
Abstract
Fibrous erionite-Na from Rome (Oregon, USA) was K-exchanged and characterized from the structural point of view. In addition, the modifications experienced after contact with a Fe(II) source were investigated for evaluating if the large potassium ions, blocking off nearly all the erionite cavity openings, might prevent the Fe(II) binding process, which is currently assumed to be one of the reasons of the toxicity of erionite. The K-exchanged sample had a 95% reduction of the BET surface area indicating that it behaves as a mesoporous material. Exchanged K is segregated at K2 and at OW sites commonly occupied by H2O. The latter K cations provide a relevant contribution to the reduction of the surface area. Surprisingly, despite the collapse of its surface area the sample preserves the tendency to bind Fe(II). Therefore, yet in the case of a peculiar and potentially hostile structural environment the Fe(II) ion-exchange process has essentially the same kinetics observed in a typical erionite sample. This is a clear evidence of the very limited effect of the chemical composition of erionite on the Fe(II) binding process and reasonably it does not play a significant role in its toxicity.
Similar content being viewed by others
Introduction
Erionite is a fairly common zeolite often occurring with fibrous habit in several geological environments1. In the recent years, a relevant number of papers has been published aimed at investigating the chemical and structural properties of this zeolite owing to its well-known link, upon inhalation, with malignant mesothelioma2. As a result, erionite has been recently classified as a Group 1 Human-Carcinogen by the International Agency for Research on Cancer (IARC)3, 4. Obviously, a crucial point to be addressed is the definition of the mechanism/s inducing carcinogenicity to devise an inactivation route of the fibres. A common view attributes genotoxicity of erionite to the production of reactive oxygen species (ROS) by Fenton reaction5, 6. Moreover, it has been shown that erionite is able to induce transformation of human mesothelial cells (MET5-A), but on the other hand, asbestos is unable to cause such transformation7. Other factors may be significant in the development of malignant mesothelioma by erionite, such as levels of exposure that are indeed dangerous for the population8 as well as genetic predisposition9.
Relevantly, erionite is a nominally Fe-free phase but it has been supposed that it is able to fix iron, provided by protein injury, owing to its cation-exchange properties10. Recently, it has been shown that erionite can act as a vehiculating media in the human body for iron as several Fe-bearing mineral species (nano-oxydes, sulphates, phyllosilicates) were found to adhere to its surface11,12,13,14. Therefore, the behaviour of erionite fibres kept in contact with iron sources has been thoroughly investigated to define the fate of both Fe(II) and Fe(III) and in particular the tendency to bind to the surface or to be fixed as extra-framework (EF) cations15,16,17. Results unequivocally showed that Fe(III) is mainly fixed to the surface at relatively high concentration, whereas at low concentrations typical of body fluids it is prevalently segregated at the Ca3 site within the erionite cavity17. Differently, Fe(II) is segregated at Ca3 at all concentrations16. Unfortunately, this picture indicates that erionite under typical physiological conditions may act as a very efficient ROS generator because iron is located at well-defined crystallographic sites and it possesses a very low nuclearity18, 19. Therefore, it appears to be of great interest to investigate different physico-chemical procedures to prevent the access of iron species to the erionite cavity as future roads of (partial) inactivation of such fibres.
Under such aspect, it is worth noting that Eberly20 first reported the adsorption properties of erionite and its cation-exchanged forms. He measured a surface area of 203 m2 g−1 and an effective pore diameter comprised between 4.5 and 5.4 Å, in substantial agreement with the minor and major axes of the largest opening (Ballirano and Cametti21: erionite-K 3.50–5.05 Å; Ballirano and Pacella22: erionite-Na 3.45–5.05 Å). The K-exchanged form experienced a dramatic reduction of 95% of the surface area resulting in a complete removal of its adsorption properties, apart from a residual adsorptive capacity for H2O20. Moreover, it has been reported that zeolites having a large K content show reduced catalytic properties23.
On this basis, the present work reports the results of a detailed crystal chemical and structural characterization of K-exchanged erionite. In addition, the modifications experienced by the K-exchanged fibres whenever in contact with a ferrous chloride solution were investigated to evaluate if the large potassium ions, blocking off nearly all the adsorption cavities, may prevent the Fe(II) binding process.
Results
After an enrichment procedure, erionite was K-exchanged by immersion in a concentrated KCl solution (see methods). ICP data indicate that erionite fibres released significant amounts of Na (581 nmol/mg) at the end of the treatment with the KCl solution. SEM-EDS data of the erionite treated with the KCl solution provide clear evidence that the K binding was successful, since the relevant increase of the K content from 2.5 (3) to 5.91 (9) atoms per formula unit (apfu) in the fibres (Table 1). Coherently with ICP data, a marked reduction of the Na content from 3.8 (3) to 1.41 (10) apfu was evidenced. The final average crystal-chemical formula of (Na1.41K5.91Mg0.44)[Al7.49Si28.51O72.35] · 30.73H2O allows a sample classification as erionite-K.
Fe(II)-loading of the K-exchanged fibres was performed by suspending erionite in a 500 µM FeCl2 solution. ICP data reveal that erionite fibres bind 290 (13) nmol/mg of Fe. Notably, the Fe amount acquired by erionite-K is perfectly comparable with that observed by Ballirano et al.16 for erionite-Na after suspension in the 500 µM FeCl2 solution [291 (1) nmol/mg]. ICP analysis of supernatants for EF cation release evidences a significant net release of K [445 (33) nmol/mg], coupled with a very minor release of Na, Ca, and Mg during the suspension of the fibres in the FeCl2 solution (Table 2). It should be pointed out that the low Ca release detected is due to the presence of traces of admixed impurities (see methods), being Ca absent in the pristine sample as indicated by SEM-EDS analysis (Table 1). The calculated charge balance evidences that the number of Fe acquired charges is comparable with that of released charges (Supplementary Table S1), highlighting that Fe(II) was mainly acquired by erionite through ion-exchange with the EF cations, as reported by Ballirano et al.16 (Supplementary Table S1). However, in the case of erionite-K, fibres fix Fe(II) within the structure through an ion-exchange process, mainly involving K, while for erionite-Na the process mainly affected Na (Supplementary Table 2). SEM-EDS analysis of the Fe(II) loaded sample shows a significant reduction of both K and Na content [from 5.91 (9) to 4.2 (4) apfu and from 1.41 (10) to 0.6 (3) apfu, respectively], counterbalanced by an upload of 0.79 (15) apfu Fe(II). However, ICP data indicated only a very marginal Na release (Supplementary Table 2). The observed discrepancy between SEM-EDS and ICP data may be attributed to the relevant Na migration occurring in the external layer of fibres during micro-analysis17, 24. Summarizing, the following crystal chemical formula (Mg0.6Fe0.7Na1.5K4.2)[Al7.5Si28.5O72]∙ 30.5H2O may be retrieved for Fe(II) erionite.
Pacella et al.17 indicated for erionite-Na (pristine material) a BET specific surface area, measured in the conventional p/p0 0.05–0.3 range, of 252 (5) m2 g−1 and of 325 (5) m2 g−1 in the p/p0 0–0.1 range, typical of microporous materials (Table 3). The BET specific surface area of the K-exchanged samples collapses to about 12 m2 g−1. It is worth noting that erionite-Na (pristine material) possesses an external surface area of 10.1 (5) m2 g−1, i.e. a value very close to the BET surface area of K-exchanged samples (erionite-K), indicating that upon treatment the sample behaves exclusively as a mesoporous material. The total volume of pores decreases from 0.151 cm3 g−1 of the pristine sample (of which 0.130 cm3 g−1 attributable to micropores) to ca. 0.02 cm3 g−1. The Fe(II) loaded sample exhibited an increase of the BET specific surface area to 73.7 (17) m2 g−1 significantly smaller than that of the pristine sample. In addition, the total volume of pores slightly increases with respect to the K-exchanged samples to 0.0495 cm3 g−1.
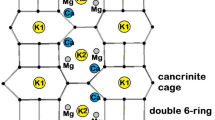
XRPD data reveal that the cell volume expansion of erionite-K is caused by the c-axis expansion only partly counterbalanced by the a-axis contraction (Supplementary Table S2). Iron binding produces a further minor contraction of both a- and c-axis leading to a cell volume smaller than that of erionite-Na (pristine material). The c/a ratio passes from ca. 1.138 to ca. 1.140 of the two treated samples (Supplementary Table S2). As expected no significant modifications of the framework (Fig. 1) were detected with the variation of <T-O> bond distances (Supplementary Table S3) and T-O-T bond angles (not shown) being confined within a few standard deviations. The slightly higher R values as calculated from Jones’ determinative curves25 with respect to those from SEM-EDS might be related to the effect of protonation at some oxygen sites of the framework progressively occurring during treatments17. On the contrary, relevant differences are observed as far as the site scattering (s.s.) of both EF and water molecule sites is referred to (Supplementary Table S4). In the case of the K-exchanged sample we observe a complete depletion of the Ca1 site, which is preferentially occupied by Mg and Na in the pristine sample (erionite-Na) and a very strong reduction of the s.s. at Ca3 (occupied by Na). Conversely, the K2 site (occupied by K) significantly increases its s.s. (Fig. 1). It is worth mentioning that the total EF s.s. drops from ca. 113 e− of the pristine sample to ca. 94 e− of the KCl treated sample apparently indicating a significant cationic discharge during the K binding process. The Fe(II) loading process induces a small refilling of the Ca1 site (ca. 4 e−), a significant increase of s.s., from ca. 5 to ca. 20 e− at Ca3, whereas K2 remains substantially unchanged. Concerning the OW sites occupied by H2O, the most relevant differences between pristine and KCl treated sample are related to a significant s.s. increase at OW7, OW10 and OW12 and a marked reduction at OW11 in the case of the KCl treated sample (Fig. 1). As a net result, the total OW sites s.s. rises from 245 (11) e− for the pristine to 282 (7) e− for the KCl treated sample. In the case of the Fe(II) loaded sample the s.s. of OW7, OW10 and OW12 reduces back and the total water molecules s.s. decreases to 268 (9) e−.

Drawing of (left) the framework of the K-exchanged form of erionite and of (right) the EF cations and water molecules location within the erionite cage. H2O residing at OW sites may be partly replaced by K cations.
Discussion
Textural analysis of the KCl treated sample indicates values of the BET surface that are ca. 5% of the pristine material. The corresponding values of ca. 10 m2 g−1 compare favourably with those observed in both amphibole and chrysotile asbestos (3–20 m2 g−1) and are consistent with the 7–9 m2 g−1 reported by Eberly20 for erionite-K from Rome, Oregon, USA. At the structural level, the abrupt abatement of the BET surface implies the occlusion of all microporosity. Erionite possesses two apertures that have been considered to act as possible ways for cation mobility, the so-called 8-member (8MR) and 6-member (6MR) rings. The first one, which is the largest, represents the preferential way for any exchange process, whereas the second one, that is shared between the cancrinite and erionite cages, albeit of small dimensions, has been claimed to act as a revolving door for the so-called “internal ionic exchange” mechanism occurring at high temperature21, 22, 26. It is worth noting that the K2 site is located approximately at the centre of 8MR. Therefore, the reported increased population at this site implies a more extended occlusion of 8MR leading to a reduction of the BET surface. Despite an exchange process involving the replacement by K (19 electrons) of (mainly) Na (11 electrons) and Mg (12 electrons), the total EF s.s (i.e. the sum of the s.s. of Ca1, Ca2, Ca3, K1, and K2 sites) decreases (Supplementary Table S4). This is a clear indication that a fraction of the EF cations was re-located at sites previously occupied by H2O (Fig. 1). This is confirmed by the simultaneous increase of the total OW sites s.s. from 245 (11) e− of the pristine sample to 282 (7) e− for the KCl treated sample mainly justified by a significant s.s. increase at OW7, OW10 and OW12 (Supplementary Table S4). Recent investigations of the thermal behaviour of erionite21, 22 have detected relevant s.s. at both OW7 and OW12 at temperatures well exceeding that of dehydration (280–330 °C thermal range). This fact has been attributed to the migration of EF cations toward those sites to obtain a favourable coordination with oxygen atoms of the framework. We may hypothesize the onset of a similar mechanism in the case of the K-binding process. Therefore, the extra 37 e− detected at the water molecule sites of the KCl treated sample with respect to the pristine one may be attributed to EF cations, preferentially K. Summing this extra s.s. to that arising from the EF cations sites a total of 131 e− is obtained in perfect agreement with that determined by SEM-EDS (Table 1). A close inspection of the spatial distribution of the OW sites indicates a few possible coordination types for EF cations residing at (near) the OW7, OW10 and OW12 sites. However, it is expected that the reported fractional coordinates of H2O (+cations) sites represent the average position of very close neighbouring sites. Analysis of the s.s. at the various EF cations sites (Table 4) seems to suggest, except for Na, an unusual occurrence of K at Ca2. This hypothesis is supported by the presence of several potential ligands in the 3.1–3.3 Å range (OW12 × 3, OW9 × 3, O5 × 3). Both s.s. and coordination are consistent with the presence of Mg at Ca3 (Table 4). It must be noted that, considering the s.s. from SEM-EDS data and those from Rietveld refinement, there is a remarkably, albeit uncommon, good agreement, the possible mismatch being attributed to alkali-metal migration23. In the present case, we may hypothesize that the almost complete pore occlusion caused by the additional K cations prevent alkalies from migration/volatilization.
It must be pointed out that, despite the collapse of the surface area and the occlusion of all microporosity, the K-exchanged form unfortunately preserved its tendency to bind Fe whenever in contact with a source of Fe(II). Regarding the Fe(II) loaded sample, a significant increase of s.s. at Ca3 may be attributed to the segregation of Fe(II) at this site (Table 4), similarly to the experiments by Ballirano et al.16. In addition, Ca1 site, previously completely depleted during the K-exchange process, upon iron-loading acquires a s.s. that reasonably matches that of Mg as determined by SEM-EDS. Mobilization of Mg from Ca3 to Ca1 is favoured by a relatively small, non-occurring, distance between the two sites of 1.3 (2) Å. Therefore, we may hypothesize that Mg occupies Ca1 whereas Na is preferentially allocated at Ca2. However, the calculated s.s. of Ca2 is significantly smaller than that retrieved from the Rietveld refinement (Table 4). We may suggest that the Na content has been underestimated in the SEM-EDS analysis due to the well-known, above mentioned, analytical effects. The absence of a coupled significant reduction of s.s. at K2 indicates that the Fe(II) → K exchange process, devised by ICP analysis, prevalently involves K ions located at the OW sites as well as those at Ca2. This hypothesis is confirmed by the reported decrease of the total s.s. of the OW sites from 282 (7) to 268 (9) e− (Supplementary Table S4). Accordingly, the renewed Na (and possibly K) cation mobility under the electron beam can be justified by the leaching of the K cations located at the OW sites. On this basis, hypothesizing the same Na content as in the case of the KCl treated sample (1.5 apfu instead of 0.6), the overall s.s. balance improves significantly. As above mentioned, the BET specific surface area fairly increases with respect to the KCl treated sample passing from ca. 10 to ca. 75 m2 g−1. Our results showed that this is prevalently due to the release of K ions allocated in the OW sites, that therefore play a primary role in governing the specific surface area of the fibres (Fig. 2).

BET surface area as a function of the K content in the OW sites.
Notably, the BET specific of the Fe(II) loaded sample measured in the p/p0 0.05–0.3 range [and in the 0–0.1 range], significantly increases from 73.7 (17) m2 g−1 [and from 75.6 (17) m2] to 308 (7) m2 g−1 if the sample is further pre-treated under vacuum at 200 °C for 1.6 hours (Table 3). The sample heating at this temperature promotes its dehydration and consequently the migration of K hosted from the water molecular OW sites. Accordingly, the sample reacquires the peculiarity of microporous material with the total volume of the sample increasing to 0.182 cm3 g−1 (of which 0.151 cm3 g−1 attributable to micropores), value comparable with that measured for the pristine sample (0.151 cm3 g−1, of which 0.130 cm3 g−1 attributable to micropores).
Conclusions
Erionite-Na experienced a dramatic reduction of the BET surface area of ca. 95% upon K-loading indicating that the sample behaves exclusively as a mesoporous material, coherently with literature data. Although the large potassium ions blocked off nearly all the adsorption cavities, erionite-K preserves its capacity of binding Fe(II) in the erionite cavity by ionic exchange, with efficiency comparable to that observed for the pristine sample (erionite-Na). Rietveld refinement highlighted that the K ions residing at the OW sites provide a significant contribution to the reduction of the BET surface area. Coherently, the Fe(II) loaded sample showed a BET specific surface reduced of ca. 25% with respect to that of the pristine sample (erionite-Na). Notably, since the well-known relationship between surface properties of fibres and their interacting ability with biological medium27,28,29,30,31, this abrupt reduction supposedly plays a fundamental role in governing the corresponding chemical reactivity of erionite. It is worth noting that our experiments testify the very strong tendency of Fe(II) to bind to the erionite structure owing to its relatively high ionic potential exceeding that of the common EF cations. In fact, it is able to enter even into a very crowded erionite cage hosting additional large K cations at sites previously occupied by water molecules. Therefore, yet in the case of a very peculiar and potentially hostile crystal chemical and structural environment for a Fe(II) ion-exchange process we found essentially the same kinetics observed in a typical erionite sample16. This is a clear evidence of the very limited effect of the chemical composition of fibres on the Fe(II) bind process and, accordingly, we may hypothesize that their chemical composition does not play a significant role in inducing toxicity. Present results are of interest in future attempts to engineer alternative roads toward the (partial) inactivation of those fibres.
Materials and Methods
Materials
We used erionite-Na from Rome, Oregon, USA that has been previously characterized from the chemical and structural point of view by Ballirano et al.15. It has a crystal chemical formula (Na3.75K2.45Mg0.42)[Al7.24Si28.76O72.10] · 29.60H2O. About 5 g of the raw sample were enriched following the protocol devised by Ballirano and Cametti32.
SEM-EDS analysis
Chemical analyses were performed using a Quanta 400 SEM equipped with an EDAX Genesis EDS system (FEI, Hillsboro, OR, USA) and operating at 15 kV accelerating voltage, 11 mm working distance and 0° tilt angle. Chemical data were collected at least at 8 analytical points and the crystal chemical formulae were calculated, after renormalization of the chemical analyses hypothesizing a water content of 18.5 wt.% (corresponding to ca. 30 apfu), on the basis of 36 (Si + Al) apfu. The balance error formula E%33 and the K content test34 were evaluated for selecting the reliable analyses.
K binding to erionite experiment
K binding was performed following the indications of Eberly20 except for rescaling of the quantity of the materials. The KCl solution was obtained by dissolving 75 g of potassium chloride (KCl anhydrous ACS ≥99%, Sigma Aldrich) in 250 mL of deionized water. About 500 mg of erionite were placed in a 50 mL FalconTM polypropylene tube with 2 mL of KCl solution at 65 °C in an oscillating bath. A non-enriched material was used for a trial to define the best operating conditions for the K-binding procedure. In detail, three different treatments of the fibres with KCl solution were done. In the first one, fibres were stirred with KCl solution for 2 hours. Then, the fibres were separated from the solution by centrifugation at 4000 rpm for 10 min. The solution was filtered with a 0.2 µm membrane filter and stored for ICP-OES analysis. The fibres were recovered on filter and repeatedly rinsed with ultrapure deionized water to remove any residues of Cl ions and K not bound. The washing procedure was repeated until the absence of Cl ions was evidenced by the absence of opalescence in the washing solution after the addition of few drops of a solution of AgNO3 (0.1N). At the end of these washing cycles the fibres were dried and stored prior to the BET surface area measurement (KCl1 sample).
In the second experiment, the fibres after having been subjected to the first treatment were added again in a fresh KCl solution and stirred for other two hours. Then, solid and solution were separated following the same procedure described for KCl1 sample and stored for BET (KCl2 sample) and ICP-OES analysis, respectively. In the third experiment, the procedure of KCl treatment was repeated three times (KCl3 sample). A blank procedure, suspending the fibres in deionized water, was also performed. Notably, BET results (Table 3 ) showed no significant differences in the surface area between samples undertaking one (KCl1) or three KCl treatments (KCl3). Accordingly, only one KCl treatment was performed on the enriched material for the K-binding experiment.
Fe(II) binding to erionite experiment
Fe(II) binding was performed using the enriched erionite sample previously treated with the KCl solution and following the procedure described in Ballirano et al.19. In detail, 25 mg of erionite fibres were suspended in 25 mL of a 500 µM FeCl2 solution, in a 50 mL FalconTM polypropylene tube. All sample suspensions were prepared in a nitrogen-filled glove-bag to prevent iron oxidation. Later, all the polypropylene tubes, closed and sealed with Parafilm®, were removed from the glove-bag and instantly placed in an oscillating bath at 37 °C for 1 hour. After 1 h the tubes were placed again in the oxygen-free glove bag and the supernatant solution was sampled and filtered, using a 0.22 µm nitrocellulose membrane filter, for ICP-OES analysis. The fibres were recovered from the tube on filter, rinsed with ultrapure deionized water to remove any residues of iron not bound, dried and stored under nitrogen prior to the SEM-EDS and XRPD investigations. Blank samples were prepared by suspending fibres in deionized water adjusted to the appropriate pH (ca. 5: i.e. the same pH of the 500 μM FeCl2 solution) by adding HCl (Ultrex II, ultrapure reagent, JT Baker). Experiments were done in triplicate.
ICP-OES analysis
To measure the concentration of released cations from the fibres suspended in both the KCl and FeCl2 solutions, the filtered solutions were properly diluted with 1% nitric acid solution and analysed by ICP-OES. All measurements were performed using an Optima 2000 DV ICP-OES spectrometer (Perkin-Elmer, Waltham, MA, USA) equipped with a cross flow nebulizer placed inside a Scott spray chamber. ICP Aristar (BDH) standard solutions in nitric acid for the investigated elements were used to prepare the calibrating solutions for ICP-OES analyses. To ensure adequate quality assurance, the measures of the standard solutions were regularly repeated after the analysis of a defined number of samples. Concerning the Fe(II) binding to erionite experiments, the amount of Fe bound to the fibres (nmol/mg) was calculated by difference between the Fe concentration measured in the initial solution and that measured in the solution after 1 h of sample incubation. The net cation release was calculated by difference between the values measured after suspension in the FeCl2 solution and those measured after suspension of fibres in water at the appropriate pH (ca. 5). In order to verify if Fe(II) was acquired by erionite through an ion exchange mechanism, a charge balance was calculated using the same approach of Eborn and Aust15. The number of charges of each element was determined multiplying the nanomoles by the valence of the corresponding element and therefore, the net charges released into solution were compared to the total charges acquired in form of Fe.
Surface area measurements
Surface area, Brunauer–Emmett–Teller (BET) multipoint method35, and textural analysis were obtained by N2 adsorption/desorption measurements at the liquid nitrogen temperature (−196 °C), using a 3-Flex analyser (Micromeritics, Norcross, GA, USA). Before analysis, samples were pre-treated under vacuum at 65 °C for 15 hours. The pores distribution was determined from the adsorption curve by the Barret–Joyner–Halenda (BJH) method36 and from the analysis of the micropore isotherm by the t-test37 taking the curve of Harkins and Jura38. The total pore volume was determined by the rule of Gurvitsch39.
X-ray powder diffraction (XRPD)
XRPD data were collected in transmission mode, using SiO2-glass capillaries, on a D8 Advance (Bruker AXS, Karlsruhe, Germany) equipped with incident-beam focussing Göbel mirrors and a PSD VÅntec-1. XRPD patterns were collected in the 6–145° 2θ angular range, 0.022° 2θ step-size and 10 s counting time. Data were evaluated by a mixed Rietveld/Pawley method described in ref. 26 using Topas V.4.240. Starting structural data were taken from Ballirano et al.16 and refinement procedures follow Ballirano41. A content of chabazite (Ca2Al4Si8O24 ∙ 12H2O) and quartz (SiO2) of ca. 3 and 0.1 wt.% have been calculated by the Rietveld method, respectively, and ≪5 wt.% of nontronite has been estimated. Miscellaneous data of the refinements are reported as Supplementary Table S5. Conventional Rietveld plots are reported as Supplementary Figure S1.
References
Tschernich, R. W. Zeolites of the world pp. 156–166 (Geoscience Press, 1992).
Baris, Y. I. et al. An outbreak of pleural mesothelioma and chronic fibrosing pleurisy in the village of Karain/Urgup in Anatolia. Thorax 33, 181–192, doi:10.1136/thx.33.2.181 (1978).
IARC IARC. Monographs on the evaluation of the carcinogenic risk to humans. Silica and some silicates. 42, 225–239 (1987).
IARC IARC. Monographs on the evaluation of the carcinogenic risk to humans. Arsenic, metals, fibres and dusts. 100, 311–316 (2011).
Fubini, B. & Mollo, L. Role of iron in the reactivity of mineral fibers. Toxicol. Letters 82–83, 951–960, doi:10.1016/0378-4274(95)03531-1 (1995).
Gazzano, E., Riganti, C., Tomatis, M., Turci, F., Bosia, A., Fubini, B. & Ghigo, D. J. Potential toxicity of nonregulated asbestiform minerals: balangeroite from the western Alps. Part 3: depletion of antioxidant defenses. Toxicol. Environ. Health 68, 41–49, doi:10.1080/15287390590523957 (2005).
Wang, Y., Faux, S., Hallden, G., Kirn, D., Houghton, C., Lemoine, N. & Patrick, G. Interleukin-1beta and tumour necrosis factor-alpha promote the transformation of human immortalised mesothelial cells by erionite. Int. J. Oncol. 25, 173–180 (2004).
De Assis, L. V. M., Locatelli, J. & Isoldi, M. C. The role of key genes and pathways involved in the tumorigenesis of Malignant Mesothelioma. Biochim. Biophys. Acta 1845, 232–247 (2014).
Dogan, A. U., Baris, Y. I., Dogan, M., Emri, S., Steele, I., Elmishad, A. G. & Carbone, M. Genetic predisposition to fiber carcinogenesis causes a mesothelioma epidemic in Turkey. Cancer Res. 66, 5063–5068, doi:10.1158/0008-5472.CAN-05-4642 (2006).
Carr, A. & Frei, B. Does vitamin C act as a pro-oxidant under physiological conditions? FASEB J. 13, 1007–1023 (1999).
Ballirano, P., Andreozzi, G. B., Dogan, M. & Dogan, A. U. Crystal structure and iron topochemistry of erionite-K from Rome, Oregon, USA. Am. Mineral. 94, 1262–1270, doi:10.2138/am.2009.3163 (2009).
Matassa, R., Famigliari, G., Relucenti, M., Battaglione, E., Downing, C., Pacella, A., Cametti, G. & Ballirano, P. A Deep Look Into Erionite Fibres: an Electron Microscopy Investigation of their Self-Assembly. Sci. Rep. 5, 16757, doi:10.1038/srep16757 (2015).
Croce, A., Allegrina, M., Rinaudo, C., Gaudino, G., Yang, H. & Carbone, M. Numerous Iron-Rich Particles Lie on the Surface of Erionite Fibers from Rome (Oregon, USA) and Karlik (Cappadocia, Turkey). Microsc. Microanal. 21, 1341–1347, doi:10.1017/S1431927615014762 (2015).
Gualtieri, A. F., Bursi Gandolfi, N., Pollastri, S., Pollok, K. & Langenhorst, F. Where is iron in erionite? A multidisciplinary study of fibrous erionite-Na from Jersey (Nevada, USA). Sci. Rep. 6, 37981, doi:10.1038/srep37981 (2016).
Eborn, S. K. & Aust, A. E. Effect of iron acquisition on induction of DNA single-strand breaks by erionite, a carcinogenic mineral fiber. Arch. Biochem. Biophys. 316, 507–514, doi:10.1006/abbi.1995.1067 (1995).
Ballirano, P. et al. Fe(II) segregation at a specific crystallographic site of fibrous erionite: A first step toward the understanding of the mechanisms inducing its carcinogenicity. Micropor. Mesopor. Mat. 211, 48–63, doi:10.1016/j.micromeso.2015.02.046 (2015).
Pacella, A., Fantauzzi, M., Atzei, D., Cremisini, C., Nardi, E., Montereali, M. R., Rossi, A. & Ballirano, P. Iron within the erionite cavity and its potential role in inducing its toxicity: Evidence of Fe(III) segregation as extra-framework cation. Microp. Mesop. Mat. 237, 168–179, doi:10.1016/j.micromeso.2016.09.021 (2017).
Schwidder, M., Santhosh Kumar, M., Klementiev, K. V., Pohl, M. M., Brückner, A. & Grünert, W. Selective reduction of NO with Fe-ZSM-5 catalysts of low Fe content I. Relations between active site structure and catalytic performance. J. Catal. 231, 314–330, doi:10.1016/j.jcat.2005.01.031 (2005).
Zecchina, A., Rivallan, M., Berlier, G., Lamberti, C. & Ricchiardi, G. Structure and nuclearity of active sites in Fe-zeolites: comparison with iron sites in enzymes and homogeneous catalysts. Phys. Chem. 9, 3483–3499, doi:10.1039/b703445h (2007).
Eberly, P. E. Adsorption properties of naturally occurring erionite and its cationic-exchanged forms. Am. Mineral. 49, 30–40 (1964).
Ballirano, P. & Cametti, G. Dehydration dynamics and thermal stability of erionite-K: Experimental evidence of the “internal ionic exchange” mechanism. Microp. Mesopor. Mat. 163, 160–168, doi:10.1016/j.micromeso.2012.06.059 (2012).
Ballirano, P. & Pacella, A. Erionite-Na upon heating: dehydration dynamics and exchangeable cations mobility. Sci. Rep. 6, 22786, doi:10.1038/srep22786 (2016).
Kokotailo, G. T., Fyfe, C. A., Feng, Y., Grondey, H., Gies, H., Marler, B. & Cox, D. E. Powder X-ray diffraction and solid state NMR techniques for zeolite structure determination. In Catalysis of Microporous Materials (eds Bayer, H. K., Nagy, J. B., Karge, H. G. & Kiricsi, I.). Studies in Surface Science and Catalysis, Elsevier, Amsterdam, pp. 78–100 (1995).
Pacella, A., Ballirano, P. & Cametti, G. Quantitative chemical analysis of erionite fibres using micro-analytical SEM-EDX method. Eur. J. Mineral. 28, 257–264, doi:10.1127/ejm/2015/0027-2497 (2016).
Jones, J. B. Al-O and Si-O tetrahedral distances in aluminosilicate framework structures. Acta Crystallogr. B24, 355–358, doi:10.1107/S0567740868002360 (1968).
Schlenker, J. L., Pluth, J. J. & Smith, J. V. Dehydrated natural erionite with stacking faults of the offretite type. Acta Crystallogr. B33, 3265–3268, doi:10.1107/S0567740877010784 (1977).
Fubini, B. The possible role of surface chemistry in the toxicity of inhaled fibres In Fibre Toxicology (ed. Warheit, D. B.) 229–257 (Academic Press, 1993).
Fubini, B. Use of physico-chemical and cell free assays to evaluate the potential carcinogenicity of fibres In Mechanisms of Fibre Carcinogenesis (eds Kane, A. B., Boffetta, P., Saracci, R. & Wilbourn, J. D.) 35–54 (IARC Sci. Publ., Lyon, 1996).
Ottaviani, M. F. & Venturi, F. Physico-chemical study on the adsorption properties of asbestos. An EPR study on the adsorption of organic radicals. J. Phys. Chem. 100, 265–269, doi:10.1021/jp9515850 (1996).
Ottaviani, M. F., Venturi, F., Pokhrel, M. R., Schmutz, T. & Bossmann, S. H. Physico-Chemical Studies on the Adsorption Properties of Asbestos 2. An EPR and Fluorescence Study on the Adsorption of Pyrene. J. Colloid Interface Sci. 238/2, 371–380, doi:10.1006/jcis.2001.7529 (2001).
Mattioli, M., Giordani, M., Dogan, M., Cangiotti, M., Avella, G., Giorgi, R., Dogan, A. U. & Ottaviani, M. F. Morpho-chemical characterization and surface properties of carcinogenic zeolite fibers. J. Haz. Mat. 306, 140–148, doi:10.1016/j.jhazmat.2015.11.015 (2016).
Ballirano, P. & Cametti, G. Crystal chemical and structural modifications of erionite fibers leached with simulated lung fluids. Am. Mineral. 100, 1003–1012, doi:10.2138/am-2015-4922 (2015).
Passaglia, E. The crystal chemistry of chabazites. Am. Mineral. 55, 1278–1301 (1970).
Cametti, G., Pacella, A., Mura, F., Rossi, M. & Ballirano, P. New morphological, chemical, and structural data of woolly erionite-Na from Durkee, Oregon, USA. Am. Mineral. 98, 2155–2163, doi:10.2138/am.2013.4474 (2013).
Gregg, S. J. & Sing, K. S. W. Adsorption, Surface Area and Porosity. Second Edition (Academic Press, 1982).
Barrett, E. P., Joyner, L. G. & Halenda, P. P. The determination of pore volume and area distributions in porous substances. I. Computations from nitrogen isotherms. J. Am. Ceram. Soc. 73, 373–380, doi:10.1021/ja01145a126 (1951).
Lippens, B. J. & de Boer, J. H. Studies on pore systems in catalysts: V. The t method. J. Catal. 4, 319–323, doi:10.1016/0021-9517(65)90307-6 (1965).
Harkins, W. D. & Jura, G. Surfaces of Solids. XIII. A vapor adsorption method for the determination of the area of a solid without the assumption of a molecular area, and the areas occupied by nitrogen and other molecules on the surface of a solid. J. Am. Chem. Soc. 66, 1366–1373, doi:10.1021/ja01236a048 (1944).
Gurvitsch, L. Physicochemical attractive force. J. Phys. Chem. Soc. Russ. 47, 805–827 (1915).
Bruker, AXS Topas V.4.2: General profile and structure analysis software for powder diffraction data. Bruker AXS, Karlsruhe, Germany (2009).
Ballirano, P. Effects of the choice of different ionisation level for scattering curves and correction for small preferred orientation in Rietveld refinement: the MgAl2O4 test case. J. Appl. Crystallogr. 36, 1056–1061, doi:10.1107/S0021889803010410 (2003).
Acknowledgements
The financial support by Sapienza Università di Roma (P.R. Ateneo 2014, Prof. Ballirano) is acknowledged.
Author information
Authors and Affiliations
Contributions
A.P. performed SEM-EDS, analysed the results, wrote and revised the manuscript; C.C., E.N. and M.R.M. engineered and performed binding experiments, carried out ICP-OES measurements, analysed the results, wrote and revised the manuscript; I.P. performed surface area measurements, analysed the results, wrote and revised the manuscript; P.B. conceived the research and conducted XRPD experiments, performed structure refinements, analysed the results, wrote and revised the manuscript.
Corresponding author
Ethics declarations
Competing Interests
The authors declare that they have no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Pacella, A., Cremisini, C., Nardi, E. et al. The mechanism of iron binding processes in erionite fibres. Sci Rep 7, 1319 (2017). https://doi.org/10.1038/s41598-017-01477-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-017-01477-x
