depth research on environmental adaptation, dietary specialization, and the evolutionary diversification of Scarabaeidae.
To enhance our understanding of adaptive evolution ecology, we propose assembling a chromosome-level genome of Kibakoganea sinica, Bouchard, 2005 (Coleoptera: Scarabaeidae), combining PacBio HiFi, Illumina, and Hi-C data. We annotated repeats, non-coding RNAs, and protein-coding genes. This high-quality genome assembly of K. sinica provides valuable insights into the evolution and ecological adaptation of the Scarabaeoidea superfamily.
Methods
Sample collection and sequencing
A K. sinica pupa was collected in Guizhou, China, on December 6, 2023, and used for genome sequencing, including Illumina, PacBio, Hi-C, and RNA sequencing. To minimize contamination, the sample was carefully rinsed in phosphate-buffered saline for 10 minutes, flash-frozen in liquid nitrogen for 20 minutes, and subsequently stored at –80 °C until further processing.
Genomic DNA and RNA were isolated from the specimen using the DNeasy Blood & Tissue Kit (Qiagen) and TRIzol Reagent (Thermo Fisher Scientific), respectively, in accordance with the manufacturers’ instructions. Short-read libraries were prepared without PCR amplification using the Illumina TruSeq DNA PCR-Free Kit, generating 150 bp paired-end reads with 350 bp inserts. For Hi-C sequencing, we implemented a standard protocol13, including DNA crosslinking, MboI digestion, end repair, and DNA purification. All short-read sequencing was conducted using an Illumina NovaSeq. 6000 system. For long-read sequencing, we constructed a 20 kb SMRTbell library (PacBio SMRTbell Express Template Prep Kit 2.0) and sequenced it on the PacBio Sequel II system in HiFi mode. Library construction and sequencing were conducted at Berry Genomics (Beijing, China). Our sequencing generated a total of 160.95 Gb of high-quality data, including 36.70 Gb (61.02 × coverage) PacBio HiFi reads, 56.09 Gb (93.26×) Illumina short reads, and 58.56 Gb (97.36×) Hi-C data (Table 1).
Genome assembly
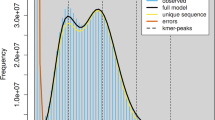
Raw Illumina reads were processed for quality control using BBTools v38.8214. Duplicate reads were first removed with “clumpify.sh”. Subsequently, bbduk.sh was applied to trim low-quality bases (Q < 20) and adapter sequences according to strict quality criteria. Specifically, sequences with quality scores below 20 were discarded, reads containing more than five Ns were filtered out, poly-A/G/C tails longer than 10 bp were trimmed, and overlapping paired reads were corrected. To estimate the genome size, heterozygosity, and repetitive sequence content in the K. sinica genome, a genome survey was conducted using GenomeScope v2.015. The estimated genome size ranged from 567.23 to 568.25 Mb, with repetitive sequences accounting for 31.57–31.58% of the genome. Additionally, the survey indicated a high heterozygosity rate, estimated at 2.11–2.13% (Fig. 1).

Genome size estimation for Kibakoganea sinica based on GenomeScope.
The initial genome assembly was generated using PacBio HiFi long reads and assembled with Hifiasm v0.19.816 under default parameters. To reduce heterozygosity, we applied Purge_Dups v1.2.517 with a haploid cutoff of 70 (-s 70) to identify and remove haplotigs. For chromosome-scale scaffolding, Hi-C reads were first quality-filtered and then aligned to the assembly using Juicer v1.6.218. Contigs were subsequently anchored and ordered into chromosomes using 3D-DNA v.18092219. The final assembly was manually verified and corrected in Juicebox v.1.11.018 to resolve potential misjoins or orientation errors. To ensure the assembly’s purity, we screened for contaminants using MMseqs. 2 v1.120 against the NCBI nucleotide (nt) and UniVec databases, removing any detected foreign sequences. Potential vector contaminants were identified using v2.11.021 against the UniVec database, with sequences showing > 90% similarity flagged as contaminants. Additional sequences exhibiting > 80% similarity were further validated through BLASTN searches against the NCBI nucleotide database (nt). All identified bacterial and fungal contaminants were thoroughly removed from the assembly scaffolds. Telomeric regions of each chromosome were identified using the TeloExplorer module in QuarTeT v1.2.122. The presence of continuous telomeric repeat motifs (TTAGG) within 10,000 base pairs at both ends of a chromosome was used as the criterion for confirming telomere localization (Table 3). The final chromosome-scale assembly of K. sinica spans 601.44 Mb, consisting of 23 scaffolds and 70 contigs, which is consistent with the genome size estimated in the genome survey. The assembly demonstrates high continuity, with scaffold and contig N50 values of 60.23 Mb and 24.49 Mb, respectively (Table 2). Notably, 99.57% of the assembly (598.84 Mb) was successfully anchored to 10 chromosomes, with individual chromosome lengths ranging from 16.33 Mb to 103.53 Mb (Table 3; Fig. 2; Fig. 3). The chromosome sequence names are assigned based on sequence length, with the longest sequence labeled as chromosome 1 (Table 3). Moreover, the BUSCO assessment revealed a genome assembly completeness of 99.2% (Table 2). Collectively, these findings demonstrate that our genome assembly achieves outstanding continuity and structural integrity.

The chromosomal heatmap visualization of Kibakoganea sinica genome assembly displays complete chromosomes in blue, with individual contigs demarcated by green borders.

The genomic features of Kibakoganea sinica are displayed in a circular layout. Moving inward from the outermost ring, the visualization depicts (1) chromosome length, (2) GC content, (3) gene density, and (4) various repetitive elements, including transposable elements (DNA, SINEs, LINEs, and LTRs), along with simple repeat sequences.
Genome annotation
The species-specific repeat library of K. sinica was generated using RepeatModeler v2.0.423 and integrated with known repeats from RepBase-2013090924 and Dfam 3.525 to construct a comprehensive repeat database. The custom repeat database was employed as input for RepeatMasker v4.1.426 to systematically identify and mask repetitive elements throughout the genome, followed by soft-masking of these regions. The analysis revealed that repetitive sequences account for 44.43% of the K. sinica genome assembly. These elements were classified into major categories, including unclassified elements (18.09%), LINE transposons (6.81%), LTR transposons (8.53%), DNA transposons (17.00%), and other repeat types (Table 4).
Non-coding RNAs (ncRNAs) in K. sinica were identified using Infernal v1.1.427 with the Rfam v14.10 database28, while tRNA detection was performed with tRNAscan-SE v2.0.929. The analysis revealed a diverse ncRNA repertoire, comprising 312 tRNAs, 101 rRNAs, 74 microRNAs, and 69 small nuclear RNAs, totaling 596 ncRNAs (Table 4).
Protein-coding gene annotation of the K. sinica genome was performed using MAKER v3.01.0330, integrating transcriptomic evidence, ab initio predictions, and protein homology data. Transcriptome sequences were aligned to the genome using HISAT2 v2.2.131, followed by genome-guided assembly with StringTie v2.1.632. For ab initio gene prediction, BRAKER v2.1.633 was employed, incorporating GeneMark-ES/ET/EP 4.68_lic34 and Augustus v3.4.035, both of which were trained using transcriptomic sequences and protein data from OrthoDB v1136. Additionally, homology-based gene prediction was conducted using GeMoMa v1.937, utilizing protein sequences from five reference species: Drosophila melanogaster (GCF_000001215.4)38, Apis mellifera (GCA_003254395.2)39, Coccinella septempunctata (GCA_907165205.1)40, Prosopocoilus inquinatus (GCA_036172665.1)41, and Tribolium castaneum (GCA_031307605.1)42 (Table 5). The annotation pipeline identified 12,940 protein-coding genes in the K. sinica genome, with an average gene length of 14,792.6 bp (Table 4). On average, each gene contained 6.3 exons, 5.3 introns, and 6.1 coding sequences (CDS). Gene structure analysis revealed mean lengths of 357.3 bp (exons), 2500.2 bp (introns), and 272.5 bp (CDS). To assess the quality of gene predictions, we evaluated gene set completeness using BUSCO with the Insecta dataset (n = 1,367). The results showed 80.6% (1,102) single-copy, 18.5% (253) duplicated, 0.1% (2) missing, and 0.8% (10) fragmented BUSCOs, confirming the high accuracy and reliability of the annotation.
Functional annotation was performed by aligning protein sequences against the UniProtKB database using DIAMOND v2.0.1143. Additionally, Gene Ontology (GO) terms, KEGG/Reactome pathways, and protein domains were annotated using eggNOGmapper v2.0.1444 and InterProScan 5.53-87.045. The InterProScan analysis integrated data from five databases: Pfam46, SMART47, Superfamily48, Gene3D49, and CDD50. Functional annotation revealed 11,414 COG categories, 10,333 GO terms, and 5,009 KEGG pathways in K. sinica, based on the integration of InterProScan and eggNOG annotations. Chromosomal features, including repeat elements, gene density, and GC content, were visualized using TBtools51.
Data Records
The sequencing data generated in this study are available under the following National Center for Biotechnology Information (NCBI) SRA accession numbers: transcriptome reads (SRR31019928)52, Hi-C data (SRR31019929)53, Illumina short reads (SRR31019930)54, and PacBio HiFi long reads (SRR31019931)55. The final genome assembly is available under NCBI accession GCA_043790905.156. Genome annotation data, including repetitive elements, gene structure predictions, and functional annotations, have been deposited in Figshare57.
Technical Validation
We evaluated genome assembly quality using two complementary approaches. First, assembly completeness was assessed with BUSCO v5.0.458 against the Insecta reference set (n = 1,367 conserved single-copy orthologs). The assembly exhibited a BUSCO completeness of 99.2%, with 95.2% of genes in single-copy, 4.0% duplicated, 0.2% fragmented, and 0.6% missing. Second, the assembly accuracy was assessed by calculating mapping rates through the alignment of PacBio, Illumina, and RNA-seq reads to the final assembly using Minimap2 v. 2.2359 and SAMtools v. 1.960. The assembly demonstrated high mapping rates for PacBio (99.85%), Illumina (89.71%), and RNA-seq (93.71%) reads. These comprehensive analyses confirm the high quality of our genome assemblies.
Code availability
No specific script was used in this work. All commands and pipelines used in data processing were executed according to the manual and protocols of the corresponding bioinformatic software.
References
Ratcliffe, B. C. & Cave, R. D. The Dynastinae (Coleoptera: Scarabaeidae) of the Bahamas with a description of a new species of Cyclocephala from Great Inagua Island. Insecta Mundi. 24, 1–10 (2008).
Breeschoten, T. et al. Phylogenetics and biogeography of the dung beetle genus Onthophagus inferred from mitochondrial genomes[J]. Mol Phylogenet Evol 105, 86–95 (2016).
Song, N. & Zhang, H. The mitochondrial genomes of phytophagous scarab beetles and systematic implications. J. Insect Sci. 18, 11 (2018).
Bohacz, C., du Harrison, J. G. & Ahrens, D. Comparative morphology of antennal surface structures in pleurostict scarab beetles (Coleoptera). Zoomorphology 139, 327–346 (2020).
Rodrigues, S. R., de Barros, A. T. M., Puker, A. & Taira, T. L. Diversity of coprophagous scarab beetles (Coleoptera, Scarabaeidae) collected with flight intercept trap in the Southern Pantanal, Brazil. Biota Neotrop 10, 123–127 (2010).
Barbero, E., Palestrini, C. & Rolando, A. Dung beetle conservation: Effects of habitat and resource selection (Coleoptera: Scarabaeoidea). J. Insect Conserv. 3, 75–84 (1999).
Bai, M. et al. Mandible evolution in the Scarabaeinae (Coleoptera: Scarabaeidae) and adaptations to coprophagous habits. Front. Zool. 12, 30 (2015).
Erichson, W. F. Naturgeschichte der Insecten Deutschlands: Erste Abtheilung. Coleoptera; Nabu Press: Charleston, SC, USA, 1847; Volume 1.
Gunter, N. L., Weir, T. A., Slipinksi, A., Bocak, L. & Cameron, S. L. If dung beetles (Scarabaeidae: Scarabaeinae) arose in association with dinosaurs, did they also suffer a mass co-extinction at the K-Pg boundary? PLoS ONE 11, e0153570 (2016).
Chandra, K. & Gupta, D. An inventory of scarab beetles (Coleoptera: Scarabaeidae) of Achanakmar-Amarkantak biosphere reserve, Chhattisgarh, India. Int. J. Sci. Nat. 3, 886–891 (2012).
Holter, P. & Scholtz, C. H. Elongated hindguts in desert-living dung beetles (Scarabaeidae: Scarabaeinae) feeding on dry dung pellets or plant litter. J Morphol. 274, 657–662 (2013).
Wang, Q., Liu, L., Zhang, S., Wu, H. & Huang, J. A chromosome-level genome assembly and intestinal transcriptome of Trypoxylus dichotomus (Coleoptera: Scarabaeidae) to understand its lignocellulose digestion ability. GigaScience. 11, giac059 (2022).
Belton, J. M. et al. Hi-C: A comprehensive technique to capture the conformation of genomes. Methods. 58, 268–276 (2012).
Bushnell, B. BBtools. Available online: https://sourceforge.net/projects/bbmap/ (accessed on 1 October 2022) (2014).
Ranallo-Benavidez, T. R., Jaron, K. S. & Schatz, M. C. GenomeScope 2.0 and Smudgeplot for reference-free profiling of polyploid genomes. Nat Commun. 11, 1432 (2020).
Cheng, H., Concepcion, G. T., Feng, X., Zhang, H. & Li, H. Haplotype-resolved de novo assembly using phased assembly graphs with hifiasm. Nat Methods. 18, 170–175 (2021).
Guan, D. et al. Identifying and removing haplotypic duplication in primary genome assemblies. Bioinformatics. 36, 2896–2898 (2020).
Durand, N. C. et al. Juicer Provides a One-Click System for Analyzing Loop-Resolution Hi-C Experiments. Cell Syst. 3, 95–98 (2016).
Dudchenko, O. et al. De novo assembly of the Aedes aegypti genome using Hi-C yields chromosome-length scaffolds. Science. 356, 92–95 (2017).
Steinegger, M. & Soding, J. MMseqs. 2 enables sensitive protein sequence searching for the analysisof massive datasets. Nat. Biotechnol. 35, 1026–1028 (2017).
Altschul, S. F., Gish, W., Miller, W., Myers, E. W. & Lipman, D. J. Basic local alignment search tool. J. Mol. Biol. 215, 403–410 (1990).
Lin, Y. et al. QuarTeT: A telomere-To-Telomere toolkit for gap-free genome assembly and centromeric repeat identification. Hortic. Res. 10, uhad127 (2023).
Flynn, J. M. et al. RepeatModeler2 for automated genomic discovery of transposable element families. Proc. Natl. Acad. Sci. USA. 117, 9451–9457 (2020).
Bao, W., Kojima, K. K. & Kohany, O. Repbase Update, a database of repetitive elements in eukaryotic genomes. Mob. Dna. 6, 11 (2015).
Hubley, R. et al. The Dfam database of repetitive DNA families. Nucleic Acids Res. 44, D81–D89 (2016).
Smit, A. F. A., Hubley, R. & Green, P. RepeatMasker Open-4.0. Available online: http://www.repeatmasker.org (accessed on 1 October 2022) (2013–2015).
Nawrocki, E. P. & Eddy, S. R. Infernal 1.1: 100-fold faster RNA homology searches. Bioinformatics. 29, 2933–2935 (2013).
Griffiths-Jones, S. et al. Rfam: annotating noncoding RNAs in complete genomes. Nucleic Acids Res. 33, D121–124 (2005).
Chan, P. P. & Lowe, T. M. TRNAscan-SE: Searching for tRNA genes in genomic sequences. Methods Mol Biol. 1962, 1–14 (2019).
Holt, C. & Yandell, M. MAKER2: An annotation pipeline and genome-database management tool for second-generation genome projects. Bmc Bioinformatics. 12, 491 (2011).
Kim, D., Langmead, B. & Salzberg, S. L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods. 12, 357–360 (2015).
Kovaka, S. et al. Transcriptome assembly from long-read RNA-seq alignments with StringTie2. Genome Biol. 20, 278 (2019).
Bruna, T., Hoff, K. J., Lomsadze, A., Stanke, M. & Borodovsky, M. BRAKER2: Automatic eukaryotic genome annotation with GeneMark-EP+ and AUGUSTUS supported by a protein database. Nar Genom. Bioinform. 3, lqaa108 (2021).
Bruna, T., Lomsadze, A. & Borodovsky, M. GeneMark-EP: Eukaryotic gene prediction with self-training in the space of genes and proteins. Nar Genom. Bioinform. 2, lqaa26 (2020).
Stanke, M., Steinkamp, R., Waack, S. & Morgenstern, B. AUGUSTUS: A web server for gene finding in eukaryotes. Nucleic Acids Res. 32, W309–W312 (2004).
Kriventseva, E. V. et al. OrthoDB v10: Sampling the diversity of animal, plant, fungal, protist, bacterial and viral genomes for evolutionary and functional annotations of orthologs. Nucleic Acids Res. 47, D807–D811 (2019).
Keilwagen, J., Hartung, F., Paulini, M., Twardziok, S. O. & Grau, J. Combining RNA-seq data and homology-based gene prediction for plants, animals and fungi. Bmc Bioinformatics. 19, 189 (2018).
Hoskins, R. A. et al. The Release 6 reference sequence of the Drosophila melanogaster genome. Genome res. 25, 445–458 (2015).
Gibbs, R. A. et al. Insights into social insects from the genome of the honeybee Apis mellifera. Nature. 443, 931–949 (2006).
Crowley, L. The genome sequence of the seven-spotted ladybird, Coccinella septempunctata Linnaeus, 1758. Wellcome open res. 6, 319 (2021).
Pang, B., Zhan, Z. & Wang, Y. A chromosome-level genome assembly of Prosopocoilus inquinatus Westwood, 1848 (Coleoptera: Lucanidae). Sci Data. 11, 808 (2024).
Herndon, N. et al. Enhanced genome assembly and a new official gene set for Tribolium castaneum. BMC Genomics. 21, 47 (2020).
Buchfink, B., Xie, C. & Huson, D. H. Fast and sensitive protein alignment using DIAMOND. Nat. Methods. 12, 59–60 (2015).
Huerta-Cepas, J. et al. Fast Genome-Wide Functional Annotation through Orthology Assignment by eggNOG-Mapper. Mol. Biol. Evol. 34, 2115–2122 (2017).
Finn, R. D. et al. InterPro in 2017—Beyond protein family and domain annotations. Nucleic Acids Res. 45, D190–D199 (2017).
El-Gebali, S. et al. The Pfam protein families database in 2019. Nucleic Acids Res. 47, D427–D432 (2019).
Letunic, I. & Bork, P. 20 years of the SMART protein domain annotation resource. Nucleic Acids Res. 46, D493–D496 (2018).
Wilson, D. et al. SUPERFAMILY—Sophisticated comparative genomics, data mining, visualization and phylogeny. Nucleic Acids Res. 37, D380–D386 (2009).
Lewis, T. E. et al. Gene3D: Extensive Prediction of Globular Domains in Proteins. Nucleic Acids Res. 46, D1282 (2018).
Marchler-Bauer, A. et al. CDD/SPARCLE: Functional classification of proteins via subfamily domain architectures. Nucleic Acids Res. 45, D200–D203 (2017).
Chen, C. et al. TBtools: An Integrative Toolkit Developed for Interactive Analyses of Big Biological Data. Mol. Plant. 13, 1194–1202 (2020).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR31019928 (2025).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR31019929 (2025).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR31019930 (2025).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR31019931 (2025).
NCBI Assembly https://identifiers.org/ncbi/insdc.gca:GCA_043790905.1 (2025).
Zhu, M. Genome annotation (repeats and protein-coding genes). figshare https://doi.org/10.6084/m9.figshare.28696565.v1 (2025).
Waterhouse, R. M. et al. BUSCO Applications from Quality Assessments to Gene Prediction and Phylogenomics. Mol. Biol. Evol. 35, 543–548 (2018).
Li, H. Minimap2: Pairwise alignment for nucleotide sequences. Bioinformatics. 34 (2018).
Dudchenko, O. et al. Twelve years of SAMtools and BCFtools. GigaScience. 10(2), giab008 (2021).
Acknowledgements
This study was supported by grants from Henan Science and Technology Research Project (252102320222).
Author information
Authors and Affiliations
Contributions
Y.J. contributed to the research design. Z.M., Z.J. and H.Y. collected the samples. Z.M. analyzed the data. Z.M. and H.Y. wrote the draft manuscript and revised the manuscript. All co-authors contributed to this manuscript and approved it.
Corresponding author
Ethics declarations
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Zhu, M., Zhang, J., Yan, J. et al. A chromosomal-level genome assembly of Kibakoganea sinica, Bouchard, 2005 (Coleoptera: Scarabaeidae). Sci Data 12, 1012 (2025). https://doi.org/10.1038/s41597-025-05347-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41597-025-05347-4
