
Abstract
Lake ecosystems play a fundamental role in the global biogeochemical cycling of essential elements such as carbon, nitrogen, and phosphorus. Microorganisms within these ecosystems mediate key processes that regulate these cycles. Metagenomic analyses provide valuable insights into the taxonomic and functional diversity of microbial communities in various environments, including freshwater habitats. Here, we present a comprehensive metagenomic dataset derived from Lake Soyang, the largest freshwater reservoir in South Korea. A total of 28 metagenomes were generated from water samples collected across two distinct sampling periods: the first set (n = 8) was obtained between April 2014 and January 2015 from two depths (1 m and 50 m) in four different seasons, while the second set (n = 20) was collected between January 2019 and November 2019 from five depths (1, 10, 20, 40, and 90 m) over four seasons. Metagenomic sequencing yielded 9.3–21.8 Gbp per sample. This dataset provides a valuable resource for future studies exploring the ecophysiological characteristics of microbial communities in pelagic freshwater environments.
Similar content being viewed by others

Background & Summary
Freshwater ecosystems play a pivotal role in global biogeochemical cycles, driving the transformation and movement of carbon and nutrients through processes such as sedimentation, atmospheric exchange, and hydrological transport1,2,3,4,5. Microorganisms are central to these processes, making the study of freshwater microbial communities essential for understanding global environmental changes6,7,8,9. Over the past few decades, advances in molecular biological techniques have significantly enhanced our knowledge of microbial diversity and function in freshwater environments10,11,12,13. High-throughput sequencing of 16S rRNA genes has revealed microbial diversity in freshwater ecosystems that were previously undetectable using traditional culture-based methods. These studies have demonstrated that microbial communities are predominantly composed of oligotrophic microorganisms14,15,16, exhibiting spatiotemporal dynamics and microdiversification in response to environmental variability17,18,19. More recently, metagenomic sequencing approaches have uncovered extensive phylogenetic and functional diversity across global freshwater biomes19,20,21,22,23,24,25,26,27, emphasizing the need for more comprehensive datasets spanning diverse geographic and ecological contexts, as exemplified by recent studies on freshwater nitrifiers28,29.
Lake Soyang is a large, deep, and oligotrophic-to-mesotrophic artificial reservoir in South Korea, with a maximum depth of approximately 118 m, making it the deepest lake in the country. It is classified as a warm monomictic lake, characterized by seasonal overturning that begins in early winter and persists until surface warming in spring. Heavy monsoonal rainfall in Korea (July–August) drives substantial inflows into the reservoir’s metalimnion30, triggering post-monsoon phytoplankton blooms through the upward transport of nutrients to the epilimnion, coupled with rising water temperatures31,32. In recent years, we have studied the microbial ecology of Lake Soyang, focusing on the isolation and characterization of diverse bacterial lineages33,34, including the acI actinobacterial clade35. Additionally, viral assemblages in the lake have been examined36,37.
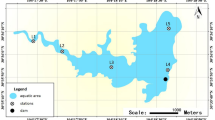
Here, we present microbial metagenomic data obtained from 28 water samples collected at varying depths from a single sampling station (Fig. 1) across two periods to capture seasonal variations. The first period set, collected from April 2014 to January 2015, comprised eight samples obtained at two depths (1 m and 50 m) at three-month intervals. The second period set, collected from January 2019 to November 2019 also at approximately three-month intervals, consisted of 20 samples obtained at five depths (1, 10, 20, 40, and 90 m). Physicochemical parameters were measured on-site (Table 1), while additional parameters such as chlorophyll a and nutrients were analyzed in the laboratory (Table 2). However, total dissolved nitrogen and dissolved organic carbon data were not available for the 2019 samples.

Location of the sampling station (a) and the sample processing scheme used in this study (b).
The eight metagenomes collected from April 2014 to January 2015 were sequenced using the Illumina HiSeq 2500 platform, yielding 14.2–21.8 Gbp of raw bases per sample (Table 3). The 20 metagenomes collected in 2019 were sequenced using the Illumina NovaSeq platform, generating 9.3–12.2 Gbp of raw bases per sample (Table 3). Taxonomic classification of high-quality metagenomic reads using the Genome Taxonomy Database38 (GTDB) identified Nanopelagicaceae (Actinomycetia), Pelagibacteraceae (Alphaproteobacteria), and Burkholderiaceae (Gammaproteobacteria) as the dominant microbial taxa in the freshwater metagenomes (Fig. 2). This study presents a new freshwater metagenome dataset alongside physicochemical characteristics, providing a foundation for further investigations into microbial community dynamics and the genomic traits of freshwater microbial assemblages.

Taxonomic classification of freshwater metagenomic reads obtained from Lake Soyang. The bubbles represent the relative abundance (%) of taxonomic groups at the family level in the samples. Taxonomic classification was performed using Kraken2 based on a database constructed using the GTDB (R207).
Methods
Sample collection
Lake water samples were collected using a Niskin sampler at depths of 1 m and 50 m in front of the dam (37.9474 N, 127.8189 E) across four seasonal intervals between April 2014 and January 2015 (Fig. 1). During the second sampling period (January−November 2019), samples were collected at five depths (1, 10, 20, 40, and 90 m) at the same location and using the same sampling method. At each time point and depth, a single 10 L lake water sample was collected. Immediately after collection, samples were stored in darkness at 4 °C and transported to the laboratory. Temperature, pH, dissolved oxygen, conductivity, and salinity were measured using a YSI 556 MPS Multiprobe System (YSI Incorporated, Yellow Springs, OH, USA) on-site. Dissolved oxygen concentrations were also analyzed using the standard Winkler titration method39. Concentrations of ammonium, nitrite, nitrate, phosphate, and silicate were analyzed after filtration through 0.45 μm pore-size membrane filters (Advantec, Toyo Roshi Kaisha, Tokyo, Japan). During the first sampling period, analyses were conducted by the National Instrumentation Center for Environmental Management (NICEM, South Korea), while during the second sampling period, Hach reagent kits (Hach, Loveland, CA, USA) were used. Dissolved organic carbon (DOC) and total dissolved nitrogen (TDN) were also measured by NICEM during the first sampling period. For chlorophyll a analysis, 1 L of each water sample was filtered through a GF/F glass microfiber filter (0.7 μm-pore-size, Whatman, Kent, UK). Chlorophyll a concentrations were quantified after acetone extraction40 using a Turner Designs™ 10-AU fluorometer (Turner Designs, Sunnyvale, CA, USA) during the first sampling period and a UV-VIS spectrophotometer (Model UV-2600, Shimadzu, Japan) during the second sampling period. For metagenome sequencing, 1 L of each water sample was sequentially pre-filtered through a 3.0 μm-pore-size mixed cellulose ester membrane filter (Advantec, Toyo Roshi Kaisha, Tokyo, Japan), followed by final filtration through a 0.2 μm-pore-size, 47 mm polyethersulfone membrane filter (Pall, NY, USA). Identical filter types and brands were used consistently for all samples throughout the entire sampling period. Filtration was completed within 12 hours of collection, and filters were stored at −80 °C until DNA extraction.
DNA extraction and metagenome sequencing
DNA extraction was performed using the entire 0.2 μm filter for each sample. For samples collected between April 2014 to January 2015, 0.2-μm-pore-size filters were placed in 5 ml screw-cap tubes. Following a manual lysis protocol described in previous studies41,42,43,44, lysozyme solution (5 μl, 10 mg ml−1) was added to the tubes along with 1 ml of lysis buffer (20 mM EDTA, 50 mM Tris, 400 mM NaCl, 0.75 M sucrose), followed by incubation at 30 °C for 30 minutes in a hybridization oven with rotation speed of 5 rpm. After incubation, proteinase K (5 μl, 20 mg ml−1) and 10% sodium dodecyl sulfate (100 μl) were added, and samples were incubated overnight at 55 °C. All solutions in 5 ml tubes were transferred to 15 ml conical tubes, and genomic DNA was extracted using the DNeasy Blood & Tissue Kit (Qiagen, Maryland, USA) following the manufacturer’s instructions, starting with the addition of RNase A and Qiagen AL buffer. For samples collected between January and November 2019, genomic DNA was extracted from 0.2-μm polyethersulfone filters using the DNeasy PowerSoil Pro Kit according to the manufacturer’s protocol. We used two different DNA extraction protocols because our laboratory procedures for extracting DNA from membrane filters changed between the two sampling periods. Given the largely consistent taxonomic profiles observed across both sampling periods (Fig. 2), any biases potentially introduced by the different protocols are considered minimal, if present at all.
Extracted DNA was used for library preparation using the TruSeq DNA Library Kit, followed by sequencing on either the Illumina HiSeq platform (250 bp paired-end; CJ Bioscience, Korea) for 2014–2015 samples or the Illumina NovaSeq platform (150 bp paired-end; CJ Bioscience, Korea) for 2019 samples. The use of two different sequencing platforms was due to practical considerations. To our knowledge, any potential bias arising from the use of different Illumina sequencing platforms is presumed to be negligible. No controls were included for DNA extraction, library construction, or sequencing.
Quality enhancement and taxonomic classification of metagenomic reads
Summary statistics of the raw sequencing data were assessed using Seqkit (v.2.8.0)45, with the “seqkit stats -a” option. Prior to taxonomic profiling, raw Illumina reads from both sampling periods (2014–2015 and 2019) were pre-processed using BBduk in BBtools46 for adapter trimming, quality filtering, and removal of phiX174 sequences. The command used for trimming and quality filtering was: bbduk.sh ref = adapters.fa ktrim = r k = 23 mink = 11 hdist = 1 qtrim = rl trimq = 10 minlen = 100. The command for phiX removal was: bbduk.sh ref = phix174_ill.ref.fa.gz k = 31 hdist = 1. Pre-processed metagenomic reads were taxonomically classified using Kraken2 (v2.1.3)47 with the GTDB database (R207)38, provided by Struo248 (http://ftp.tue.mpg.de/ebio/projects/struo2/GTDB_release207/), using the “--paired” option. The resulting taxonomic abundance data were processed using custom scripts and visualized with the R package ‘tidyverse’49. The processed taxonomic classification tables and scripts used to generate Fig. 2 are available in the project’s GitHub repository: https://github.com/SuhyunInha/SYMETA.
Data Records
The raw Illumina sequencing reads (fastq format) of the metagenomes obtained in this study have been deposited in the European Nucleotide Archive (ENA) under project accession number PRJEB2757850 (samples from 2014 and 2015) and in GenBank under project accession number PRJNA108710551 (samples from 2019).
Technical Validation
All experiments were conducted in a strictly controlled environment to prevent potential sample contamination. Before sampling and experiments, all equipment that directly contacted water samples, including filter units, carboys, bottles, and flasks, was immersed in HCl solution (10% v/v) for at least 24 hours, followed by rinsing with Milli-Q water five times. After washing, the labware was autoclaved, thoroughly dried, and stored with dust caps or wrapped in foil until use. The Niskin sampler was first washed in the laboratory and then rinsed with lake water at the sampling station at least three times before use. Water samples were transferred to sterile 20 L or 4 L carboys, placed in a cold-storage box, and immediately transported to the laboratory to prevent bacterial overgrowth. No field blanks or negative controls were included during sample collection; however, the risk of contamination was minimized through strict adherence to field protocols. Physicochemical parameters, including temperature, dissolved oxygen, pH, and conductivity, were measured on-site. Sample filtration and additional water chemistry analyses were performed on the same day as sampling. DNA extraction from the filters was performed using the same batch of the Qiagen DNeasy Blood & Tissue Kit for the 2014–2015 samples, and DNeasy PowerSoil Pro Kit for the 2019 samples. DNA extractions for each sample set were performed on the same day, respectively, within a dedicated laboratory space.
Code availability
The software, version information, and commands used in the analyses are described in the Methods section. Custom scripts for data visualization and taxonomic analysis are available at https://github.com/SuhyunInha/SYMETA and archived on Zendo52.
References
Cole, J. J. et al. Plumbing the global carbon cycle: integrating inland waters into the terrestrial carbon budget. Ecosystems 10, 171–184, https://doi.org/10.1007/s10021-006-9013-8 (2007).
Seitzinger, S. et al. Denitrification across landscapes and waterscapes: a synthesis. Ecol. Appl. 16, 2064–2090, https://doi.org/10.1890/1051-0761(2006)016[2064:DALAWA]2.0.CO;2 (2006).
Small, G. E. et al. Rates and controls of nitrification in a large oligotrophic lake. Limnol. Oceanogr. 58, 276–286, https://doi.org/10.4319/lo.2013.58.1.0276 (2013).
Callieri, C., Coci, M., Eckert, E. M., Salcher, M. M. & Bertoni, R. Archaea and bacteria in deep lake hypolimnion: in situ dark inorganic carbon uptake. J. Limnol. 73, 47–54, https://doi.org/10.4081/jlimnol.2014.937 (2014).
Wu, Z. et al. Imbalance of global nutrient cycles exacerbated by the greater retention of phosphorus over nitrogen in lakes. Nat. Geosci. 15, 464–468, https://doi.org/10.1038/s41561-022-00958-7 (2022).
Williamson, C. E., Dodds, W., Kratz, T. K. & Palmer, M. A. Lakes and streams as sentinels of environmental change in terrestrial and atmospheric processes. Front. Ecol. Environ. 6, 247–254, https://doi.org/10.1890/070140 (2008).
Salcher, M. M., Posch, T. & Pernthaler, J. In situ substrate preferences of abundant bacterioplankton populations in a prealpine freshwater lake. ISME J. 7, 896–907, https://doi.org/10.1038/ismej.2012.16 (2013).
Eckert, E. M., Salcher, M. M., Posch, T., Eugster, B. & Pernthaler, J. Rapid successions affect microbial N-acetyl-glucosamine uptake patterns during a lacustrine spring phytoplankton bloom. Environ. Microbiol. 14, 794–806, https://doi.org/10.1111/j.1462-2920.2011.02639.x (2012).
Tada, Y. & Grossart, H. P. Community shifts of actively growing lake bacteria after N-acetyl-glucosamine addition: improving the BrdU-FACS method. ISME J. 8, 441–454, https://doi.org/10.1038/ismej.2013.148 (2014).
Newton, R. J., Jones, S. E., Eiler, A., McMahon, K. D. & Bertilsson, S. A guide to the natural history of freshwater lake bacteria. Microbiol. Mol. Biol. Rev. 75, 14–49, https://doi.org/10.1128/MMBR.00028-10 (2011).
Parfenova, V. V., Gladkikh, A. S. & Belykh, O. I. Comparative analysis of biodiversity in the planktonic and biofilm bacterial communities in Lake Baikal. Microbiology 82, 91–101, https://doi.org/10.1134/S0026261713010128 (2013).
Satinsky, B. M. et al. Metagenomic and metatranscriptomic inventories of the lower Amazon River, May 2011. Microbiome 3, 39, https://doi.org/10.1186/s40168-015-0099-0 (2015).
Savio, D. et al. Bacterial diversity along a 2600 km river continuum. Environ. Microbiol. 17, 4994–5007, https://doi.org/10.1111/1462-2920.12886 (2015).
Allgaier, M., Bruckner, S., Jaspers, E. & Grossart, H. P. Intra- and inter-lake variability of free-living and particle-associated Actinobacteria communities. Environ. Microbiol. 9, 2728–2741, https://doi.org/10.1111/j.1462-2920.2007.01385.x (2007).
Newton, R. J., Jones, S. E., Helmus, M. R. & McMahon, K. D. Phylogenetic ecology of the freshwater Actinobacteria acI lineage. Appl. Environ. Microbiol. 73, 7169–7176, https://doi.org/10.1128/Aem.00794-07 (2007).
Salcher, M. M., Pernthaler, J. & Posch, T. Seasonal bloom dynamics and ecophysiology of the freshwater sister clade of SAR11 bacteria ‘that rule the waves’ (LD12). ISME J. 5, 1242–1252, https://doi.org/10.1038/ismej.2011.8 (2011).
Okazaki, Y. et al. Ubiquity and quantitative significance of bacterioplankton lineages inhabiting the oxygenated hypolimnion of deep freshwater lakes. ISME J. 11, 2279–2293, https://doi.org/10.1038/ismej.2017.89 (2017).
Paver, S. F., Newton, R. J. & Coleman, M. L. Microbial communities of the Laurentian Great Lakes reflect connectivity and local biogeochemistry. Environ. Microbiol. 22, 433–446, https://doi.org/10.1111/1462-2920.14862 (2020).
Okazaki, Y. et al. Microdiversity and phylogeographic diversification of bacterioplankton in pelagic freshwater systems revealed through long-read amplicon sequencing. Microbiome 9, 24, https://doi.org/10.1186/s40168-020-00974-y (2021).
Okazaki, Y., Nakano, S., Toyoda, A. & Tamaki, H. Long-read-resolved, ecosystem-wide exploration of nucleotide and structural microdiversity of lake bacterioplankton genomes. mSystems 7, e0043322, https://doi.org/10.1128/msystems.00433-22 (2022).
Santos-Júnior, C. D., Logares, R. & Henrique-Silva, F. Microbial population genomes from the Amazon River reveal possible modulation of the organic matter degradation process in tropical freshwaters. Mol. Ecol. 31, 206–219, https://doi.org/10.1111/mec.16222 (2022).
Buck, M. et al. Comprehensive dataset of shotgun metagenomes from oxygen stratified freshwater lakes and ponds. Sci. Data 8, 131, https://doi.org/10.1038/s41597-021-00910-1 (2021).
Cabello-Yeves, P. J. et al. Microbiome of the deep Lake Baikal, a unique oxic bathypelagic habitat. Limnol. Oceanogr. 65, 1471–1488, https://doi.org/10.1002/lno.11401 (2020).
Rohwer, R. R. et al. Two decades of bacterial ecology and evolution in a freshwater lake. Nat. Microbiol. 10, 246–257, https://doi.org/10.1038/s41564-024-01888-3 (2025).
Cheng, M. Y. et al. A genome and gene catalog of the aquatic microbiomes of the Tibetan Plateau. Nat. Commun. 15, 1438, https://doi.org/10.1038/s41467-024-45895-8 (2024).
Tran, P. Q. et al. Depth-discrete metagenomics reveals the roles of microbes in biogeochemical cycling in the tropical freshwater Lake Tanganyika. ISME J. 15, 1971–1986, https://doi.org/10.1038/s41396-021-00898-x (2021).
Chiriac, M. C., Haber, M. & Salcher, M. M. Adaptive genetic traits in pelagic freshwater microbes. Environ. Microbiol. 25, 606–641, https://doi.org/10.1111/1462-2920.16313 (2023).
Podowski, J. C., Paver, S. F., Newton, R. J. & Coleman, M. L. Genome streamlining, proteorhodopsin, and organic nitrogen metabolism in freshwater nitrifiers. mBio 13, e0237921, https://doi.org/10.1128/mbio.02379-21 (2022).
Ngugi, D. K. et al. Postglacial adaptations enabled colonization and quasi-clonal dispersal of ammonia-oxidizing archaea in modern European large lakes. Sci. Adv. 9, eadc9392, https://doi.org/10.1126/sciadv.adc9392 (2023).
Kim, Y. & Kim, B. Application of a 2-dimensional water quality model (CE-QUAL-W2) to the turbidity interflow in a deep reservoir (Lake Soyang, Korea). Lake Reserv Manage 22, 213–222, https://doi.org/10.1080/07438140609353898 (2006).
Kim, B., Choi, K., Kim, C., Lee, U. H. & Kim, Y. H. Effects of the summer monsoon on the distribution and loading of organic carbon in a deep reservoir, Lake Soyang, Korea. Water Res. 34, 3495–3504, https://doi.org/10.1016/S0043-1354(00)00104-4 (2000).
Jung, S. et al. The effects of Asian summer monsoons on algal blooms in reservoirs. Inland Waters 6, 406–413, https://doi.org/10.5268/Iw-6.3.967 (2016).
Park, M. et al. Flavobacterium lacicola sp. nov., isolated from a freshwater lake. Int. J. Syst. Evol. Microbiol. 68, 1565–1570, https://doi.org/10.1099/ijsem.0.002712 (2018).
Nam, G. G. et al. Flavobacterium soyangense sp nov., a psychrotolerant bacterium, isolated from an oligotrophic freshwater lake. Int. J. Syst. Evol. Microbiol. 67, 2440–2445, https://doi.org/10.1099/ijsem.0.001987 (2017).
Kim, S., Kang, I., Seo, J. H. & Cho, J. C. Culturing the ubiquitous freshwater actinobacterial acI lineage by supplying a biochemical ‘helper’ catalase. ISME J. 13, 2252–2263, https://doi.org/10.1038/s41396-019-0432-x (2019).
Moon, K., Kang, I., Kim, S., Kim, S. J. & Cho, J. C. Genome characteristics and environmental distribution of the first phage that infects the LD28 clade, a freshwater methylotrophic bacterial group. Environ. Microbiol. 19, 4714–4727, https://doi.org/10.1111/1462-2920.13936 (2017).
Moon, K., Kim, S., Kang, I. & Cho, J. C. Viral metagenomes of Lake Soyang, the largest freshwater lake in South Korea. Sci. Data 7, 349, https://doi.org/10.1038/s41597-020-00695-9 (2020).
Parks, D. H. et al. A complete domain-to-species taxonomy for Bacteria and Archaea. Nat. Biotechnol. 38, 1098–1098, https://doi.org/10.1038/s41587-020-0539-7 (2020).
Winkler, L. Methods for measurement of dissolved oxygen. Ber Dtsch Chem Ges 21, 2843–2855 (1888).
American Public Health Association, American Water Works Association & Water Environment Federation. Standard Methods for the Examination of Water and Wastewater, 19th edn (American Public Health Association, 1995).
Giovannoni, S. J., Rappé, M. S., Vergin, K. L. & Adair, N. L. 16S rRNA genes reveal stratified open ocean bacterioplankton populations related to the Green Non-Sulfur bacteria. Proc. Natl Acad. Sci. USA 93, 7979–7984, https://doi.org/10.1073/pnas.93.15.7979 (1996).
Yang, S.-J., Kang, I. & Cho, J.-C. Expansion of cultured bacterial diversity by large-scale dilution-to-extinction culturing from a single seawater sample. Microb. Ecol. 71, 29–43, https://doi.org/10.1007/s00248-015-0654-4 (2016).
Kim, S., Islam, M. R., Kang, I. & Cho, J. C. Cultivation of dominant freshwater bacterioplankton lineages using a high-throughput dilution-to-extinction culturing approach over a 1-year period. Front. Microbiol. 12, 700637, https://doi.org/10.3389/fmicb.2021.700637 (2021).
Lim, Y., Yang, S. J., Kang, I. L. & Cho, J. C. Metagenomic data from surface seawater of the east coast of South Korea. Sci. Data 10, 647, https://doi.org/10.1038/s41597-023-02556-7 (2023).
Shen, W., Sipos, B. & Zhao, L. SeqKit2: a Swiss army knife for sequence and alignment processing. iMeta 3, e191, https://doi.org/10.1002/imt2.191 (2024).
Bushnell, B. BBTools. Available online: https://sourceforge.net/projects/bbmap/ (accessed in 2024) (2014).
Wood, D. E., Lu, J. & Langmead, B. Improved metagenomic analysis with Kraken 2. Genome Biol. 20, 257 (2019).
Youngblut, N. D. & Ley, R. E. Struo2: efficient metagenome profiling database construction for ever-expanding microbial genome datasets. PeerJ 9, e12198, https://doi.org/10.7717/peerj.12198 (2021).
Wickham, H. et al. Welcome to the tidyverse. J. Open Source Softw. 4, 1686, https://doi.org/10.21105/joss.01686 (2019).
ENA European Nucleotide Archive https://identifiers.org/ena.embl:ERP109673 (2020).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRP494926 (2025).
Kim, S. SYMETA: Microbial metagenomes of Lake Soyang. Zenodo https://doi.org/10.5281/zenodo.15469394
Acknowledgements
This study was supported by the Mid-Career Research Program (NRF-2022R1A2C3008502 to J.-C.C.) and the Sejong Science Fellowship (NRF-2022R1C1C2004070 to S.K.) through the National Research Foundation (NRF) funded by the Ministry of Sciences and ICT of the Korea government.
Author information
Authors and Affiliations
Contributions
S.K. and M.S.P. collected the samples and performed the bioinformatic analyses. I.K. and J.-C.C. designed the project and supervised computational work. S.K., M.S.P., I.K. and J.-C.C. wrote the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Kim, S., Park, M.S., Kang, I. et al. Microbial metagenomes from Lake Soyang, the largest freshwater reservoir in South Korea. Sci Data 12, 1201 (2025). https://doi.org/10.1038/s41597-025-05569-6
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41597-025-05569-6
