
Abstract
The significant role of microRNAs (miRNAs) in various biological processes and diseases has been widely studied and reported in recent years. Several computational methods associated with mature miRNA identification suffer various limitations involving canonical biological features extraction, class imbalance, and classifier performance. The proposed classifier, miRFinder, is an accurate alternative for the identification of mature miRNAs. The structured-sequence features were proposed to precisely extract miRNA biological features, and three algorithms were selected to obtain the canonical features based on the classifier performance. Moreover, the center of mass near distance training based on K-means was provided to improve the class imbalance problem. In particular, the AdaBoost-SVM algorithm was used to construct the classifier. The classifier training process focuses on incorrectly classified samples, and the integrated results use the common decision strategies of the weak classifier with different weights. In addition, the all mature miRNA sites were predicted by different classifiers based on the features of different sites. Compared with other methods, the performance of the classifiers has a high degree of efficacy for the identification of mature miRNAs. MiRFinder is freely available at https://github.com/wangying0128/miRFinder.
Similar content being viewed by others

Introduction
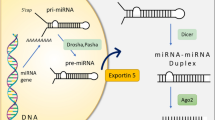
MicroRNAs (miRNAs) are an ~22 nucleotide (nt) long, conserved class of noncoding RNAs that play key regulatory roles in diverse biological processes and diseases1,2, especially cancers, by modulating the gene expression3. Therefore, identification of miRNAs has important significance for mining the association of miRNAs and diseases. In mammals, mature miRNAs derive from the hairpin in primary transcripts by two cleavages process: primary miRNA (pri-miRNA) is processed and canonically cleaved to precursor miRNA (pre-miRNA) by Drosha4, then pre-miRNA is exported by Exportin 55 and cytoplasmically processed into a miRNA:miRNA* duplex by Dicer6,7. Afterwards, one strand of the duplex product becomes a mature miRNA and the other degrades8,9,10. In individual cases, both strands are entirely functional.
Because pre-miRNAs are conservative and have the typical features involving sequence, structure and free energy, much software has been development to identify the mature miRNAs from their pre-miRNAs based on computational methods. Table 1 displays the main algorithms and software for mature miRNA identification.
MatureBayes11 identifies the starting sites of mature miRNAs for mice and humans based on Naïve Bayes. As a result, the method finds that 7, 8 and 9 nt from the starting position have the typical biological features to distinguish mature miRNAs. Microprocessor SVM12, MiRpara13, MaturePred14 and matPred15 are developed using the SVM algorithm. Microprocessor SVM is proposed based on 686 features that are associated with sequence and structure, the accuracy of this method is 50%, and 90% of its predictions are within a 2 nt deviation. Due to too many features, the results are not easy to use for biological analysis. In addition, the forecasting ability for the 3′ arm of miRNA is relatively weak. MiRpara is trained for animals and plants, and upon filtering these features based on the greedy algorithm and the SPSS method, its accuracy reached 80%. MaturePred uses 160 features to identify mature plant miRNAs, and it extracts features from the miRNA:miRNA* duplex and the flanking region, and then 86 features were selected using the gain information algorithm. Thus, the method achieves a higher prediction performance. MiRRim216 is designed based on phastcons and phylop scores, which are extracted from position 20 downstream and upstream the Drosha processing sites. Twelve submodels were trained according to the scores and base pair to predict mature human miRNA in their pre-miRNA. As a result, its sensitivity and positive predictive value exceeded 0.4. MiRmat and miRdup were designed based on the random forest algorithm. MiRmat17 is based on the molecular interaction and miRNA biogenesis for vertebrates and consists of two parts: Drosha and Dicer sites prediction. Using the random forest algorithm, MiRmat reached 77.8% and 92.8% accuracy, respectively, for the Dosha and Dicer sites. MiRdup18 predicted the mature miRNAs based on 100 features from five lineages of cleavage sites on the miRNA:miRNA* duplexes using the random forest algorithm integrated with adaptive boost (Adaboost). MirExplorer19 was designed using transition probability matrices and miRNA biogenesis vectors using the Adaboost method for 16 species, whereas with earlier methods, it obtained a specificity of 95.03% and a sensitivity of 93.71% on human data. In particular, matPred was proposed in our previous work. MatPred is a highly effective method for identifying mature miRNAs within novel pre-miRNA transcripts based on the SVM for humans. It significantly outperformed three other widely used methods. Recently, many up-to-date methods have been designed for pre-miRNA identification, and they have a detailed research on feature selection and algorithm optimization, such as iMiRNA-PseDPC20, gapped kernels21, methods based on structure status22 and miRNA-dis23. In addition, powerful web servers have been specifically used for extracting the features from the RNA sequences, such as repRNA24 and Pse-in-One25. They solve problems of various types in pre-miRNA identification and improve the performance of classification.
Although many methods are available for mature miRNA location prediction, they suffer from various limitations. The canonical biological features extraction from the characteristic hairpins directly affects the accuracy of algorithms. In the second structure of pri-miRNA, the default information always corresponds to ‘bulge’, ‘interloop’ and ‘multibranch loop’, which are associated with the change in free energy, but this biological feature receives little attention. Otherwise, the computational methods of mature miRNA identification remain the primary focus on the start sites of mature miRNAs, but the end sites of mature miRNAs have important functions, especially for the heterogeneous miRNAs finding that always occurs in the end of mature miRNAs26,27,28. More importantly, because the true and pseudo-mature miRNAs have a very high sequence similarity, the accuracy of mature miRNAs identification methods remains low, no deviation identification accuracy is less than 50%, and the 5 nt deviation identification accuracy is less than 90%.
In this study, we introduce a computational method, matFinder, that uses an AdaBoost-SVM algorithm to predict all the process sites of the mature miRNA in a pre-miRNA transcript. The structured-sequence features that focus on the default information in the secondary structure of pre-miRNAs are presented. For all the processing sites, corresponding models are trained based on their biological characters. More importantly, we design a mature miRNAs identification method using the AdaBoost and SVM algorithms. Because the AdaBoost algorithm adjusts the data set of SVMs based on the incorrectly classified samples, the accuracy of our method is promoted instead of that of the method based on SVM. In addition, comparison with existing tools suggests that MatFinder achieves the highest prediction accuracy.
The schematic of the overall method is illustrated in Fig. 1.

The schematic of the overall method. MiRFinder consists of five steps, namely, dataset construction, construction of structured-sequence, biological-related features extraction, feature selection, class imbalance problem and classifier construction. For features selection, three algorithms including information gain, chi-square and relief were investigated. For class imbalance problem, the AdaBoost algorithm was adopted. Moreover, for classifier construction, the AdaBoost and SVM were used. SVM is used as the weak classifier, and AdaBoost is used as the strong classifier.
Materials and Methods
Data
The training and test datasets were obtained from the miRBase V2129. The pre-miRNAs that have a secondary structure including multibranch loops were excluded. The process of dataset construction is shown in Fig. 2.

The process of dataset construction.
First, the human pre-miRNAs were downloaded from miRBase V20 and divided into 5_data and 3_data based on the location of the mature miRNAs. If the mature miRNA of pre-miRNA is located in the 5′ arm, then the pre-miRNA is divided into 5_data. Otherwise, if the mature miRNA of pre-miRNA is located in the 3′ arm, then the pre-miRNA is divided into 3_data. In a similar way, the pre-miRNAs that only belong to version 21, named miRBase V21_new, were downloaded from miRBase V21 and divided into 5_newdata and 3_newdata. Second, Test set1 and Test set2 for testing the 5′ arm mature miRNAs models were randomly selected from 5_data and 5_newdata. Between them, Test set2 was randomly selected from the newest dataset that only belongs to version 21. Moreover, Test set3 and Test set4 for testing the 3′ arm mature miRNAs models were randomly selected from 3_data and 3_newdata, and between them, Test set4 was randomly selected from the newest dataset that only belongs to version 21. Finally, except for the Test set, the other dataset was used to construct the training data. As a result, we constructed the 5_for_train and the 3_for_train datasets to train the models for identifying the 5′ arm and 3′ arm mature miRNAs, respectively. The 3_for_train and 5_for_train datasets consist of 1118 and 1071 pre-miRNAs sequences, respectively. Test set1, Test set2, Test set3 and Test set4 consist of 100 pre-miRNAs.
The positive and negative datasets were constructed based on the above training and test datasets. Taking hsa-mir-19a as an example, the structure-based dataset construction method is shown in Fig. 3.

Structure-based dataset construction method.
RNAfold30 was used to predict the secondary structure of each pre-miRNA, and then the stem-loop of each pre-miRNA was constructed based on the sequence and structure. Furthermore, the stem of the pre-miRNA was complemented by a 10 base pair (bp) double strand and a right base pair (bp). The double strand regions, which include 22 nt nucleotides in the 5′ arm, were extracted from the first to the last nucleotide using the sliding window method. Finally, the double strand, which was consistent with the starting position of the true mature miRNA, was selected as the positive example, whereas the other sequences were defined as the negative example.
The extraction of feature set
Taking has-miR-19a as an example, the process of feature extraction is shown in Fig. 4. To extract the features of mature miRNAs, we predicted the secondary structure of each pre-miRNA using ViennaRNA software, and then constructed the structured-sequence based on the sequence, structure and the alignment information. Depending on the requirements of feature extraction, strand regions of different lengths were selected. Based on the sequence, structure, structured-sequence and these strand regions, the features were extracted.

The process of the feature extraction.
The absence information, ‘−’, in the structured-sequence is important. It always means the change in free energy, sequence and structure associated with a ‘bulge’, ‘interloop’ and ‘multibranch loop’. To compare the performance of the structured-sequence with the absence information, we also extracted features without the absence information.
Feature selection
The feature set of mature miRNA recognition includes hundreds of feature vectors, and such high-dimensional data can cause “dimension disaster”. The effective feature selection algorithm is an important part of data preprocessing. Feature selection can remove the uncorrelated redundant features, reduce the data set dimension, optimize and classify the high correlation characteristics, avoid dimension disaster in the machine learning method, and improve the generalization performance of the model. Importantly, by analyzing the characteristics of mature miRNAs, the typical characteristics of true and pseudo-mature miRNAs that differentiate mature miRNAs and other short nucleotide sequences are identified, which has important value and biological significance for miRNA recognition and its functional research.
In this paper, three feature selection algorithms are used to select the feature selection, namely, the information gain (IG), CHI (CHI) and relief algorithms. On the basis of a performance test, we selected the optimal feature selection algorithm.
-
(1)
The information gain (IG) selects characteristics by calculating the difference in information entropy. It defines class Ci, and the information gain of characteristic t is defined as:
$${\rm{IG}}({\rm{t}},{{\rm{C}}}_{{\rm{i}}})=-\,{\rm{P}}({{\rm{C}}}_{{\rm{i}}}){\rm{logP}}({{\rm{C}}}_{{\rm{i}}})+{\rm{P}}({\rm{t}}){\rm{P}}({{\rm{C}}}_{{\rm{i}}}|{\rm{t}}){\rm{logP}}({{\rm{C}}}_{{\rm{i}}}|{\rm{t}})+{\rm{P}}(\bar{{\rm{t}}}){\rm{P}}({{\rm{C}}}_{{\rm{i}}}|\bar{{\rm{t}}}){{\rm{logPC}}}_{{\rm{i}}}|\bar{{\rm{t}}})$$(1)where t means t nonexistence. Then, the IG of t is defined as:
$${\rm{IG}}({\rm{t}})={\sum }_{{\rm{i}}}{\rm{IG}}({\rm{t}},{{\rm{C}}}_{{\rm{i}}})$$(2) -
(2)
Chi-square statistic (CHI) selects features between the representation variables by calculating the correlation. The larger the statistical value of CHI is, the more important the feature is. For any type of C_i, the CHI value of characteristic t is:
$${\rm{CHI}}({{\rm{C}}}_{{\rm{i}}},{\rm{t}})=\frac{{\rm{P}}({{\rm{C}}}_{{\rm{i}}},{\rm{t}}){\rm{P}}({\bar{{\rm{C}}}}_{{\rm{i}}},\bar{{\rm{t}}})-{\rm{P}}({\bar{{\rm{C}}}}_{{\rm{i}}},{\rm{t}}){\rm{P}}({{\rm{C}}}_{{\rm{i}}},\bar{{\rm{t}}})}{{\rm{P}}({{\rm{c}}}_{{\rm{i}}}){\rm{P}}({\rm{t}}){\rm{P}}({\bar{{\rm{C}}}}_{{\rm{i}}}){\rm{P}}(\bar{{\rm{t}}})}$$(3) -
(3)
Relief algorithm.
The relief algorithm selects the nearest neighbor according to the weight by calculating the distance between the samples. Set \({\rm{X}}=\{{{\rm{X}}}_{1},{{\rm{X}}}_{2},\ldots ,{{\rm{X}}}_{{\rm{n}}}\}\) as the sample dataset, and Xi = [Xi1, Xi2, …XiN]T is the Nth character of the ith sample. The weight of the sample on each characteristic is defined as:
where H(x) and M(x) are the nearest neighbors of the same and different class of X, respectively, and diff is defined as:
Weak classifier training based on SVM with probability
Mature miRNAs identification identifies the true mature miRNAs from many short sequences that are constructed from one pre-miRNAs. Therefore, for each pre-miRNA, we cannot identify whether a short sequence is a mature miRNA, but the possibility exists that it is a mature miRNA, so here we introduced the SVM method based on probability to train a mature miRNA classifier. The SVM algorithm based on a probabilistic model is as follows:
Define mature miRNA training samples \({\rm{T}}=\{({{\rm{x}}}_{1},{{\rm{y}}}_{1}),({{\rm{x}}}_{2},{{\rm{y}}}_{2}),\ldots \ldots .,({{\rm{x}}}_{{\rm{N}}},{{\rm{y}}}_{{\rm{s}}})\}\), where xi is the character value of the sample, ys ∈ {1, −1}, and the number of samples is N, which includes Na positive samples and Nb negative samples. Set each sample to having M characters, \({{\rm{x}}}_{{\rm{i}}}=\{{{\rm{x}}}_{{\rm{i}}}^{1},{{\rm{x}}}_{{\rm{i}}}^{2},\ldots \ldots ,{{\rm{x}}}_{{\rm{i}}}^{{\rm{M}}}\}\), and the class functions are defined as follows:
where xi is a character vector of some sample, x is the prediction sample, ∝i(0 ≤ ∝i ≤ C) is a trainable coefficient, C is a penalty parameter, and <x, xi> is the inner product of x and xi. The kernel function is used to calculate the inner product, and it solves the problem of the data mapping from the original space to the high dimensional linear nonseparable problem. In particular, the radial basis function (RBF) is defined as follows:
where δ is the conventional control parameter, which determines the weight of the feature.
The output of traditional SVM is binary. This means that for each point, it either belongs to a positive or negative class. In this way, for a pre-miRNA, all the candidate mature miRNA sequences on one arm are predicted to be pseudo-sequences, or the predicted mature miRNAs are greater than or equal to two sequences. Obviously, this is not biological. The mature miRNAs prediction needs to find the only mature miRNA in one arm of a pre-miRNA, so the SVM based on probability output was used to solve this problem.
For the sample χ, the posttest probability is:
The sum of the probability of the sample belonging to two classes is 1. Therefore, the constraint conditions of equation (8) are:

Set rij to the probability estimate of two types of problems. According to (9), (10) proposes the following solution:
(11) is calculated as follows:
where
The matrix Q is a semipositive definite matrix, so equation (11) is a convex quadratic programming problem with linear constraints. If P is the optimal solution to the quadratic programming problem, the following conditions are met:
The solution to equation (11) can be obtained by solving linear equations.
Using the above method, the probability of each short sequence of a pre-miRNA is estimated, and the probability of the true mature miRNA is given. In the process of model training, it is necessary to optimize the two parameters of the planning factor C and the Gaussian width g. For the planning factor, if C → ∞, it is shown that the classification rules for satisfying all the constraint conditions will reduce the generalization ability and improve training complexity, so the C range must be as wide as possible to satisfy the classifier generalization performance. For parameter g, the optimization algorithm is used for adjusting, and the software grid py is used for training.
Strong classifier constructed based on Adaboost
When SVM is combined with AdaBoost, on the one hand, the SVM algorithm makes up for the error in AdaBoost in processing high-dimensional data. On the other hand, the SVM algorithm follows the structure risk minimization principle, and the parameter optimization of the RBF_SVM classifier can improve the classification performance of a weak classifier. By selecting appropriate parameters C and g, it can avoid overfitting. In addition, the AdaBoost algorithm is also a process of data transfer in the integration process, and the research on the solution of the class imbalance problem is a direction worth exploring.
For the selection of data sets, the AdaBoost algorithm implements the training subset sample selection according to the continuous adjustment of the sample parameters. The method first sets the initial weight value, then adjusts the sample weight through the sample error rate during each progressive training process and adjusts the weight of the weak classifier accordingly. In the entire process, the weight of the wrong subsample is divided into emphasis to improve the recognition rate. Our method is based on AdaBoost, and the weak classifier adopts the SVM algorithm based on the probabilistic mode, and in each round of weak classifier training, the parameter optimization is performed. The Adaboost-SVM algorithm is described as follows:
Set the training dataset S = {(xi, yi)|i = 1,2, … n}, where xi ∈ X is the mature miRNA sample, and yi ∈ Y = {+1, −1} is the class of the samples.
Set the weight of xi in the training set, which in the sample of training dataset S is \({{\rm{D}}}_{{\rm{t}}}({\rm{i}})\,\)in round t, and the first round of sample weight is initialized to:
We use the adjustable parameters of the SVM based on probability as a weak classifier. In the process of training, through the parameter adjustment, the optimal classification plane is selected, and for each of the pre-miRNA, a given probability of each sample that it is the true mature miRNA is given. The class of the candidate that has the largest probability is set as +1, the class of other candidates are set as −1, and the classifier Gt(X): X → {−1, +1}.
Set the training round T.
Define the weight distribution of training set S:
where Dt is the vector that is constructed by all samples. From the training set, train the subset St based on the sample weight.
The error rate of the training subset is calculated by setting Gt(X):{X → Y}. The sample error rate can be described as:
The classifier weight is:
Then, the weight of the sample can be update as:
where formula (19)can be shown as,
In formula (20), zt is defined as the weight of the next round t of the training set, which is a generalized constant defined as:
Finally, the integrated classifier is defined according to the weak classifier \({{\rm{G}}}_{{\rm{t}}}({\rm{X}})\) and its weight:
Improving classification by solving the class imbalance problem
The ratio of the positive dataset and negative dataset of these approaches is usually larger than 1:10. To improve the classification performance with respect to the imbalanced dataset including the positive and negative dataset, we designed the algorithm to solve the class-imbalance problem of mature miRNAs prediction.
Set the training dataset S = {Sneg, Spos}. The K-means algorithm was used to cluster the positive samples. Given the negative dataset including n samples, define a threshold that is the number of clusters. Because the ratio of the positive and the negative dataset is 1:10, the threshold was set as 10. This means that the negative samples were divided into 10 clusters, namely, K1, K2, ……, K10, where Ki ∈ D, Kj ∈ D, Ki ∩ Kj = ϕ.
For any cluster Ki, define km as the center of mass, which is obtained by calculating the average value of the features, and the distance of any sample kn and the center of mass is defined as dist(kn, km). The mass E is defined as the sum of all squared sample features in the cluster and the center of mass, as follows:
By calculating the optimal distance distribution, the distance between samples in the cluster is minimized, but the distance between samples and other clusters is maximized and reaches the maximum degree of mutually independence between clusters.
The pseudocode for constructing the sample balance method of the center of mass near distance training based on the K-means algorithm is described as:
Algorithm: The sample balance method of the center of mass near distance training based on K-means
Input: negative dataset, cluster threshold
Output: negative subset
The processing flow:
Input negative dataset;
The threshold of the cluster;
Ten cluster masses were selected in the negative dataset.
The distances between all samples and the center of mass in the negative dataset were calculated and divided into 10 clusters.
The mean of each cluster was recalculated and was taken as the new center of mass of the cluster.
If the centers of mass are the same, the next step is taken. Otherwise, the center of mass is calculated circularly until the centers of mass are the same.
After determining the center of mass, one tenth of the samples that are close to the center of mass was selected as a subset of the negative subset.
The above method is used to determine the initial training subset S1. When we trained the first weak classifier, the training dataset was classified, and the misclassified subset Sincor1 and S1 were combined to construct the training subset S2, which was used to train the next classifier. Therefore, the training subset Si is defined as:
Classifier performance estimation
To evaluate the classification performance of the classifier, two indices are adopted, namely, the prediction accuracy and position deviation.
The prediction accuracy was defined as the percentage of the correct mature miRNA and total mature miRNA. For N pre-miRNA sequences, there are M mature miRNA candidate sequences in the ith sequence. Assuming that T (s) is the true mature miRNA and P (T) is the true mature miRNA, the prediction accuracy, Acc, can be described as:
The average position deviation (APD) is the absolute value of the difference between the predicted mature miRNA position and the true mature miRNA position. It is defined as follows:
where ti is the nucleotide position of the ith pre-miRNA, and si is the position of the ith predicted mature miRNA.
Results and Discussion
Comparison of the structured-sequence-based and sequence-based methods
To investigate the classifier performance of the structured-sequence-based characters and sequence-based characters, we designed the mature miRNA identification method based on sequence-based characters. Taking P5_5 as an example, the two methods are compared in Tables 2–5:
As shown in Tables 2–5, taking training set 1 as an example, we compared two classifiers that are trained based on sequence-based characters and structured-sequence-based characters: the first candidate prediction accuracy are 17% and 30%, respectively, the latter being 13% higher than the former. The position deviation predicted accuracy with the 5 nt position deviation was 67% and 90%, respectively. The latter is 33% higher than the former. The position deviation predicted accuracy of the top five candidates are 46% and 72% respectively, as the latter increased by 26%. The predicted accuracy with the 5 nt position deviation is 98% and 100%. Therefore, according to various indicators, the performance of the classifier based on the structured-sequence-based characters is greatly improved.
To investigate the efficiency of our tool as it varies in the presence of polynucleotide regions in the bulge of the pre-miRNAs that form the spacer in the pre-miRNA coding frame, we test it based on two test datasets. The components of bulge (absence information, “−”) were counted, and the relationship between the amount of absence information and identification accuracy are shown in Fig. 5 and Table 6.

The relationship between the amount of absence information and identification accuracy. (A) The relationship between the amount of absence information and identification accuracy of the first candidate. (B) The relationship between the amount of absence information and identification accuracy of the top five candidates.
When the amount of absence information is 0–2, the identification accuracy of the first candidate is 51.2%, and it is the highest. When the amount of absence information is 3–5, the identification accuracy of the first candidate is 33.8%, and it is the lowest. Overall, the identification accuracy decreased as the amount of absence information increased. This result can be analyzed in two ways. On the one hand, it illustrates that the identification of our tool can be affected by the amount of absence information, and with the amount of absence information increasing, the identification accuracy decreased. On the other hand, it indicates that the capture of biological characters were affected by the amount of absence information, and with the amount of absence information increasing, biological characteristics become less typical.
Comparison of the feature selection methods
We examined three feature selection algorithms: the information gain algorithm, chi-square statistics and the relief method. First, we used all feature sets to train the classifier to obtain the accuracy of the position offset prediction of the first candidate. Then, we used these three algorithms to filter feature sets. The information gain method sorts features according to the information gain, whereas the chi-square statistic method provides a measure of the correlation between features and the categories of measurements. The relief method sorts the features according to the sample weight. According to the results of the algorithms, features that contribute a value of less than zero are deleted, and then the feature selection algorithm and the feature set are confirmed.
The position deviation predicted accuracy of the first candidate based on all features is shown in Table 7, and the position deviation predicted accuracy of the first candidate based on the chi-square and relief algorithms is shown in Tables 8 and 9. The position deviation predicted accuracy of the first candidate based on IG is shown in Table 10.
The position deviation predicted accuracy of the first candidate of the all features, chi-square Relief and IG algorithms were 24%, 12%, 13%, and 30%, respectively. The position deviation predicted accuracy of the first candidate with 5 nt deviation was 79%, 71%, 66% and 90%, respectively. The information gain method obtained the highest prediction performance, and when the feature subset selected 110 features, we obtained the maximum prediction precision. Compared with the all features method, the position deviation predicted accuracy of the first candidate of the two test sets were 30% and 59%, and improved 6% and 11%, respectively.
Comparison of methods before and after using the balance algorithm
To investigate the effect of balance algorithm on the classifier performance, taking Test1 as an example, we compared the methods before and after using the balance algorithm. The position deviation predicted accuracy and APD of the first candidate and the top five candidates are shown in Tables 11 and 12, respectively.
The position deviation predicted accuracy of the first candidate is 31% and 33% before and after using the balance algorithm, respectively, and the position deviation predicted accuracy with the 5 nt position deviation are 90% and 100%. The position deviation predicted accuracy of the top five candidates is 66% and 68%, and the position deviation predicted accuracy with the 5 nt position deviation is 99% and 100%. After using the balance algorithm, the APD of the top five candidates are 1.62 nt and 1.24 nt. Therefore, from the point of view of various indicators, the performance of the classifier using the balance method has been greatly improved.
Comparison with other methods
Taking Test1 as an example, four methods that were developed to identify the mature miRNAs were compared with our method. The position deviation predicted accuracy of MiRPara, MatureByes, MiRdup, MatPred and MatFinder is shown in Fig. 6 and Table 13.

The position deviation predicted accuracy of MiRPara, MatureByes, MiRdup, MatPred and MatFinder.
The prediction accuracy of MatFinder, MiRPara, MatureByes, MiRdup and MatPred, for the first candidate was 4%, 9%, 26%, 30% and 33%, respectively. Our method, MatFinder was 29%, 24%, 7% and 3% higher than that of the other three methods. With the 5 nt position deviation, the prediction accuracy was 37%, 84%, 81%, 90% and 100%, and MatFinder was higher than the other three methods. In addition, the average position deviation is 5.43 nt, 4.65 nt, 2.67 nt, 2.45 nt and 2.05 nt, respectively. Above all, the MatFinder method is significantly superior to the other methods in the various indices.
The performance of classifiers on all sites identification
Due to the excellent performance of MatFinder for the P5_5 site, other sites identification methods were trained based on different datasets to accomplish all sites identification. The performance of the P5_3 classifier was test based on Test1 and Test2, and the performance of the P3_3 and P3_5 classifiers were investigated using Test2 and Test3.
The position deviation predicted accuracy of the top five candidates of mature miRNA for all sites is shown in Table 14. The position deviation predicted accuracy of the P5_3, P3_5 and P3_3 classifiers was 66%, 55% and 67%, respectively. With the increase in the deviation nucleotide distance, the accuracy improved greatly. Within a 1 nt deviation, the accuracy was 92%, 89% and 84%, respectively. Within a 5 nt deviation, the recognition accuracy reached 100%, 100% and 98%, respectively.
Discussion and Conclusion
MiRBase data sets consists computational predictions as well, but the computational predictions had the canonical biological characters, they will not affect our prediction results. Therefore, in our train and test dataset, we choose all the pre-miRNAs from miRBase, the experimental and computational annotations in miRBase are not separate out in our data processing.
The results show that our method, Matfinder, is superior to the other methods in identification accuracy and average position deviation. These results can be explained as follows:
The feature extraction methods based on sequence and structured-sequence were designed to investigate the biological significant of absence information. On basis of the comparison results, the structured-sequence-based method obtained the better classification results. This shows that structured-sequence-based features can represent mature miRNAs biological characteristics. It also illustrates the importance of the miRNAs structural biological characteristics. The secondary structure of miRNAs is divided into two parts, one part is the base complement of each other, the other part is isolated from the double helix region without base pairs, namely, the loop, which primarily includes: hairpin loops, the inner loops and multibranch loops. The extracted-features capture these characteristics of loops to a greater extent, so the identification accuracy is improved to a certain extent.
MatFinder is proposed based on the integration method, which is a strong classifier using AdaBoost and a weak classifier using the adjustable parameter SVM algorithm. Aiming to improve the data imbalance problem, the K-means algorithm is used to construct the center of mass of closer samples. To achieve the balanced training subsets of the data, in the process of the integrated classifier, and focus on incorrectly classified samples, the integrated results use common decision strategies of the weak classifier with different weights. This method not only solves the problem of monotone sequence diversity in mature miRNA identification but also improves the performance of the classifier.
The weak classifier is constructed using the SVM algorithm based on adjustable probability parameters. First, the SVM algorithm solves the problem of “overlearning” of the AdaBoost algorithm. Second, the SVM algorithm is based on the probability model, and the results provided five mature miRNA candidates. Importantly, in the process of integration, multiple weak classifiers based on SVM can be regulated by adjusting C and g to learn the complexity and classifier performance, which overcomes the effect of the static parameters on the performance of classifiers. The weak classifiers were trained alone with independent parameters.
In addition, we trained the P5_3, P3_5 and P3_3 classifiers based on the different biological characters. These classifiers are more accurate in identifying the first five candidates.
References
Roberts, T. C. The MicroRNA Biology of the Mammalian Nucleus. Molecular therapy. Nucleic acids 3, e188, https://doi.org/10.1038/mtna.2014.40 (2014).
Jiang, Q. et al. miR2Disease: a manually curated database for microRNA deregulation in human disease. Nucleic acids research 37, D98–104, https://doi.org/10.1093/nar/gkn714 (2009).
Romero-Cordoba, S. L., Salido-Guadarrama, I., Rodriguez-Dorantes, M. & Hidalgo-Miranda, A. miRNA biogenesis: biological impact in the development of cancer. Cancer biology & therapy, 0, https://doi.org/10.4161/15384047.2014.955442 (2014).
Nam, J. W. et al. Human microRNA prediction through a probabilistic co-learning model of sequence and structure. Nucleic acids research 33, 3570–3581, https://doi.org/10.1093/nar/gki668 (2005).
Tijsterman, M. & Plasterk, R. H. Dicers at RISC; the mechanism of RNAi. Cell 117, 1–3 (2004).
Lee, Y. S. et al. Distinct roles for Drosophila Dicer-1 and Dicer-2 in the siRNA/miRNA silencing pathways. Cell 117, 69–81 (2004).
Brennecke, J., Stark, A., Russell, R. B. & Cohen, S. M. Principles of microRNA-target recognition. PLoS biology 3, e85, https://doi.org/10.1371/journal.pbio.0030085 (2005).
Kruger, J. & Rehmsmeier, M. RNAhybrid: microRNA target prediction easy, fast and flexible. Nucleic acids research 34, W451–454, https://doi.org/10.1093/nar/gkl243 (2006).
Stark, A., Brennecke, J., Russell, R. B. & Cohen, S. M. Identification of Drosophila MicroRNA targets. PLoS biology 1, E60, https://doi.org/10.1371/journal.pbio.0000060 (2003).
Okamura, K. et al. The regulatory activity of microRNA star species has substantial influence on microRNA and 3′ UTR evolution. Nat Struct Mol Biol 15, 354–363, https://doi.org/10.1038/Nsmb.1409 (2008).
Gkirtzou, K., Tsamardinos, I., Tsakalides, P. & Poirazi, P. MatureBayes: a probabilistic algorithm for identifying the mature miRNA within novel precursors. PLoS One 5, e11843, https://doi.org/10.1371/journal.pone.0011843 (2010).
Helvik, S. A., Snove, O. Jr. & Saetrom, P. Reliable prediction of Drosha processing sites improves microRNA gene prediction. Bioinformatics 23, 142–149, https://doi.org/10.1093/bioinformatics/btl570 (2007).
Wu, Y., Wei, B., Liu, H., Li, T. & Rayner, S. MiRPara: a SVM-based software tool for prediction of most probable microRNA coding regions in genome scale sequences. BMC bioinformatics 12, 107, https://doi.org/10.1186/1471-2105-12-107 (2011).
Xuan, P., Guo, M., Huang, Y., Li, W. & Huang, Y. MaturePred: efficient identification of microRNAs within novel plant pre-miRNAs. PLoS One 6, e27422, https://doi.org/10.1371/journal.pone.0027422 (2011).
Li, J. et al. MatPred: Computational Identification of Mature MicroRNAs within Novel Pre-MicroRNAs. BioMed research international 2015, 546763, https://doi.org/10.1155/2015/546763 (2015).
Terai, G., Okida, H., Asai, K. & Mituyama, T. Prediction of Conserved Precursors of miRNAs and Their Mature Forms by Integrating Position-Specific Structural Features. Plos One 7, e44314, https://doi.org/10.1371/journal.pone.0044314.g001 (2012).
He, C. et al. Mature microRNA Sequence Prediction. Plos One 7, e51673, 10.1371/ (2012).
Leclercq, M., Diallo, A. B. & Blanchette, M. Computational prediction of the localization of microRNAs within their pre-miRNA. Nucleic acids research 41, 7200–7211, https://doi.org/10.1093/nar/gkt466 (2013).
Guan, D. G., Liao, J. Y., Qu, Z. H., Zhang, Y. & Qu, L. H. mirExplorer: detecting microRNAs from genome and next generation sequencing data using the AdaBoost method with transition probability matrix and combined features. RNA biology 8, 922–934, https://doi.org/10.4161/rna.8.5.16026 (2011).
Liu, B., Fang, L., Liu, F., Wang, X. & Chou, K. C. iMiRNA-PseDPC: microRNA precursor identification with a pseudo distance-pair composition approach. Journal of biomolecular structure & dynamics 34, 223–235, https://doi.org/10.1080/07391102.2015.1014422 (2016).
Liu, B. et al. Identification of microRNA precursor with the degenerate K-tuple or Kmer strategy. Journal of theoretical biology 385, 153–159, https://doi.org/10.1016/j.jtbi.2015.08.025 (2015).
Liu, B. et al. Identification of real microRNA precursors with a pseudo structure status composition approach. PloS one 10, e0121501, https://doi.org/10.1371/journal.pone.0121501 (2015).
Liu, B., Fang, L., Chen, J., Liu, F. & Wang, X. miRNA-dis: microRNA precursor identification based on distance structure status pairs. Molecular bioSystems 11, 1194–1204, https://doi.org/10.1039/c5mb00050e (2015).
Liu, B., Liu, F., Fang, L., Wang, X. & Chou, K. C. repRNA: a web server for generating various feature vectors of RNA sequences. Molecular genetics and genomics: MGG 291, 473–481, https://doi.org/10.1007/s00438-015-1078-7 (2016).
Liu, B. et al. Pse-in-One: a web server for generating various modes of pseudo components of DNA, RNA, and protein sequences. Nucleic acids research 43, W65–71, https://doi.org/10.1093/nar/gkv458 (2015).
Yates, L. A., Norbury, C. J. & Gilbert, R. J. The long and short of microRNA. Cell 153, 516–519, https://doi.org/10.1016/j.cell.2013.04.003 (2013).
Starega-Roslan, J. & Krzyzosiak, W. J. Analysis of microRNA length variety generated by recombinant human Dicer. Methods in molecular biology 936, 21–34, https://doi.org/10.1007/978-1-62703-083-0_2 (2013).
Tan, G. C. et al. 5′ isomiR variation is of functional and evolutionary importance. Nucleic Acids Res 42, 9424–9435, https://doi.org/10.1093/nar/gku656 (2014).
Kozomara, A. & Griffiths-Jones, S. miRBase: integrating microRNA annotation and deep-sequencing data. Nucleic Acids Res 39, D152–157, https://doi.org/10.1093/nar/gkq1027 (2011).
Auyeung, V. C., Ulitsky, I., McGeary, S. E. & Bartel, D. P. Beyond secondary structure: primary-sequence determinants license pri-miRNA hairpins for processing. Cell 152, 844–858, https://doi.org/10.1016/j.cell.2013.01.031 (2013).
Acknowledgements
This work was supported by the Youth Science Fund of Heilongjiang Province of China (No. QC2017079). This work was also supported in part by grants from The Fundamental Research Funds in Heilongjiang Provincial Universities (Nos: 135109246, 135209208) and the Science and Technology Project of Qiqihar (GYGG-201513, GYGG-201709).
Author information
Authors and Affiliations
Contributions
Jian Zhang contributed to developing the workflows and data analysis. Ying Wang developed the programs; Jidong Ru collected and collated data; Yueqiu Jiang participated in the writing the manuscript and interpreting the results. All authors read and approved the final manuscript and agreed to the submission.
Corresponding author
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Wang, Y., Ru, J., Jiang, Y. et al. Adaboost-SVM-based probability algorithm for the prediction of all mature miRNA sites based on structured-sequence features. Sci Rep 9, 1521 (2019). https://doi.org/10.1038/s41598-018-38048-7
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-018-38048-7
This article is cited by
-
Identification of novel rhesus macaque microRNAs from naïve whole blood
Molecular Biology Reports (2019)
